PENJAJARAN GLOBAL SEKUEN DNA MENGGUNAKAN
ALGORITME NEEDLEMAN-WUNSCH
AGUNG WIDYO UTOMO
DEPARTEMEN ILMU KOMPUTER
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM
INSTITUT PERTANIAN BOGOR
PERNYATAAN MENGENAI SKRIPSI DAN
SUMBER INFORMASI SERTA PELIMPAHAN HAK CIPTA
Dengan ini saya menyatakan bahwa skripsi berjudul Penjajaran Global Sekuen DNA Menggunakan Algoritme Needleman-Wunsch adalah benar karya saya dengan arahan dari komisi pembimbing dan belum diajukan dalam bentuk apa pun kepada perguruan tinggi mana pun. Sumber informasi yang berasal atau dikutip dari karya yang diterbitkan maupun tidak diterbitkan dari penulis lain telah disebutkan dalam teks dan dicantumkan dalam Daftar Pustaka di bagian akhir skripsi ini.
Dengan ini saya melimpahkan hak cipta dari karya tulis saya kepada Institut Pertanian Bogor.
Bogor, Juni 2013 Agung Widyo Utomo
ABSTRAK
AGUNG WIDYO UTOMO. Penjajaran Global Sekuen DNA Menggunakan Algoritme Needleman-Wunsch. Dibimbing oleh WISNU ANANTA KUSUMA.
Tujuan penjajaran global sekuen DNA adalah mencari kemiripan dua buah sekuen DNA dengan memeriksa kecocokan seluruh nukleotida dari dua buah sekuen DNA tersebut. Penelitian ini mengimplementasikan penjajaran global menggunakan algoritme Needleman-Wunsch pada sekuen genom lengkap dari mitokondria Ancylostoma duodenale (NC_003415.1) dan sekuen genom lengkap dari mitokondria Necator americanus (NC_003416.2). Hasil penjajaran memperlihatkan bahwa kemiripan antara dua sekuen DNA tersebut adalah 83.7 % dengan gap sebesar 6.5%. Pengujian selanjutnya dilakukan pada sekuen genom lengkap dari Human papillomavirus type 134 (NC_014956.1) dan sekuen genom lengkap dari Human papillomavirus type 132 (NC_014955.1). Hasil penjajaran menunjukkan bahwa kemiripan antara dua sekuen tersebut adalah 62.9% dengan gap sebesar 23.5%. Kedua hasil penjajaran tersebut menunjukkan bahwa penjajaran dengan menggunakan algoritme Needleman-Wunsch menghasilkan nilai kemiripan yang lebih tinggi dibandingkan dengan menggunakan algoritme penjajaran global GSA tree dan super pairwise alignment.
Kata kunci: penjajaran global, penjajaran sekuen, Needleman-Wunsch
ABSTRACT
AGUNG WIDYO UTOMO. Global Alignment of DNA Sequence Using Needleman-Wunsch Algorithm. Supervised by WISNU ANANTA KUSUMA.
Global alignment of DNA sequence aims to determine similarity between two DNA sequences by measuring the matching region which involves the overall nucleotides of two DNA sequences. This research implements the global alignment using Needleman-Wunsch algorithm on the sequence of Ancylostoma duodenale mitochondrion, complete genome (NC_003415.1) and the sequence of Necator americanus mitochondrion, complete genome (NC_003416.2). The result shows that the similarity of these sequences is 83.7% with 6.5% gaps . The second experiment is performed using the sequence of Human papillomavirus type 134, complete genome (NC_014956.1 ) and the sequence of Human papillomavirus type 132, complete genome (NC_014955.1). The result shows that the similarity is 62.9% and gaps of 23.5%. Both results conclude that the Needleman-Wunsch could obtain higher similarity than those of GSA tree and super pairwise alignment.
Skripsi
sebagai salah satu syarat untuk memperoleh gelar Sarjana Komputer
pada
Departemen Ilmu Komputer
PENJAJARAN GLOBAL SEKUEN DNA MENGGUNAKAN
ALGORITME NEEDLEMAN-WUNSCH
AGUNG WIDYO UTOMO
DEPARTEMEN ILMU KOMPUTER
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM
INSTITUT PERTANIAN BOGOR
Judul Skripsi : Penjajaran Global Sekuen DNA Menggunakan Algoritme Needleman-Wunsch
Nama : Agung Widyo Utomo
NIM : G64104022
Disetujui oleh
Dr Wisnu Ananta Kusuma, ST, MT Pembimbing
Diketahui oleh
Dr Ir Agus Buono, MSi, MKom Ketua Departemen
PRAKATA
Puji dan syukur penulis panjatkan ke hadirat Allah subhanahu wata’ala, yang telah memberikan nikmat yang begitu banyak, sehingga penulis dapat menyelesaikan penelitian dan tulisan ini. Shalawat dan salam penulis sampaikan kepada Nabi Muhammad shallallahu ‘alaihi wasallam, keluarganya, sahabatnya, serta umatnya hingga akhir zaman. Tulisan ini merupakan hasil penelitian yang penulis lakukan sejak Agustus 2012 hingga Mei 2013. Tulisan ini mengambil topik Bioinformatika, dan bertujuan menerapkan algoritme Needleman-Wunsch pada penjajaran global sekuen DNA.
Tak lupa penulis mengucapkan terima kasih kepada seluruh pihak yang telah berperan dalam penelitian ini, yaitu:
1 Istri tercinta Ida Hayati Sholihah, Ayahanda Budi Wiratno, Ibunda Sulastri, serta kakak Estiwi Retno Utami atas kasih sayang, doa, semangat, dan dorongan kepada penulis agar dapat segera menyelesaikan penelitian ini. 2 Bapak Dr Wisnu Ananta Kusuma, ST, MT selaku dosen pembimbing, yang
telah memberikan banyak ide, masukan, dan dukungan kepada penulis. 3 Bapak Irman Hermadi, SKom, MS dan Bapak Toto Haryanto, SKom, MSi,
yang telah bersedia menjadi penguji.
4 Bapak Drs Priyambodo Rahardjo selaku atasan yang telah memberikan izin, mendukung dan memotivasi dalam menyelesaikan pendidikan di alih jenis Ilmu Komputer IPB.
6 Rekan satu bimbingan: Fariz Azhar, Bernita Sinurat, Alharis Tamsin, Fitria, dan Galih yang saling berbagi ide dan saling memotivasi selama pengerjaan skripsi.
7 Rekan-rekan DKM Alghifari IPB, FOKUS IPB, FRASE, ROHIS 58 dan ROHIS 9 Jakarta atas doa dan dukungannya.
8 Rekan-rekan BPPT Enjiniring atas perhatian dan motivasinya. 9 Pihak-pihak lain yang tidak dapat penulis sebutkan satu persatu.
Penulis berharap penelitian dan tulisan ini dapat memberikan manfaat untuk kemajuan masyarakat Indonesia.
Bogor, Juni 2013
DAFTAR ISI
DAFTAR TABEL vi
DAFTAR GAMBAR vi
DAFTAR LAMPIRAN vi
PENDAHULUAN 1
Latar Belakang 1
Tujuan Penelitian 2
Manfaat Penelitian 2
Ruang Lingkup Penelitian 2
METODE 2
Penyiapan Data 3
Algoritme Penjajaran Global Needleman-Wunsch 4
Analisis dan Evaluasi 6
Lingkungan Pengembangan 6
HASIL DAN PEMBAHASAN 6
Implementasi Algoritme Penjajaran Global Needleman-Wunsch 6
Analisis dan Evaluasi 7
SIMPULAN DAN SARAN 9
Simpulan 9
Saran 10
DAFTAR PUSTAKA 10
LAMPIRAN 11
RIWAYAT HIDUP 16
DAFTAR TABEL
1 Hasil pengujian pertama 7
2 Hasil pengujian kedua 7
DAFTAR GAMBAR
1 Contoh penjajaran global dan penjajaran lokal 1
2 Diagram alur metode penelitian 3
3 Contoh data berformat FASTA 3
4 Contoh inisialisasi matriks penskoran 4
5 Contoh matriks penskoran yang telah terisi 5
6 Contoh traceback 5
7 Perbandingan pengujian pertama algoritme Needleman-Wunsch
dengan GSA tree, SPA, dan aplikasi EMBOSS 8
8 Perbandingan pengujian kedua algoritme Needleman-Wunsch dengan
GSA tree, SPA, dan aplikasi EMBOSS 8
9 Grafik waktu eksekusi terhadap panjang sekuen 9
DAFTAR LAMPIRAN
1 Pseudocode algoritme Needleman-Wunsch 11
2 User interface aplikasi penjajaran global 12 3 Perbandingan hasil pengujian pertama dengan metode lain 13 4 Perbandingan hasil pengujian kedua dengan metode lain 14
PENDAHULUAN
Latar Belakang
Salah satu bagian terpenting dari sel yang menentukan karakteristik makhluk hidup adalah DNA (deoxyribo nucleic acid). DNA merupakan rantai ganda dari molekul sederhana (nukleotida) yang diikat bersama-sama dalam struktur helix yang dikenal dengan double helix. Nukleotida-nukleotida tersebut tersusun atas empat basa nitrogen yaitu adenine, cytosine, guanine dan thymine yang dinotasikan dalam abjad A, C, G, dan T (Annibal 2003).
Salah satu cara untuk menganalisis DNA adalah melalui penjajaran sekuen (sequence alignment). Tujuan penjajaran sekuen adalah mencari sebanyak mungkin kecocokan pada setiap subsekuen yang identik, sehingga dapat dianalisis dan disimpulkan kemiripan dua sekuen tersebut melalui nilai penjajarannya.
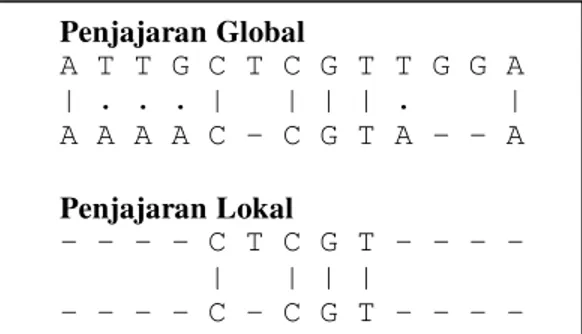
Penjajaran sekuen DNA dapat dilakukan dengan dua cara yaitu penjajaran global (global alignment) dan penjajaran lokal (local alignment). Penjajaran global dilakukan dengan melibatkan keseluruhan nukleotida dalam sekuen DNA. Adapun penjajaran lokal hanya hanya melibatkan daerah tertentu dari sekuen DNA yang memberikan nilai penjajaran paling tinggi. Perbedaan penjajaran global dan penjajaran lokal dapat dilihat pada Gambar 1.
Gambar 1 Contoh penjajaran global dan penjajaran lokal
Penelitian penjajaran global sekuen DNA telah banyak dilakukan sebelumnya, antara lain penelitian yang dilakukan oleh Safi’i (2011) dan Pantua (2011). Data yang digunakan untuk percobaan pertama adalah sekuen genom lengkap dari mitokondria Ancylostoma duodenale (NC_003415.1), dan sekuen genom lengkap dari mitokondria Necator americanus (NC_003416.2). Data yang digunakan untuk percobaan kedua adalah sekuen genom lengkap dari Human papillomavirus type 134 (NC_014956.1) dan sekuen genom lengkap dari Human papillomavirus type 132 (NC_014955.1). Hasil penjajaran dari kedua penelitian tersebut divalidasi oleh aplikasi EMBOSS (European Molecular Biology Open Software Suite) dengan tipe Needle yang menggunakan algoritme Needleman-Wunsch (Palmenberg dan Sgro 2008).
Safi’i (2011) menggunakan metode dengan pendekatan heuristic yang terdiri atas tiga bagian yaitu algoritme penjajaran sederhana, algoritme perluasan untuk pencarian substring umum terpanjang, dan graphical simple alignment tree (GSA tree) yang menggunakan penelusuran post-order traversal (Qi et al. 2010). Simpulan yang diperoleh adalah metode GSA tree tidak selalu memberikan hasil
2
penjajaran global yang optimal karena dipengaruhi oleh inisialisasi awal penjajaran sekuen dan pemilihan parameter.
Sementara itu, Pantua (2011) menggunakan metode SPA (super pairwise alignment) yang menggabungkan metode probabilitas dan analisis kombinatorial (Shen et al. 2002). Simpulan yang diperoleh adalah penggunaan metode SPA memiliki kelemahan dalam pemilihan parameter yang mengakibatkan hasil berbeda-beda dan tidak optimal.
Penelitian ini akan menerapkan algoritme klasik penjajaran global yaitu algoritme Needleman-Wunsch dan membandingkannya dengan metode GSA tree dan SPA. Diharapkan dengan penilitian ini diperoleh hasil penjajaran yang lebih optimal.
Tujuan Penelitian
Tujuan dari penelitian ini adalah untuk :
1 Menerapkan algoritme Needleman-Wunsch pada penjajaran global sekuen DNA.
2 Membandingkan performa algoritme Needleman-Wunsch dengan metode GSA tree dan SPA.
Manfaat Penelitian
Manfaat dari penelitian ini adalah untuk mendukung penelitian di bidang Bioinformatika yang memerlukan informasi dari hasil penjajaran global sekuen DNA seperti perancangan primer, pengidentifikasian single nucleotide polymorphism (SNP), dan lain-lain.
Ruang Lingkup Penelitian
Pada penelitian ini dilakukan pembatasan masalah antara lain:
1 Data yang digunakan adalah data penelitian yang digunakan oleh Safi’i (2011) dan Pantua (2011).
2 Data yang digunakan berformat FASTA.
3 Ukuran masing-masing sekuen DNA yang digunakan maksimal 15 000 bp (base pair).
METODE
3
Gambar 2 Diagram alur metode penelitian
Penyiapan Data
Data yang digunakan pada penelitian ini berasal dari GenBank yang diunduh dari situs resmi National Centre for Biotechnology Information (NCBI). Data tersebut berformat FASTA seperti contoh pada Gambar 3. Data berformat FASTA terdiri atas baris pertama berupa simbol “ > ” yang diikuti identifier dari sekuen dan baris kedua berupa data sekuen.
Gambar 3 Contoh data berformat FASTA
Untuk membaca file berformat FASTA, diperlukan proses parsing. Pada proses ini, program akan mengidentifikasi keberadaan simbol “>” sebagai tanda bahwa baris tersebut merupakan baris identifier. Kemudian program akan
>gi|19073878|ref|NC_003415.1| Ancylostoma duodenale mitochondrion, complete genome
4
mengidentifikasi keberadaan baris baru sebagai tanda dimulainya baris data sekuen. Data sekuen inilah yang akan digunakan untuk penjajaran global sekuen DNA.
Penelitian ini menggunakan data yang sama dengan penelitian Safi’i (2011) dan Pantua (2011) yaitu sekuen genom lengkap dari mitokondria Ancylostoma duodenale (NC_003415.1) dengan panjang sekuen 13 721 bp, mitokondria Necator americanus (NC_003416.2) dengan panjang sekuen 13 605 bp, Human papillomavirus type 134 (NC_014956.1) dengan panjang sekuen 7 309 bp, dan Human papillomavirus type 132 (NC_014955.1) dengan panjang sekuen 7 125 bp.
Algoritme Penjajaran Global Needleman-Wunsch
Algoritme ini ditemukan oleh Needleman dan Wunsch (1970) yang digunakan untuk menemukan penjajaran global yang memiliki nilai optimal dari dua buah sekuen. Algoritme Needleman-Wunsch menghitung semua informasi yang terdapat pada dua sekuen sehingga jika kedua sekuen itu berukuran n, maka kompleksitas waktunya adalah O(n2). Selain itu algoritme ini menyimpan seluruh matriks pada memori sehingga kompleksitas ruangnya juga kuadratik (Annibal 2003). Untuk mencari penjajaran global terbaik pada algoritme ini digunakan matriks penskoran (scoring matrix). Algoritme ini dibagi menjadi 3 tahap, yaitu : 1 Inisialisasi
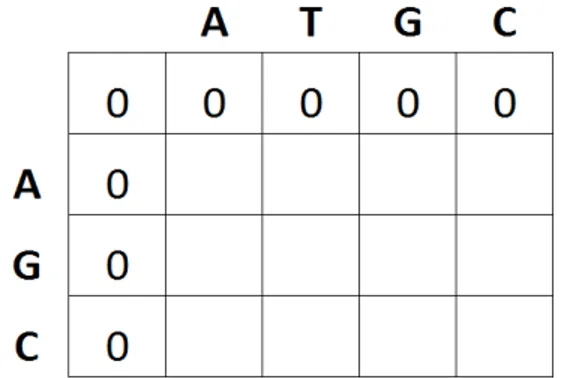
Pada tahap ini dilakukan pemberian nilai awal pada matriks penskoran M[i,j]. Jika panjang query sequence adalah m dan panjang reference sequence adalah n, maka matriks penskoran M[i,j] tersebut berukuran (m+1)×(n+1). Selanjutnya baris dan kolom pertama disi dengan nilai gap penalty. Gap penalty adalah nilai yang diperoleh ketika membandingkan karakter dengan karakter kosong (gap). Pada penelitian ini ditentukan gap penalty bernilai 0. Contoh inisialisasi awal matriks penskoran dapat dilihat pada Gambar 4.
Gambar 4 Contoh inisialisasi matriks penskoran 2 Pengisian Matriks
5 M[i-1,j] + w
M[i,j] = Max M[i,j-1] + w M[i-1,j-1] + S[i,j]
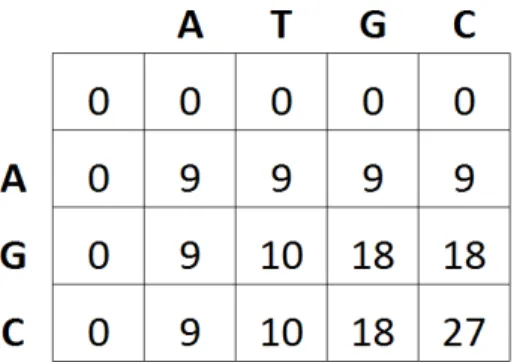
di mana S[i,j] adalah match/mismatch score, w adalah konstanta gap penalty dan M[i,j] adalah matriks penskoran yang akan diisi nilai yang diperoleh dari ketentuan di atas. Nilai penskoran yang digunakan pada penelitian ini adalah match = 9, mismatch = 1, dan gap = 0. Karena akan dilakukan perbandingan dengan metode yang digunakan oleh Safi’i (2011) dan Pantua (2011) maka nilai match dan mismatch yang digunakan sama dengan yang digunakan pada penelitian Safi’i (2011) dan Pantua (2011). Contoh matriks penskoran yang telah terisi dapat dilihat pada Gambar 5.
Gambar 5 Contoh matriks penskoran yang telah terisi 3 Traceback
Traceback merupakan tahap menyusun jalur dari matriks penskoran (scoring matrix) yang telah berisi nilai-nilai pada langkah sebelumnya. Jalur tersebut disusun dari matriks M(m+1, n+1) sampai dengan M(0,0) sehingga memiliki nilai penskoran yang maksimum. Contoh traceback dapat dilihat pada Gambar 6.
Gambar 6 Contoh traceback
6
1 Length, yaitu panjang penjajaran yang terbentuk.
2 Similaritiy yang merepresentasikan jumlah sekuen yang match dan persentase kemiripan dua sekuen yang dijajarkan dari panjang penjajaran yang terbentuk.
3 Gaps yang mewakili persentase kemunculan gap dari panjang alignment yang terbentuk.
4 Score, yaitu total nilai hasil penjajaran.
5 Execution time, yaitu waktu yang dibutuhkan untuk mengeksekusi algoritme Needleman-Wunsch.
6 Visualisasi hasil penjajaran dua sekuen.
7 Grafik yang menampilkan hubungan match antara reference sequence dengan query sequence.
Analisis dan Evaluasi
Hasil yang diperoleh dianalisis dan dievaluasi kinerjanya dengan membandingkan dengan hasil penelitian lain yang menggunakan penjajaran global yaitu penelitian Safi’i (2011) dan Pantua (2011). Analisis berikutnya dilakukan pada penjajaran sekuen DNA menggunakan Needleman-Wunsch yang datanya dibangkitkan secara acak dari karakter A, C, G, dan T. Dari penjajaran tersebut dibandingkan panjang sekuen dengan waktu eksekusinya untuk melihat hubungannya dengan kompleksitas algoritme.
Lingkungan Pengembangan
Penelitian ini dibangun dengan menggunakan bahasa pemrograman Visual Basic .NET dan didukung perangkat lunak dan perangkat keras dengan spesifikasi sebagai berikut :
Perangkat Lunak :
Sistem operasi Microsoft Windows 7 Microsoft Visual Studio 2005
Perangkat Keras :
Intel Atom Dual-Core N570 @1.67 GHz Memory 2 GB RAM
Harddisk dengan kapasitas sisa 100 GB Monitor resolusi 1366 x 768 pixel Mouse dan keyboard
HASIL DAN PEMBAHASAN
Implementasi Algoritme Penjajaran Global Needleman-Wunsch
7 Lampiran 1. User interface aplikasi yang telah dibangun dapat dilihat pada Lampiran 2. Pengujian pada aplikasi dilakukan sebanyak dua kali.
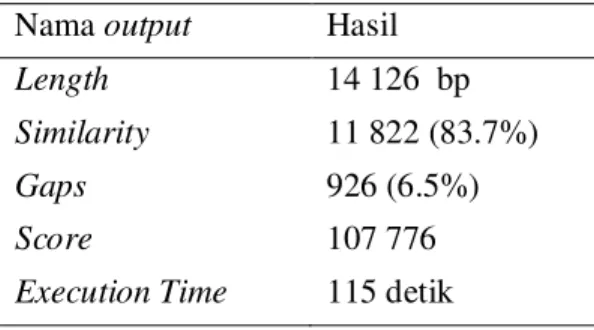
Pengujian pertama dilakukan penjajaran sekuen genom lengkap dari mitokondria Ancylostoma duodenale (NC_003415.1) sebagai reference sequence dengan panjang sekuen 13 721 bp dan sekuen genom lengkap dari mitokondria Necator americanus (NC_003416.2) sebagai query sequence dengan panjang sekuen 13 605 bp. Hasil dari penjajaran tersebut dapat dilihat pada Tabel 1.
Tabel 1 Hasil pengujian pertama
Pengujian kedua dilakukan penjajaran sekuen genom lengkap dari Human papillomavirus type 134 (NC_014956.1) sebagai reference sequence dengan panjang 7 309 bp dan sekuen genom lengkap dari Human papillomavirus type 132 (NC_014955.1) sebagai query sequence dengan panjang 7 125 bp. Hasil pengujian kedua dapat dilihat pada Tabel 2.
Tabel 2 Hasil pengujian kedua Nama output Hasil
Length 8 180 bp Similarity 5 146 (62.9%) Gaps 1 926 (23.5%)
Score 47 422
Execution Time 29 detik
Analisis dan Evaluasi
Pada tahap ini hasil pengujian dibandingkan dengan hasil penjajaran global yang telah dilakukan oleh Safi’i (2011) dan Pantua (2011). Safi’i (2011) menggunakan metode GSA tree dan Pantua (2011) menggunakan metode SPA. Kedua penelitian tersebut menerapkan dua kombinasi parameter yang berbeda di setiap pengujian. Kemudian kedua metode tersebut divalidasi oleh aplikasi EMBOSS Needle. Sayangnya penelitian Safi’i (2011) dan Pantua (2011) hanya menyajikan hasil similaritas dan gaps saja sehingga hanya kedua hal tersebut yang dapat dibandingkan. Hasil perbandingan Needleman-Wunsch dengan GSA tree, SPA, dan aplikasi EMBOSS secara lengkap disajikan pada Lampiran 2 untuk
Nama output Hasil Length 14 126 bp Similarity 11 822 (83.7%)
Gaps 926 (6.5%)
Score 107 776
8
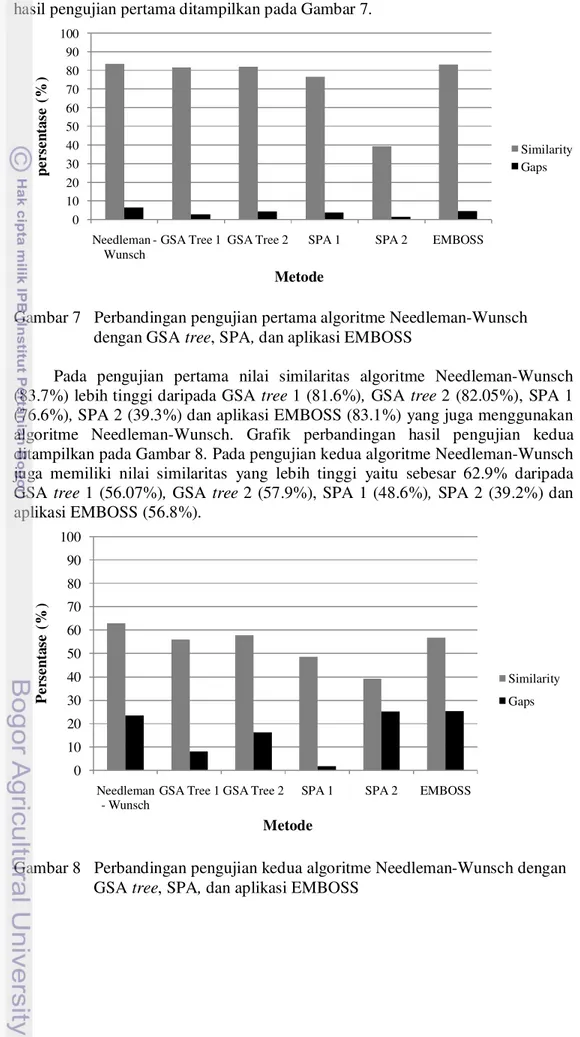
pengujian pertama dan Lampiran 3 untuk pengujian kedua. Grafik perbandingan hasil pengujian pertama ditampilkan pada Gambar 7.
Gambar 7 Perbandingan pengujian pertama algoritme Needleman-Wunsch dengan GSA tree, SPA, dan aplikasi EMBOSS
Pada pengujian pertama nilai similaritas algoritme Needleman-Wunsch (83.7%) lebih tinggi daripada GSA tree 1 (81.6%), GSA tree 2 (82.05%), SPA 1 (76.6%), SPA 2 (39.3%) dan aplikasi EMBOSS (83.1%) yang juga menggunakan algoritme Needleman-Wunsch. Grafik perbandingan hasil pengujian kedua ditampilkan pada Gambar 8. Pada pengujian kedua algoritme Needleman-Wunsch juga memiliki nilai similaritas yang lebih tinggi yaitu sebesar 62.9% daripada GSA tree 1 (56.07%), GSA tree 2 (57.9%), SPA 1 (48.6%), SPA 2 (39.2%) dan aplikasi EMBOSS (56.8%).
Gambar 8 Perbandingan pengujian kedua algoritme Needleman-Wunsch dengan GSA tree, SPA, dan aplikasi EMBOSS
9 Kedua pengujian tersebut menunjukkan algoritme Needleman-Wunsch memiliki similaritas yang lebih unggul dari GSA tree dan SPA. Dengan demikian dapat dikatakan bahwa algoritme Needleman-Wunsch adalah yang paling optimal dalam penjajaran global sekuen DNA. Hal ini terjadi karena algoritme Needleman-Wunsch menggunakan seluruh informasi yang terdapat pada dua sekuen sehingga hasil penjajaran lebih optimal. Namun karena algoritme ini melakukan penjajaran dengan melibatkan seluruh nukleotida, maka waktu eksekusinya menjadi lambat.
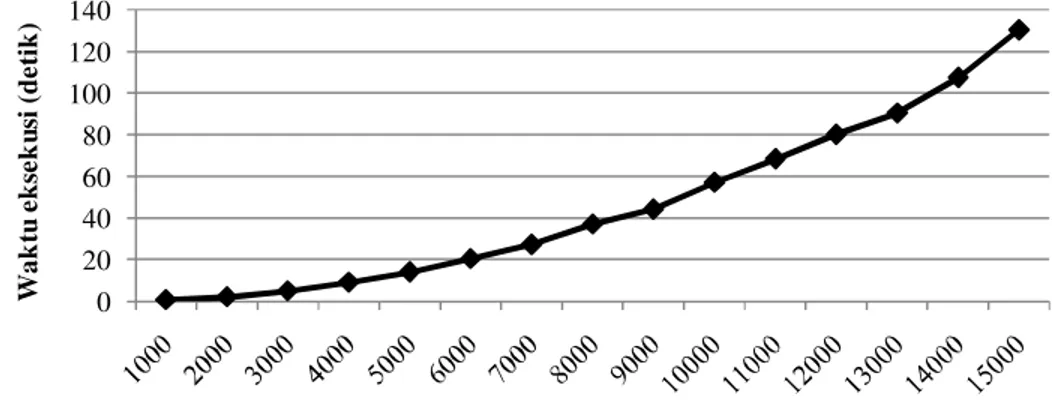
Waktu eksekusi algoritme Needleman-Wunsch pada sekuen yang datanya dibangkitkan secara acak dapat dilihat pada Lampiran 5. Perbandingan antara banyak sekuen dan waktu eksekusi dapat dilihat pada Gambar 9. Grafik tersebut menunjukkan bahwa bertambahnya panjang sekuen mengakibatkan waktu eksekusi meningkat dengan tendensi kuadratik. Hal ini sesuai dengan kompleksitas waktu algoritme Needleman-Wunsch yaitu O(n2).
Gambar 9 Grafik waktu eksekusi terhadap panjang sekuen
SIMPULAN DAN SARAN
Simpulan
Dari penelitian yang telah dilakukan dalam penjajaran global sekuen DNA menggunakan algoritme Needleman-Wunsch ini dapat disimpulkan sebagai berikut :
1 Penerapan algoritme Needleman Wunsch pada penjajaran global sekuen DNA mendapatkan hasil similaritas yang tertinggi dibandingkan GSA tree, SPA (super pairwise alignment), dan aplikasi EMBOSS. Dengan demikian dapat dikatakan algoritme Needleman-Wunsch memiliki hasil penjajaran paling optimal.
2 Bertambahnya panjang sekuen mengakibatkan waktu eksekusi meningkat secara kuadratik sesuai dengan kompleksitas waktu algoritme Needleman-Wunsch.
10
Saran
Untuk penelitian selanjutnya disarankan sebagai berikut:
1 Menggunakan algoritme Needleman-Wunsch yang telah dikembangkan, menerapkan matriks BLOSUM, parameter gap opening dan gap extension. 2 Membandingkan waktu eksekusi dengan metode lain.
3 Membuat phylogenetic tree dari multiple alignment menggunakan algoritme Needleman-Wunsch dipadukan dengan metode center star.
DAFTAR PUSTAKA
Annibal S. 2003. Sequence alignment algorithms [tesis]. London (GB): School of Physical Sciences and Engineering King's College.
Needleman SB, Wunsch CD. 1970. A general method applicable to search for similarities in the amino acid sequence of two proteins. Journal of Molecular Biology. 48:443-453.
Palmenberg A, Sgro JY. 2008. Biochemistry 711 : EMBOSS Software for Sequence Analysis. Madison (US) : University of Wisconsin.
Pantua A. 2011. Implementasi super pairwise alignment pada global alignment [skripsi]. Surabaya (ID): Institut Teknologi Sepuluh November.
Qi ZH, Qi XQ, Liu CC. 2010. New method for global alignment of 2 DNA sequences by the tree data structure. Journal of Theoretical Biology. 263(2):227-236. doi: 10.1016/j.jtbi.2009.12.012.
Safi’i M. 2011. Implementasi pensejajaran global sekuen DNA menggunakan GSA tree [skripsi]. Surabaya (ID): Institut Teknologi Sepuluh November. Shen SY, Adam Y, Hwang PI, Yang J. 2002. Super pairwise alignment (SPA): an
11 Lampiran 1 Pseudocode algoritme Needleman-Wunsch
/*Inisialisasi for i=0 to length(seqA) M(i,0) ← 0
for j=0 to length(seqB) M(0,j) ← 0
12
13 Lampiran 3 Perbandingan hasil pengujian pertama dengan metode lain
Metode Parameter Hasil
Needleman-Wunsch Match = 9 Mismatch = 1 Gap = 0
Similarity: 11 822 (83.7%) Gaps: 926 (6.5%)
14
Lampiran 4 Perbandingan hasil pengujian kedua dengan metode lain
Metode Parameter Hasil
Needleman-Wunsch Match = 9 Mismatch = 1
15 Lampiran 5 Panjang sekuen terhadap waktu eksekusi
Reference sequence
Query sequence
Waktu eksekusi (detik)
16
RIWAYAT HIDUP
Penulis dilahirkan di Jakarta pada tanggal 25 Mei 1989 sebagai anak kedua dari pasangan Budi Wiratno dan Sulastri. Penulis merupakan lulusan SMA Negeri 58 Jakarta (2004 - 2007), SLTP Negeri 9 Jakarta (2001 - 2004) dan SD Negeri 03 Ciracas Pagi (1995 - 2001).
Pada tahun 2007 penulis diterima sebagai mahasiswa Diploma III Program Keahlian Manajemen Informatika, Direktorat Program Diploma Institut Pertanian Bogor angkatan 44 melalui jalur Undangan Seleksi Masuk IPB (USMI). Setelah menyelesaikan pendidikan Diploma III pada tahun 2010, penulis kembali melanjutkan pendidikan Strata 1 (S1) melalui jalur Alih Jenis dan diterima sebagai mahasiswa Departemen Ilmu Komputer, Fakultas Matematika dan Ilmu Pengetahuan Alam, Institut Pertanian Bogor.