PARALELISASI MULTIPLE SEQUENCE ALIGNMENT
ASRIL ADI SUNARTO
SEKOLAH PASCASARJANA INSTITUT PERTANIAN BOGOR
PERNYATAAN MENGENAI TESIS DAN
SUMBER INFORMASI SERTA PELIMPAHAN HAK CIPTA
Dengan ini saya menyatakan bahwa tesis berjudul Paralelisasi Multiple Sequence Alignment adalah benar karya saya dengan arahan dari komisi pembimbing dan belum diajukan dalam bentuk apa pun kepada perguruan tinggi mana pun. Sumber informasi yang berasal atau dikutip dari karya yang diterbitkan maupun tidak diterbitkan dari penulis lain telah disebutkan dalam teks dan dicantumkan dalam Daftar Pustaka di bagian akhir tesis ini.
Dengan ini saya melimpahkan hak cipta dari karya tulis saya kepada Institut Pertanian Bogor.
Bogor, Januari 2014 Asril Adi Sunarto
RINGKASAN
ASRIL ADI SUNARTO. PARALELLIZATION MULTIPLE SEQUENCE ALIGNMENT. Dibimbing oleh WISNU ANANTA KUSUMA dan HERU SUKOCO.
Sequence alignment (penjajaran sekuens) adalah salah satu problem biologi molekuler yang dapat dipecahkan dengan bantuan program komputer. Tujuan dari penjajaran sekuens adalah untuk mengidentifikasi daerah kemiripan pada Deoxyribonucleic acid (DNA), Ribonucleic acid (RNA), atau protein. Salah satu metode penjajaran sekuens adalah metode Needleman-Wunsch di mana penjajaran yang dilakukan melibatkan seluruh sekuen.
Metode Needleman-Wunsch mempunyai kompleksitas yang sangat tinggi yaitu O(n2). Dengan cara yang sama, multiple sequence alignment (MSA) melibatkan k sekuens yang diproses satu demi satu menggunakan metode Star Alignment yang memiliki kompleksitas O(k2 n2). Oleh karena itu, masalah utama dalam MSA adalah waktu komputasi. Namun, meskipun waktu komputasinya tinggi, metode Star Alignment tetap memberikan hasil penjajaran yang akurat. Tingginya waktu komputasi ini dapat diatasi dengan menerapkan prinsip komputasi paralel yang diimplementasikan pada beberapa komputer yang terhubung satu sama lain sebagai suatu sistem jaringan komputer.
Penelitian ini bertujuan untuk mempercepat proses MSA menggunakan metode Star Alignment dengan menerapkan prinsip komputasi paralel. Teknik komputasi paralel yang diusulkan diimplementasikan menggunakan Message Passing Interface (MPI). Selanjutnya kinerja metode ini dievaluasi dengan mengukur speed up. Hasil percobaan menunjukan bahwa paralelisasi MSA dengan metode Star Alignment dapat meningkatkan speed up 4-6 kali lipat lebih cepat dibandingkan dengan hanya menggunakan komputer dengan pemroses tunggal.
SUMMARY
ASRIL ADI SUNARTO. PARALELLIZATION MULTIPLE SEQUENCE ALIGNMENT. Supervised by WISNU ANANTA KUSUMA and HERU SUKOCO.
Sequence alignment (sequence alignment) is one of the problems in molecular biology that can be solved with the help of computer programs. The purpose of an alignment is to identify similarity region of sequence in Deoxyribonucleic acid (DNA), Ribonucleic acid (RNA), or protein. One of sequence alignment methods is Needleman-Wunsch which includes all sequences in its alignment.
The Needleman-Wunsch method has high computational complexity with value of O(n2). Similarly, multiple sequence alignment (MSA) that processes k sequences one by one by using Star Alignment method has complexity of O(k2n2). Therefore, the main problem in MSA using Star Alignment method is computational time. However, the Star Alignment method can still obtain high accuracy. The time consuming problem faced by Star Alignment method can be solved by applying parallel computing technique implemented using multiple computers working together as a network system.
This research focuses on finding a faster method to process MSA based on Star Alignment method using parallel computing. The proposed method is implemented using Message Passing Interface (MPI). The evaluation is conducted by calculating speed up. The experiment results show that the paralellization of the Star Alignment method can increase speed up 4-6 times faster than that of using single CPU.
© Hak Cipta Milik IPB, Tahun 2014
Hak Cipta Dilindungi Undang-Undang
Dilarang mengutip sebagian atau seluruh karya tulis ini tanpa mencantumkan atau menyebutkan sumbernya. Pengutipan hanya untuk kepentingan pendidikan, penelitian, penulisan karya ilmiah, penyusunan laporan, penulisan kritik, atau tinjauan suatu masalah; dan pengutipan tersebut tidak merugikan kepentingan IPB
Tesis
sebagai salah satu syarat untuk memperoleh gelar Magister Komputer
pada
Program Studi Ilmu Komputer
PARALELISASI MULTIPLE SEQUENCE ALIGNMENT
SEKOLAH PASCASARJANA INSTITUT PERTANIAN BOGOR
BOGOR 2014
Judul Tesis : Paralelisasi Multiple Sequence Alignment Nama : Asril Adi Sunarto
NIM : G651110131
Disetujui oleh Komisi Pembimbing
Dr Wisnu Ananta Kusuma, ST MT Ketua
Dr Heru Sukoco, SSi MT Anggota
Diketahui oleh
Ketua Program Studi Ilmu Komputer
Dr Wisnu Ananta Kusuma, ST MT
Dekan Sekolah Pascasarjana
Dr Ir Dahrul Syah, MScAgr
Tanggal Ujian:
(tanggal pelaksanaan ujian tesis)
Tanggal Lulus:
(tanggal penandatanganan tesis oleh Dekan Sekolah
PRAKATA
Puji dan syukur penulis panjatkan kepada Allah Subhanahu Wa Ta’ala atas segala karunia dan rahmat-Nya karya ilmiah ini berhasil diselesaikan. Tema yang dipilih dalam penelitian yang dilaksanakan sejak bulan November 2012 ini ialah komputer paralel pada Bioinformatika, dengan judul Paralelisasi Multiple Sequence Alignment.
Terima kasih penulis ucapkan kepada Bapak Dr Wisnu Ananta Kusuma, ST MT dan Dr Heru Sukoco, SSi MT selaku pembimbingyang telah banyak memberi saran. Disamping itu, penghargaan penulis sampaikan kepada keluarga besar mahasiswa Ilmu Komputer IPB angkatan 13 dan para staf tata usaha yang tidak bisa disebutkan satu demi satu yang telah membantu selama pengumpulan data. Ungkapan terima kasih juga disampaikan kepada ayah, ibu, serta seluruh keluarga, atas segala doa dan kasih sayangnya.
Semoga karya ilmiah ini bermanfaat.
DAFTAR ISI
DAFTAR TABEL vi
DAFTAR GAMBAR vi
DAFTAR LAMPIRAN vi
1 PENDAHULUAN 1
Latar Belakang 1
Perumusan Masalah 2
Tujuan Penelitian 2
Manfaat Penelitian 2
Ruang Lingkup Penelitian 2
2 TINJAUAN PUSTAKA 3
Sequence Alignment 3
Multiple Sequence Alignment (Star Alignment) 5
Komputer Paralel 6
3 METODE 7 Pembagian (partition) 7
Pengiriman sequence 8
Pair Wise Alignment 8
Center Star 8
Bahan 8
Alat 8
4 HASIL DAN PEMBAHASAN 9
Hasil 9
Pembahasan 16
5 SIMPULAN DAN SARAN 20
Simpulan 20
Saran 20
DAFTAR PUSTAKA 21
13
DAFTAR TABEL
1 Hasil Sequence Alignment antara Chlamydophila caviae dan
Alicyclobacillus acidocaldarius pada fragmen DNA 1-10 ... 3
2 Keterangan sequence alignment ... 4
3 Multiple sequence dari NCBI ... 8
4 Pemetaan pembagian sequence ... 9
5 Pemetaan pasangan sequence... 10
6 Hasil pair wise alignment ... 13
7 Rekapitulasi skor tiap sequence ... 13
8 Perbandingan kompleksitas processor tunggal dan multi processor ... 14
9 Perbandingan waktu antara satu processor dengan multi processor ... 15
10 Speed up program paralel ... 15
11 Efficiency program paralel ... 15
12 Pemetaan biaya komunikasi ... 17
13 Waktu eksekusi antara satu processor dengan beberapa processor ... 18
14 Pengukuran kinerja speed up dan efficiency desain ke dua ... 18
DAFTAR GAMBAR
1 Deoxyribonucleic Acid (DNA) ... 32 Algoritma Star Alignment (Gusfield, 1991) ... 5
3 Metode penelitian Paralelisasi MSA ... 7
4 Ilustrasi pembagian sequence ... 9
5 Langkah-langkah inisiasi matrik pair-wise alignment ... 11
6 Langkah-langkah pengisian matrik ... 11
7 Hasil akhir pengisian matrik ... 12
8 Traceback dan hasil sequence alignment ... 12
9 Speed up dari kompleksitas paralel ... 14
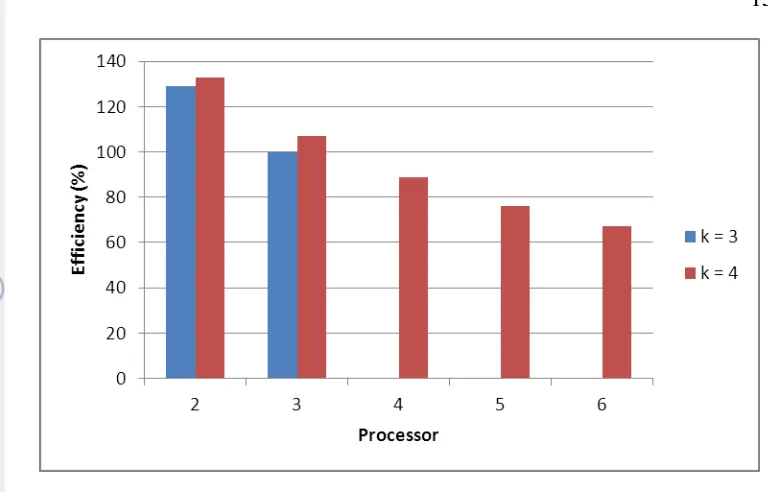
10 Efficiency dari kompleksitas paralel ... 15
11 Grafik speed up dari program paralel ... 16
12 Grafik efficiency dari program paralel ... 16
13 Paralelisasi MSA desain kedua ... 17
14 Grafik Speed up dan Efficiency program paralel desain ke dua ... 18
1
PENDAHULUAN
Latar Belakang
Sequence alignment (penjajaran sekuens) adalah salah satu problem biologi molekuler yang dapat dipecahkan dengan bantuan program komputer. Tujuan dari penjajaran sekuens adalah untuk mengidentifikasi daerah kemiripan pada Deoxyribonucleic acid (DNA), Ribonucleic acid (RNA), atau protein. Selanjutnya dari hasil identifikasi tersebut dapat ditentukan hubungan fungsional, struktural homologi antara dua sekuens atau lebih (Sing et al, 2011). Selain itu pada level aplikatif, hasil dari penjajaran banyak sekuens dapat digunakan untuk membangun phylogenetic tree, mengidentifikasi makna DNA seperti Single Nucleotide Polymorphysm (SNP) yang berfungsi untuk mendukung pemulian, penemuan obat (payne, 2007), perbaikan gen (Bronner et al, 1994, Papadopoulos et al, 1994).
Salah satu teknik penting dalam penjajaran sekuens yaitu pairwise alignment yang dilakukan engan menjajarkan suatu sekuens yang ingin dianalisis dengan sekuens referensi. Dua algoritme telah dikembangkan untuk menyelesaikan permasalahan pairwise alignment, yaitu Needleman-Wunsch untuk penjajaran global dan Smith-Waterman untuk penjajaran lokal. Penjajaran global bertujuan untuk menemukan kemiripan dari dua sekuens secara keseluruhan (Needleman dan Wunsch, 1970). Adapun penjajaran lokal hanya mempertimbangkan kemiripan subsekuens dari dua sekuens yang dijajarkan (Smith dan Waterman, 1984).
Metode Smit-Waterman dan Needleman-Wunsch, keduanya menggunakan dynamic programming. Dynamic programming merupakan metode yang mempunyai strategi memecah masalah menjadi lebih kecil dan menggunakan solusi yang lebih kecil tadi guna membangun solusi yang lebih besar (Jones dan Pevzner, 2004). Kompleksitas waktu menggunakan dynamic programming ini mencapai O(n2). Pairwise alignment ini menjadi dasar untuk melakukan Multiple Sequence Alignment (MSA) yang melibatkan banyak sekuens. Salah satu metode yang digunakan dalam MSA adalah Star Alignment. Metode yang diperkenalkan Gusfield (1991) ini melakukan pairwise alignment terhadap k sekuens untuk mencari sekuens yang memiliki tingkat kemiripan paling tinggi dengan sekuens lainnya. Sekuens ini menjadi star yang selanjutnya menjadi acuan untuk melakukan penjajaran dengan sekuens-sekuens lainnya. Kompleksitas metode ini masih tinggi yaitu O(k2 n2). Selanjutnya beberapa metode dikembangkan antara lain metode progresif yang diterapkan pada Clustal W dan Clustal X versi 2.0 (Larkin et al, 2007), MAFFT yang menerapkan Fast Fourier Transform (Katoh et al, 2002), metode Novel Center Star oleh Zou, Shan, dan Jiang (2012) dan metode probabilistic consistency menggunakan Pair Hidden Markov Model (Do et al, 2013).
2
X version 2.0 (Larkin et al, 2007). Tool ini merupakan pengembangan dari program sebelumnya yang diperkenal oleh Thompson, Higgins, Gibbon, dan Sharp(1988, 1994). Selain itu, Katoh et al (2002) juga memperkenalkan MAFFT yang menggunakan metode Fast Fourier Transform. Selanjutnya, MAFFT juga mengalami pengembangan dengan memparalelkan metode MAFF sebelumnya (Katoh and Toh, 2010). Tools terbaru yang disebut PAGAN dikemukakan oleh Loytynoja, Vilella, dan Goldman (2012) dengan metode progressive dan Novel Center star yang dikemukan oleh Zou, Shan, dan Jiang (2012) yang mengklaim 900 kali lipat dari hasil tool yang dihasilkan oleh Clustal.
Tingginya kompleksitas dalam memproses MSA ini yang mencapai O(k2 n2) menjadi masalah baru dalam pemrosesan penjajaran sekuens. Sama seperti Katoh dan Toh (2010), paralelisasi pada Star Alignment layak untuk dikembangkan. Salah satu bentuk paralelisasi yang menggunakan banyak processor yang disebut komputer paralel. Komputer paralel adalah teknik melakukan komputasi secara bersamaan dengan memanfaatkan beberapa komputer independen dan terpusat (Quinn, 2003). Pada penelitian ini, paralelisasi pada MSA dilakukan dengan melibatkan sebanyak enam unit komputer yang terhubung satu sama lain. Hal ini untuk mengetahui seberapa cepat dan efektif metode paralelisasi pada MSA ini.
Perumusan Masalah
Metode Star Alignment adalah salah satu metode MSA yang dibangun berdasarkan teknik pairwise alignment. Pada metode Star, Pair wise alignment dilakukan pada seluruh sekuens untuk menemukan sekuens yang akan menjadi Star. Hal ini menyebabkan kompleksitas metode Star cukup tinggi, yaitu O(k2 n2). Kompleksitas yang tinggi berdampak pada waktu eksekusi yang tinggi. Untuk mendapatkan proses yang efisien dapat dilakukan melalui dua cara, yaitu memodifikasi algoritme atau menerapkan komputasi paralel. Penelitian ini bermaksud menemukan solusi yang efisien untuk mengimplementasikan metode Star pada lingkungan paralel.
Tujuan Penelitian
Pada penelitian ini akan dikembangkan teknik komputasi paralel untuk MSA dengan metode Star Alignment. Diharapkan dengan penerapan konsep komputasi paralel ini dapat mereduksi waktu komputasi MSA dengan metode Star.
Manfaat Penelitian
Adapun manfaat dari penelitian yang dilakukan adalah agar pihak-pihak yang berkepentingan dalam analisa DNA dapat memanfaatkan aplikasi ini untuk mendukung proses analisis agar lebih efisien.
Ruang Lingkup Penelitian
3
2
TINJAUAN PUSTAKA
Sequence Alignment
Penjajaran sekuens adalah cara mengatur urutan DNA, RNA, atau protein untuk mengidentifikasi daerah kemiripan yang mungkin menjadi konsekuensi dari hubungan fungsional, struktural, atau evolusi antara dua urutan (Sing et al, 2011). Salah satu proses penjajaran sekuens adalah dalam memproses DNA. DNA merupakan asam nukleat yang terdapat dalam inti sel atau nucleus berisi mengenai informasi genetik pada suatu organisme yang merupakan untai ganda (double helix) yang disatukan oleh ikatan hidrogen adenin (A), sitosin (C), guanin (G), dan timin (T) (Jones dan Pevzner 2004). Gambaran umum mengenai DNA dapat dilihat pada Gambar 1.
Sumber :IPA Educational Material Archive (http://www2.edu.ipa.go.jp/gz/) Gambar 1 Deoxyribonucleic Acid (DNA)
Sedangkan gambaran umum mengenai penjajaran sekuens DNA dapat dilihat pada Tabel 1 antara Chlamydophila caviae dan Alicyclobacillus acidocaldarius frame 1-10 yang diambil dari National Center for Biotechnology Information (NCBI).
Tabel 1 Hasil Penjajaran sekuens antara Chlamydophila caviae dan Alicyclobacillus acidocaldarius pada fragmen DNA 1-10
Organisme Frame
1 2 3 4 5 6 7 8 9 10 11 12
Chlamydophila caviae T C A C C C A C C T -
-Alicyclobacillus acidocaldarius T - - T G C T T G T G G
4
ke dua dan tiga delesi (deletion). Selain itu, pada frame ke empat, lima, tujuh, delapan, dan sembilan mengalami mutasi (unmatch). Sedangkan pada frame satu, enam dan sepuluh merupakan conserved / match yang artinya terdapat kecocokan / kesamaan string. Keterangan tersebut menunjukan adanya similarity dan homology. Similarity adalah tingkat kesamaan antara satu sekuens dengan sekuens yang lain, sedangkan homology adalah menunjukan hubungan evolusi antara dua sekuens atau lebih (Baxevanis dan Ouellete 2001). Contoh hasil penjajaran sekuens di atas merupakan contoh hanya untuk mendapatkan skor similarity, sedangkan homology tidak dapat disimpulkan, karena hanya memuat sebagian DNA saja. Secara garis besar berikut keterangan penjajaran sekuens yang diambil dari Tabel 1 yang dapat dilihat pada Tabel 2:
Tabel 2 Keterangan penjajaran sekuens
Terdapat dua hal dalam menentukan skor similarity yaitu secara global dan lokal. Metode Needlemen–Wunsch merupakan salah satu metode untuk penjajaran sekuens yang mendefinisikan cara menemukan similarity global dari dua sekuens (Needleman-Wunsch, 1970). Algoritma similarity secara lokal hanya mencari subsekuenss, dan perbandingan tunggal dapat menghasilkan beberapa subsekuens yang berbeda (Smith dan Waterman, 1984). Baik lokal maupun global, keduanya membandingkan dua / pasangan sekuens secara langsung yang disajikan oleh matrik dua dimensi sehingga kompleksitasnya O(n2) proses ini dinamakan dengan pairwise alignment. Juga keduanya menggunakan dynamic programming dalam memproses secara pairwise alignment. Dynamic programming merupakan metode yang mempunyai strategi memecah masalah menjadi lebih kecil dan menggunakan solusi yang lebih kecil tadi guna membangun solusi yang lebih besar (Jones dan Pevzner, 2004). Matrik yang dibangun dalam memproses penjajaran sekuens menggunakan metode Needlemen–Wunsch dengan cara seperti :
5
Multiple Sequence Alignment (Star Alignment)
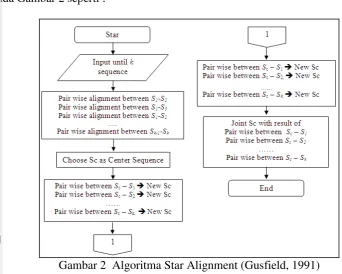
Multiple sequence alignment (MSA) adalah metode membandingkan sekuens sebanyak k > 2 (Gusfield, 1991). MSA yang dikemukakan oleh Gusfield (1991) merupakan proses membandingkan beberapa sekuens yang terlibat satu demi satu. Metode ini dikenal dengan Star Alignment yang mempunyai perhitungan jumlah sekuens yang dibandingkan sebanyak:
k = Jumlah sekuens
Jika terdapat k sekuens maka tiap sekuens mempunyai pengulangan untuk dibandingkan sebanyak k-1 atau total perhitungannya k2. Setiap pasangan yang dibandingkan secara pairwise alignment mempunyai kompleksitas O(n2) dan kompleksitas kombinasi sekuens adalah O(k2). Sehingga kompleksitas keseluruhan menjadi O(k2 n2). Pusat sekuens (center star) yang dibangun merupakan sekuens yang mempunyai skor yang paling tinggi di antara banyak sekuens yang dibandingkan. Adapun algoritma dari star aligment ini dapat dilihat pada Gambar 2 seperti :
Gambar 2 Algoritma Star Alignment (Gusfield, 1991)
6
Setelah mengetahui sekuens tertinggi, tidak serta merta center star diketahui mengingat tiap sekuens dibandingkan sebanyak k-1 dengan sekuens lainnya. Sehingga kompleksitas pencarian center sekuens ini adalah seperti di:
O(k-1) (4)
Center sekuens yang dipilih kemudian dijajarkan kembali dengan sekuenss yang di masukan. Proses ini dilakukan sebanyak 2 kali. Secara keseluruhan, kompleksitas dari metode yang diusulkan menggunakan processor tunggal (Ts) yang dapat dilihat seperti :
Ts = O(k2 n2) (5)
Komputer Paralel
Komputer pararel adalah sistem komputer dengan banyak processor yang mendukung paralel programming. Paralel programming adalah bahasa pemograman yang memungkinkan seorang programmer dapat membagi tugas yang sama atau berbeda kepada komputer-komputer yang terlibat guna menyelesaikan sesuai tugas yang diberikan (Quinn, 2003). Terdapat klasifikasi terbaik dalam komputer paralel yang dinamakan dengan Flynn’s Taxonomy. Klasifikasi ini bergantung pada pengetahuan pada alur perintah dan alur data. Klasifikasi mesin komputasi paralel (Flynn and Rudd, 1996) dapat dikatagorikan sebagai Single Instruction, Single, Single Instruction, Multiple Data, Multiple Instruction, Single Data, Multiple Instruction Multiple Data.
Tahapan selanjutnya adalah bagaimana komputer paralel tersebut didesain sedemikian rupa sehingga waktu eksekusi meningkat. Desain algoritma komputer paralel menggunakan metode Ian Foster. Metode yang dijelaskan oleh Ian Foster dibagi menjadi 4 tahapan (Foster, 1995), yaitu partitioning, communication, agglomeration, and mapping. Fokus utama penggunaan program paralel adalah untuk mempercepat waktu eksekusi. Untuk memastikan progaram paralel lebih unggul dibanding dengan program sekuensial, maka diperlukan perbandingan speed up (S) dan perhitungan efisiensi (E) yang ada dari kinerja komputer paralel. Speed up (S) merupakan perbandingan antara waktu eksekusi yang dimiliki oleh satu prorcessor dengan waktu ekseksusi yang dimiliki oleh multi processor. Formula Speed up (S) dapat dilihat seperti :
7 Dalam komputer paralel, terdapat dua tipe arsitektur yang bisa dibuat, yaitu share memory dan distributed memory (Wilkinson dan Allen, 2010). Shared memory merupakan sebuah lingkungan yang mana setiap processor dapat mengakses memory module sehingga setiap program dapat mengakes data jika diperlukan. Meskipun begitu lingkungan ini terbatas pada jumlah processor yang ada yang dapat berpengaruh pada waktu pemrosesan MSA. Arsitektur lain, yaitu distributed memory yang dibangun menggunakan seperangkat komputer yang dihubungkan oleh jaringan komputer. Hal ini memungkinkan penambahan data maupun penambahan processor dapat lebih leluasa.
3
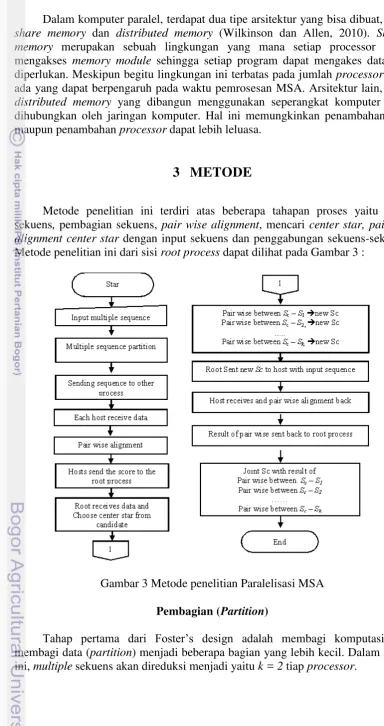
METODE
Metode penelitian ini terdiri atas beberapa tahapan proses yaitu input sekuens, pembagian sekuens, pair wise alignment, mencari center star, pairwise alignment center star dengan input sekuens dan penggabungan sekuens-sekuens. Metode penelitian ini dari sisi root process dapat dilihat pada Gambar 3 :
Gambar 3 Metode penelitian Paralelisasi MSA
Pembagian (Partition)
8
Pengiriman Sequence
Tahap selanjutnya dari Foster’s design adalah berkaitan erat dengan komunikasi data, aglomerasi, dan mapping yang berhubungan dalam pengiriman data.. Pengiriman data dalam pemograman paralel ini menggunakan parameter MPI_Send() disisi pengirim dan parameter MPI_Recv() disisi penerima. Kedua parameter ini merupakan komunikasi point to point.
Pair wise Alignment
Pair wise Alignment menggunakan metode Needleman-Wunsch dengan parameter yang dipakai match = 7, mismatch = -3, dan gap = -2.
Center Star
Proses MSA menghasilkan skor similarity tiap sekuens satu sama lain. Selanjutnya adalah menentukan center star yang merupakan sekuens yang paling dekat / mirip dengan sekuens lainnya.
Bahan
Bahan yang diperlukan dalam penelitian ini menggunakan multiple sekuens yang menjadi sekuens masukan diambil dari database Genbank NCBI dengan rincian seperti pada Tabel 3:
Tabel 3 Multiple sekuens dari NCBI
No. Sekuens Panjang
1 Candidatus Riesia pediculicola 7737 2 Alicyclobacillus acidocaldarius 7909
3 Clostridium difficile 7881
4 Chlamydophila caviae 7966
Alat
Perangkat yang diperlukan demi terlaksananya peneltian ini terdiri dari perangkat hardware dan software. Berikut rincian keduan perangkat tersebut:
a. Software
- Sistem Operasi : Window Xp Profesional Service Pack 2 - Software Development : Code Block 12.11
b. Hardware
- Processor : Pentium(R) Intel Atom 1.66 GHz - Memory : RAM 2 GB
9
4
HASIL DAN PEMBAHASAN
Hasil
Tujuan utama beralih kepada program paralel salah satunya adalah untuk mendapatkan kinerja yang lebih baik dibanding program sekuensial. Hal yang perlu dipertimbangkan dalam paralelisasi ini adalah beban kerja dan biaya komunikasi. Secara garis besar, idenya adalah bagaimana menyeimbangkan beban kerja dan meminimalkan biaya komunikasi. Untuk mencapai keduanya, maka Foster’s design diimplementasikan dalam lingkungan paralel. Langkah pertama adalah tahap pembagian (partition) data. Pada tahap ini harus diperhatikan jum lah processor yang terlibat dengan jumlah kombinasi sekuens. Jumlah kombinasi sekuens menggunakan rumus pada Persamaan 2 menjadi :
Idealnya jumlah processor sama dengan jumlah kombinasi sekuens. Karena beban kerja menjadi sama tiap processor. Namun, bukan berarti tidak samanya jumlah processor dengan jumlah sekuens menjadi program tidak bisa. Hanya saja beban kerja menjadi tidak seimbang di salah satu processor. Hal ini dapat di lihat pada Gambar 4.
Gambar 4 Ilustrasi pembagian sekuens. a) Distribusi sekuens dengan jumlah processorsama dengan jumlah kombinasi sekuens. b) Distribusi sekuens dengan
jumlah processor yang tidak sama dengan jumlah kombinasi sekuens Tahap kedua adalah tahap komunikasi data. Pasangan sekuens akan segera dikirimkan kepada host yang telah di set sebelumnya. Berikut pemetaan pasangan sekuens yang dikirim yang dapat dilihat pada Tabel 4 :
Tabel 4 Pemetaan pembagian sekuens
10
*root process
Berdasarkan Tabel 4, kompleksitas komunikasi data dari root-host-root menjadi:
Tahap Ketiga adalah tahap agglomerasi (agglomeration). Tahapan aglomerasi adalah tahap yang menggabungkan beberapa sekuens dengan tujuan host yang sama. Sehingga ini dapat meminimalkan biaya komunikasi. Terakhir adalah tahap pemetaan (mapping). Pada tahap pemetaan ini menggunakan parameter MPI_Send() dan Parameter MPI_Recv() untuk menerima data. Setelah sekuens diterima oleh proses yang terlibat, maka selanjutnya melakukan pair wisealignment menggunakan metode Needleman-Wunsch. Pada penelitian ini, parameter yang digunakan untuk kecocokan residu (string) adalah match = 7, mismatch = -3, dan gap = -2. Kompleksitas pair wise alignment terdapat pada komputer paralel adalah :
Data yang akan dibandingkan di masukan sebanyak k > 2. Banyaknya data yang terlibat di dekomposisi. Data-data tersebut kemudian dipasangkan sedemikian rupa dengan jumlah pasangan sesuai dengan Formula 2, sehingga memudahkan dalam partition. Proses pembagian ini dilakukan pada processor 1 (root process). Setelah diurai menjadi lebih kecil, selanjutnya dipetakan (mapping) ke prosesor tujuan. Data yang telah dipetakan juga harus dipikirkan cara komunikasi (communication) yang dibutuhkan. Berikut pembagian data dari organisme yang terlibat dipetakan pada processor tujuan yang dapat dilihat pada Tabel 5 :
Tabel 5 Pemetaan pasangan sekuens
Processor Sekuens
1* Candidatus Riesia pediculicola Alicyclobacillus acidocaldarius 2 Candidatus Riesia pediculicola Clostridium difficile
3 Candidatus Riesia pediculicola Chlamydophila caviae 4 Alicyclobacillus acidocaldarius Clostridium difficile 5 Alicyclobacillus acidocaldarius Chlamydophila caviae 6 Clostridium difficile Chlamydophila caviae
*
= root process
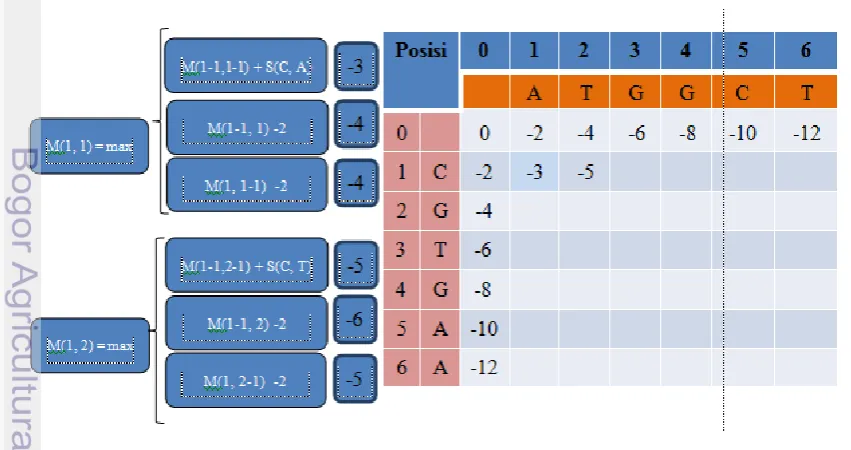
11 a. Inisiasi matrik
Inisiasi matrik dibangun berdasarkan parameter gap yang dipilih. Secara utuh parameter digunakan adalah match = 7, mismatch = -3, dan gap = -2. Untuk memudahkan gambaran mengenai pair wise alignment, maka diperlukan contoh sekuens yang lebih kecil. Misalkan terdapat sekuens “ATGGCT” dan “CGTGAA” yang akan dibandingkan. Berikut Gambar 5 mengenai inisiasi matrik :
Posisi
0 1 2 3 4 5 6
A T G G C T
0 0 -2 -4 -6 -8 -10 -12
1 C -2 2 G -4 3 T -6 4 G -8 5 A -10 6 A -12
Gambar 5 Langkah-langkah inisiasi matrik pair wise alignment
b. Pengisian matrik
Untuk pengisian matrik diperlukan Formula 1. Berikut proces pengisian matrik pada Gambar 6 yang menggunakan Formula 1:
12
Secara keseluruhan hasil pair wise ini dapat dilihat pada Gambar 7 :
Posisi
Gambar 7 Hasil akhir pengisian matrik c. Runut Balik (Trace Back)
Trace back dilakukan bergerak secara diagonal dengan mencari nilai maksimum dimulai dari titik M[i, j] sampai titik M[1, 1]. Hasil trace back ini menentukan hasil penjajaran sekuens. Berikut Gambar 8 yang memperlihatkan trace back dan hasil penjajaran sekuens.
Gambar 8 Trace back dan hasil penjajaran sekuens Skor similarity yang dihasilkan dari penjajaran sekuens adalah:
13 Secara ringkas, pada Tabel 6 menunjukan hasil pair wise alignment yang berada di tiap processor dengan ketentuan C(k, 2) = p:
Tabel 6 Hasil pair wise alignment
Processor Sekuens Panjang Skor
1 Candidatus Riesia pediculicola
Alicyclobacillus acidocaldarius 9314 51 2 Candidatus Riesia pediculicola
Clostridium difficile 9476 54
3 Candidatus Riesia pediculicola
Chlamydophila caviae 9436 53
4 Alicyclobacillus acidocaldarius
Clostridium difficile 9242 49
5 Alicyclobacillus acidocaldarius
Chlamydophila caviae 9587 50
6 Clostridium difficile
Chlamydophila caviae 9387 53
Skor pair wise alignment ini kemudian dikirimkan ke root process. Biaya komunikasi pada proses ini sama seperti Formula 9. Root process menerima skor ini untuk ditindak lanjuti guna mencari center sekuens. Untuk mencari center sekuens diperlukan rekapitulasi nilai tiap sekuens seperti pada Tabel 7:
Tabel 7 Rekapitulasi skor tiap sekuens
No. Sekuens Hasil Candidatus Riesia pediculicola yang dijajarkan dengan Clostridium difficile. Kemudian langkah selanjutnya adalah pair wise alignment kembali antara center star yang sudah dipilih dengan sekuens yang dinputkan. Kompleksitas yang dimiliki dalam proses ini dapat dilihat seperti :
O ((k-1) n2) (12) Proses ini kembali dilakukan, bedanya pada proses pair wise antara new center star dengan sekuens awal dilakukan di host process. Kompleksitas pengiriman data dari root-host-root menjadi :
O ((k-1) 4n) (13)
14
Keterangan :
Perbadingan kompleksitas antara menggunakan processor tunggal (Persamaan 5) dan multi processor (Persamaan 13) dapat dilihat pada Tabel 8:
Tabel 8 Perbandingan kompleksitas antara processor tunggal dengan multi processor
n k p pasangan
Kompleksitas
Speed up
Efficiency (%)
Tunggal Multi
processor
8,000 3 2 3 576,000,000 224,096,000 2.570 128.516
8,000 3 3 3 576,000,000 192,128,000 2.998 99.933
8,000 4 2 6 1,024,000,000 384,192,000 2.665 133.267 8,000 4 3 6 1,024,000,000 320,224,000 3.198 106.592 8,000 4 4 6 1,024,000,000 288,224,000 3.553 88.820 8,000 4 5 6 1,024,000,000 269,024,000 3.806 76.127 8,000 4 6 6 1,024,000,000 256,256,000 3.996 66.600
p = jumlah processor n = jumlah karakter sekuens k = jumlah sekuens
Berdasarkan Tabel 8, berikut grafik speed up dan efficiency yang dapat dilihat pada Gambar 9 dan Gambar 10:
15
Gambar 10 Efficiency dari kompleksitas paralel
Sedangkan hasil program paralel yang menggunakan enam buah processor dapat dilihat pada Tabel 9 seperti :
Tabel 9 Perbandingan waktu antara satu processor dengan multi processor
Jumlah Sekuens
Waktu eksekusi (s)
p = 1 p = 2 p = 3 p = 4 p = 5 p = 6
3 45.203 32.781 39.281
4 97.141 52.735 56.766 52.719 47.157 39.062 Berdasarkan Tabel 13 pengukuran kinerja program paralel mengenai speed up dan efficiency yang dihitung menggunakan Persamaan (6) dan Persamaan (8) ditampilkan pada Tabel 10 dan Tabel 11 :
Tabel 10 Speed up program paralel
Jumlah Sekuens
Speed Up
p = 2 p = 3 p = 4 p = 5 p = 6
3 1.38 1.15
4 1.84 1.71 1.84 2.06 2.49
Tabel 11 Efficiency program paralel
Jumlah Sekuens
Efficiency (%)
p = 2 p = 3 p = 4 p = 5 p = 6
3 68.95 38.36
16
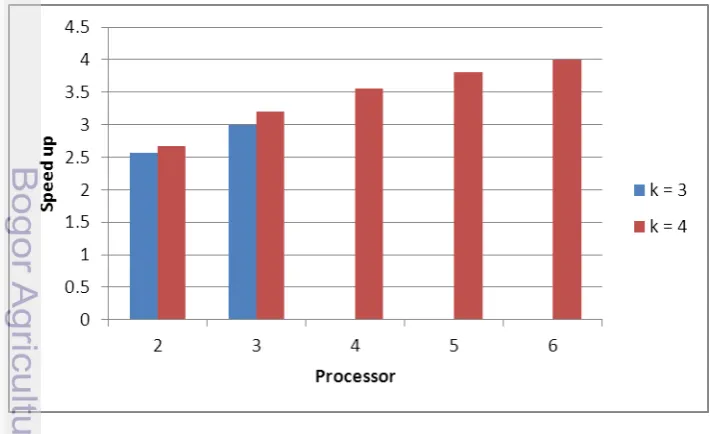
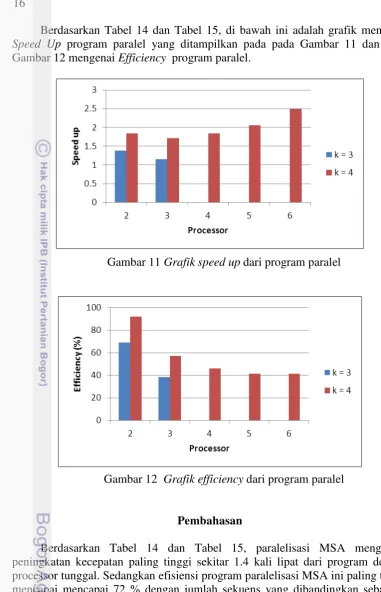
Berdasarkan Tabel 14 dan Tabel 15, di bawah ini adalah grafik mengenai Speed Up program paralel yang ditampilkan pada pada Gambar 11 dan pada Gambar 12 mengenai Efficiency program paralel.
Gambar 11 Grafik speed up dari program paralel
Gambar 12 Grafik efficiency dari program paralel
Pembahasan
17 Logika mudahnya semakin banyak processor maka semakin cepat pula proses MSA selesai. Namun ternyata tidak demikian. Salah satu faktor yang mempengaruhi kondisi ini adalah biaya komunikasi. Hal ini sesuai dengan Tabel 4 yang memperlihatkan biaya komunikasi yang ditimbulkan oleh tiga buah processor lebih tinggi daripada biaya komunikasi yang ditimbulkan oleh dua buah processor. Berikut Tabel 12 yang memperlihatkan perbandingan biaya komunikasi tiap setiap processor pada satu kali pengiriman.
Tabel 12 Pemetaan biaya komunikasi
*root process n = panjang sekuens
Pada penelitian ini juga, terdapat metode paralelisasi MSA desain kedua yang berbeda dengan yang digambarkan pada Gambar 3. Berikut bentuk paralelisasi desain kedua yang dapat dilihat pada Gambar 13 seperti :
18
Metode paralelisasi pada Gambar 5 ini telah dipublikasikan di http://ieeexplore.ieee.org dengan Digital Object Identifier (DOI) 10.1109/ICICI-BME.2013.6698486. Berikut hasil perhitungan waktu antara penggunaan processor tunggal dengan enam buah processor dapat dilihat pada Tabel 13 :
Tabel 13 Waktu eksekusi antara satu processor dengan beberapa processor
Percobaan Jumlah
Berdasarkan Tabel 14 pengukuran kinerja program paralel dapat dihitung menggunakan Formula (6) dan Formula (8) dan ditampilkan pada Tabel 14 seperti :
Table 14 Pengukuran kinerja speed up dan efficiency desain ke dua
Perbaan Number
Berikut grafik Speed up dan efficiency dari penelitian yang diperlihatkan pada Gambar 14 seperti :
19 dapat sama karena jumlah masalah dan jumlah processor sama banyaknya. Pada program paralel desain pertama, root process membagi data dan mengatur pemetaan distribusi data seperti pada Tabel 4 dan Tabel 12. Proses ini mempunyai kompleksitas kuadratik, sehingga waktu tunggu yang dialami oleh host process menjadi lama. Gambaran waktu tunggu ini dijabarkan oleh Wilkinson (2010) sebagai nilai startup dan dapat dilihat pada Gambar 15 seperti :
Gambar 15 Waktu tunggu (idle time) yang dialami oleh processor yang terlibat Pada program paralel desain kedua, pasangan yang sudah dipetakan langsung dikirim ke host tujuan sebanyak p-1 processor. Sehingga setiap processor mendapat pasangan secara berurutan tanpa ada proses round robin di dalamnya. Hal inilah yang mengakibatkan perbedaan waktu eksekusi selesai,
Pada penelitian yang dilakukan menggunakan enam processor untuk memproses sekuens alignment. Jumlah ini disesuaikan dengan kombinasi sekuens yang ada. Hal ini dilakukan untuk menyeimbangkan beban kerja sesuai dengan karakteristik paralel desain kedua. Baik program paralel desain pertama dan kedua, keduanya masih dalam tahap pengembangan. Kedua program ini men set processor 0 sebagai root process yang bertugas sebagai mengumpulkan dan membagi data disamping mengerjakan penjajaran sekuens. Perlu diperhatikan juga, penelitian ini masih memproses MSA dari DNA-DNA pendek. Diharapkan selanjutnya dapat dijalankan dalam lingkungan DNA yang panjang dan jumlah processor yang lebih banyak lagi.
Process 1
Process 2
Process
3 Time
20
5
SIMPULAN
Simpulan
Mempercepat waktu eksekusi tidak selalu memperbanyak jumlah processor. Banyaknya jumlah processor yang terlibat, mengakibatkan biaya komunikasi menjadi lebih besar. Dalam memparalelkan MSA, bukan hanya jumlah data yang harus dipertimbangkan, namun jumlah processor pun layak untuk diperhitungkan. Dalam perhitungan kompleksitas maupun dalam pengujian program didapat bahwa speed up bergerak naik tidak diimbangi dengan nilai efficiency yang cenderung turun dengan menggunakan banyak processor. Hal ini menjadi keputusan selanjutnya bagi yang akan menggunakan program ini, apakah speed up atau efficiency yang diperlu di utamakan.
Agar tercipta kondisi dengan beban kerja yang sama (scalable), maka setiap penambahan processor idealnya mendapatkan minimalnya satu pasangan sekuens. Semakin banyak jumlah sekuens yang terlibat dan jumlah karakter tiap sekuens semakin panjang maka kinerja program ini menjadi lebih bermanfaat. Meskipun pada kenyataannya bahwa komputer mempunyai keterbatasan ruang memory (kompleksitas ruang) yang ada.
Saran
21 Homologue Hmlh1 Is Associated With Hereditary Non-Polyposis Colon Cancer. Nature Mar 17;368(6468):258-61.
Chalker Alison F, Heather W, Minehart, Nicky J. Hughes, Kristin K Koretke, Michael A Lonetto, Kerry K Brinkman, Patrick V Warren, Andrei Lupas, Michael J Stanhope et al. 2001. Systematic Identification of Selective Essential Genes in Helicobacter pyloriby Genome Prioritization and Allelic Replacement Mutagenesis. Journal Of Bacteriology. 1259-1268. doi: 10.1128/JB.183.4.1259–1268.
Do Chuong B, Mahathi S.P. Mahabhashyam, Michael Brudno, Serafim Batzoglou. 2005. ProbCons: Probabilistic consistency-based multiple sequence alignment. Genome 15: 330-340. doi: 10.1101/gr.2821705. 2013. Feng Dai Fei and Russell F. Doolittle. 1987. Progressive Sequence Alignment as a
Prerequisite to Correct Phylogenetic Trees. J Mol Evol 25:351-360.
Flynn Michael J and Kevin W. Rudd. 1996. Parallel Architectures. Stanford University.
Foster Ian. Designing and Building Parallel Programs: Concept and Tools for Parallel Software Engineering. Reading, MA: Addison - Wesley.1995.
Giegerich Robert, Carsten Meyer, Peter Steffen. A discipline of dynamic programming over sequence data. Elsevier BV Science of Computer Programming 51 (2004) 215 - 263 doi: 10.1016/j.scico.2003.12.005. 2004. Gusfield Dan. 1991. Efficient methods for multiple sequence alignment with
guaranteed error bounds. Computer Science Division University of California.
Jones Neil C. and Pavel A Pevzner. 2004. An Introduction to Bioinformatics Algorithms. Massachusetts Institute of Technology, USA.
Katoh Kazutaka dan Hiroyuki Toh. 2010. Parallelization of the MAFFT multiple sequence alignment Program. Bioinformatics Vol. 26 no. 15 2010, pages 1899–1900doi:10.1093/bioinformatics/btq224.
Katoh Kazutaka, Kazuhara Misawa, Kei-ichi Kuma dan Takashi Miyata. 2002. MAFFT:a Novel Method for Rapid mutiple Sequence alignment Based on Fast Fourier Transform. Nucleic Acids Research Vol. 30 No. 14 3059-3066. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam
H, Valentin F, Wallace IM, Wilm A et al. 2007. Clustal W and Clustal X version 2.0. Bioinformatics, 23, 2947-2948.
Löytynoja Ari, Albert J. Vilella, dan Nick Goldman. 2012. Accurate Extension Of Multiple Sequence Alignments Using A Phylogeny-Aware Graph Algorithm.
Bioinformatics Vol. 28 no. 13 pages 1684–
1691doi:10.1093/bioinformatics/bts198.
22
Papadopoulos N et al. 1994. Mutation Of A Mutl Homolog In Hereditary Colon Cancer. ScienceVol. 263 no. 5153 pp. 1625-1629 DOI: 10.1126/science.8128251.
Payne David J, Michael N. Gwynn, David J. Holmes dan David L. Pompliano. 2006. Drugs For Bad Bugs: Confronting The Challenges Of Antibacterial Discovery. Nature Reviews Drug Discovery6:29-40.doi:10.1038/nrd2201. Quinn Michael J. 2003. Parallel Programming In C And With MPI OpenMP.
McGraw-Hill. New York - USA.
Sing et al. 2011. Role Of Bioinformatics In Agriculture And Sustainable Development. Banaras Hindu University, India.
Smith and Waterman. 1981. Identification of Common Molecular Subsequences. J. Mol. Bwl. (1981), 147, 195-197.
Tajima F and M Nei. 1984. Estimation of evolutionary distance between nucleotide sequences. Mol Biol Evol 1 (3): 269-285.
Thompson JD, Higgins DG, and Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Research, 22:4673-4680.
Wilkinson Barry and Michael Allen. 2010. Parallel Programming (Techniques and Applications Using a Network of Workstations and Parallel Computers). Andi Yogyakarta.