This chapter was originally published in the book International Review of Neurobiology, Vol. 115
published by Elsevier, and the attached copy is provided by Elsevier for the author's benefit and for the benefit of the author's institution, for non-commercial research and educational use including without limitation use in instruction at your institution, sending it to specific colleagues who know you, and providing a copy to your institution’s administrator.
All other uses, reproduction and distribution, including without limitation commercial reprints, selling or licensing copies or access, or posting on open internet sites, your personal or institution’s website or repository, are prohibited. For exceptions, permission may be sought for such use through Elsevier's permissions site at:
http://www.elsevier.com/locate/permissionusematerial
From Adrian Zhubi, Edwin H. Cook, Alessandro Guidotti and Dennis R. Grayson, Epigenetic Mechanisms in Autism Spectrum Disorder. In: Subhash C. Pandey, editor, International Review of Neurobiology, Vol.
115, Burlington: Academic Press, 2014, pp. 203-244. ISBN: 978-0-12-801311-3
Epigenetic Mechanisms in Autism
Spectrum Disorder
Adrian Zhubi*, Edwin H. Cook†, Alessandro Guidotti*, Dennis R. Grayson*,1
*The Psychiatric Institute, Department of Psychiatry, University of Illinois at Chicago, Chicago, Illinois, USA
†Institute for Juvenile Research, Department of Psychiatry, University of Illinois at Chicago,
Chicago, Illinois, USA
1
Corresponding author: e-mail address: [email protected]
Contents
1. Introduction 204
2. Molecular Aspects of Epigenetic Mechanisms 206
2.1 Histones 206
2.2 DNA methylation 209
2.3 DNA hydroxymethylation 212
3. Genetic Defects with Epigenetic Implications 213
3.1 Methyl-CpG-binding protein 2 (MECP2) 214
3.2 DNA topoisomerase 216
3.3 Chromodomain helicase DNA-binding protein 8 217
4. Epigenetic Dysregulation of ASD Candidate Genes 218
4.1 GABAergic genes 218
4.2 GAD67 (GAD1) 219
4.3 Reelin 220
4.4 GABAβ3 222
4.5 Oxytocin receptor (OXTR) 223
4.6 Brain-derived neutrophic factor (BDNF) 223
4.7 Ubiquitin–protein ligase E3A (UBE3A) 224
4.8 Engrailed-2 (EN-2) 225
4.9 SH3 and multiple ankyrin repeat domains (SHANK3) 226
5. Environmental Model of Autism 227
6. Conclusions 229
Acknowledgments 231
References 231
Abstract
Autism spectrum disorder (ASD) is a neurodevelopmental condition characterized by impaired social interactions, language deficits, as well as restrictive or repetitive behav-iors. ASD is clinically heterogeneous with a complex etiopathogenesis which may be
International Review of Neurobiology, Volume 115 #2014 Elsevier Inc.
ISSN 0074-7742 All rights reserved.
http://dx.doi.org/10.1016/B978-0-12-801311-3.00006-8
conceptualized as a dynamic interplay between heterogeneous environmental cues and predisposing genetic factors involving complex epigenetic mechanisms. Inherited andde novocopy number variants provide novel information regarding genes
contrib-uting to ASD. Epigenetic marks are stable, yet potentially reversible, chromatin modifi-cations that alter gene expression profiles by locally changing the degree of nucleosomal compaction, thereby opening or closing promoter access to the transcrip-tional machinery. Here, we review progress on studies designed to provide a better understanding of how epigenetic mechanisms impact transcriptional programs oper-ative in the brain that contribute to ASD.
1. INTRODUCTION
Autism spectrum disorder (ASD) is a neurodevelopmental disorder characterized by symptoms that include deficits in social interactions, under-developed communication skills, and restrictive or repetitive behav-iors. Population-wide prevalence of ASD is approximately 1% and it is more common in males than females (4:1) (Abrahams & Geschwind, 2008; Bailey et al., 1995; Baron-Cohen et al., 2009; O’Roak & State, 2008; Veenstra-Vanderweele & Cook, 2004). ASD is clinically and etiologically heteroge-neous with many of the diagnostic symptoms showing considerable variation in severity. Approximately 5–20% of cases involve large effect
de novo copy number variants. Genome-wide association studies are fre-quently used to compare the frequencies of single-nucleotide polymor-phisms (SNPs) in ASD DNA samples. While no replicable single SNP variants have been identified, the cumulative contributions of inherited genetic variation over many small effect loci has recently been estimated to be as high as 40% (Klei et al., 2012).
intermediate between SZ and population controls (Stefansson et al., 2014). Moreover, CNVs do not all affect the same cognitive domains and vary con-siderably from one mutation to another (Stefansson et al., 2014). Two important goals of clinical geneticists are (1) to improve CNV detection and (2) to improve the phenotyping of CNV carriers that also exhibit psy-chotic symptoms. Recent analyses of topological networks derived from ASD CNVs and mouse functional genomics are being used to unveil highly detailed ASD-associated interaction networks that allow testing of novel hypotheses regarding cellular signaling and biological function (Noh et al., 2013; Veenstra-Vanderweele & Blakely, 2012).
One of the more frequently observed CNVs associated with high pen-etrance for ASD is the maternal duplication of chromosome 15q11-13. A second well-known CNV is a 600 kb microdeletion/microinsertion at chromosome 16p11.2 which occurs in some 1% of sporadic ASD cases (Cook & Scherer, 2008). Syndromic ASD, which occurs in 10% of cases, is observed when diagnostic behaviors are comorbid with a recognized syn-drome. Some of the monogenic (syndromic) conditions associated with the ASD phenotype are Rett (RTT) syndrome (MECP2 gene; Amir et al., 1999), fragile X syndrome (FXS, FMR1), tuberous sclerosis (TSC1 and
TSC2; Wiznitzer, 2004), neurofibromatosis (NF1), Timothy syndrome (CACNA1;Splawski et al., 2006), and cortical dysplasia–focal epilepsy syn-drome (CNTNAP2; Strauss et al., 2006). Each of the above conditions exhibits phenotypes that overlap with ASD and hence offer important insight into how the corresponding genes contribute to pathogenesis.
Several twin studies have reported concordance rates between MZ twins at between 70% and 90%, while DZ twin concordance rates vary from 6% to 10%, with a more than 20-fold increased risk for siblings (Bailey et al., 1995; Eapen, Crncˇec, & Walter, 2013; Steffenburg et al., 1989). Phenotypic dis-cordance between MZ twins is often associated with de novo mutations (Hallmayer et al., 2011; Zafeiriou, Ververi, Dafoulis, Kalyva, & Vargiami, 2013) and numerous nonshared environmental factors including in utero
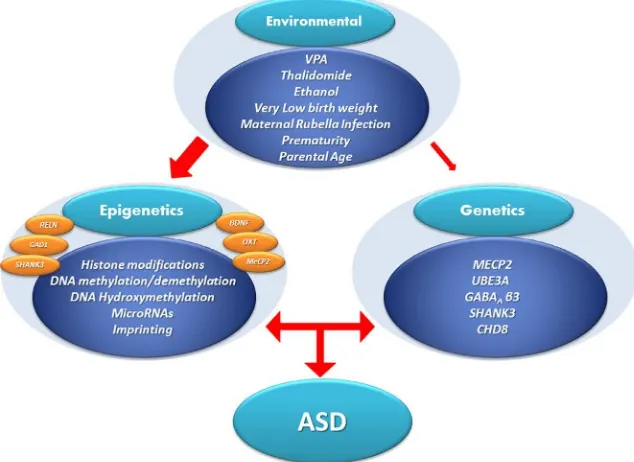
allelic origin (imprinting) or the effects of DNA methylation/hydro-xymethylation on the epigenome. Moreover, epigenetic factors also mani-fest at multiple additional levels including histone tail modifications, variations in methyl DNA-binding proteins, etc. Recently, there has been a growth in interest in studies of the involvement of epigenetic mechanisms in the pathophysiology of ASD. The overlapping phenotypic features of ASD that are shared with related neurodevelopmental disorders may be explained through altered gene expression profiles filtered through various epigenetic mechanisms (Lasalle, 2013; Miyauchi & Voineagu, 2013; Voineagu et al., 2011). In other words, as researchers begin to compare the gene expression networks between different neurodevelopmental disor-ders, differences in specific biological pathways and brain regions that reflect the phenotypes associated with each disease should become apparent (Hoerder-Suabedissen et al., 2013; Parikshak et al., 2013; Willsey et al., 2013). In summary, one plausible mechanistic approach to understanding the etiology of ASD supports the concept that environmental/epigenetic perturbations incurred during early nervous system development operate on and enhance the contributions of a large number of susceptibility genes identified as either inherited or de novo mutations which themselves may influence epigenetic regulation (seeFig. 6.1). Here, we summarize progress on epigenetic mechanisms operative in brain with the goal of understanding the distinct or overlapping features of these mechanisms in the etiopathogenesis of ASD. We provide a basic overview of histone modifi-cations, DNA methylation, and hydroxymethylation before proceeding onto subsequent topics. While we have not discussed the impact of micro-RNAs (mimicro-RNAs), long noncoding micro-RNAs (ncmicro-RNAs), and enhancer micro-RNAs on transcriptional regulation, there have been several recent reviews on these subjects (Melios & Sur, 2012; Peschansky & Wahlestedt, 2014; Roberts, Morris, & Weinberg, 2014; Velmeshev, Magistri, & Faghihi, 2013; Wilkinson & Campbell, 2013).
2. MOLECULAR ASPECTS OF EPIGENETIC MECHANISMS
2.1. Histones
these states accompany changes in transcription. Histones make contact with the DNA at multiple points, although amino terminal histone tails protrude from the histone core. Nucleosomal structures support the DNA in a mobile and interactive environment that provides a scaffold for the reversible epigenetic markings that induce local structural transitions. These epigenetic marks support DNA inside the nucleus in clusters of open (euchromatin), closed/restricted chromatin (heterochromatin), and several intermediate states associated with transitioning between these two (Gra¨ff, Kim, Dobbin, & Tsai, 2011; Grayson & Guidotti, 2013; Houston et al., 2013). The various epigenetic marks allow tran-scription factors, RNA polymerase II (PolII), and ancillary proteins access to cis-acting regulatory sequences proximal (promoters) to transcriptional start sites (TSSs). In addition, they facilitate long-range interactions between protein-bound distal enhancer elements and transcription factor-associated proximal promoter domains through the formation of topological loops organized in three dimensions (Cavalli & Misteli, 2013; Mitchell et al., 2013).
modifications and increases the predictability of transcription at the corresponding sites (Karlic´, Chung, Lasserre, Vlahovicek, & Vingron, 2010). For example, promoters with a high GC content accompanied by H3K27ac and H4K20me1 are associated with active transcription (Karlic´ et al., 2010). In contrast, low GC content promoters correlate with tran-scription most often when accompanied by H3K4me1, H3K79me1, and H3K9me3 (Houston et al., 2013; Karlic´ et al., 2010). There is an enormous diversity and growing complexity in the numbers of enzymes that (1) cat-alyze histone modifications (epigenetic writers), (2) remove these groups (erasers), and (3) proteins that recognize these modifications (readers) to effect downstream responses (Kouzarides, 2007).
2.2. DNA methylation
Methylation of DNA is an important epigenetic mechanism involved in numerous processes including X-chromosome inactivation, imprinting, and the suppression of foreign DNA such as transposable elements, proviruses, and other sequences (Bestor, 1990). The role of methylation in regulating gene expression has been increasing in interest in recent years (Grayson & Guidotti, 2013). Methylation consists of the addition of a methyl group to the C5 position of the cytosine base, an enzymatic reaction catalyzed by members of a family of evolutionarily related DNA methyltransferases (DNMTs) that include DNMT1, DNMT3A, and DNMT3B. DNMTs mediate the methyl group transfer from the donor, S-adenosyl methi-onine, to cytosines in DNA producing 5-methylcytosine (5-mC) and
CpG islands are features of vertebrate genomes that were initially thought to be associated with the 50-flanking regions of housekeeping
and many tissue-specific genes (Illingworth & Bird, 2009). They are stretches of DNA that have a higher than normal GC content over some 300 bp or more and are found to be proximal to 60–70% of gene pro-moters. These contiguous CpG-rich stretches are asymmetrically distributed throughout the genome (Gardiner-Garden & Frommer, 1987) and are largely refractory to DNA methylation. Thus, CpG island-containing pro-moters and enhancers are generally undermethylated (hypomethylated) consistent with a negative role for methylation in regulating transcription (Chen & Riggs, 2011). Based on data from recent genome-wide studies, CpG islands themselves appear to function as promoters and the high GC content plays a regulatory role in nucleosome positioning or phasing around TSSs (Fenouil et al., 2012). This concept is consistent with the large num-bers of CpG islands located proximal to promoters. It has also been demon-strated that CpG islands delineate the borders of open chromatin domains, hence allowing transcription factor and RNA Pol II access to these open regions of DNA (Fenouil et al., 2012).
Recently, the zinc finger-CXXC (ZF-CXXC) domain family of pro-teins was shown to specifically recognize CpGs at nonmethylated DNA and recruit chromatin-modifying activities to CpG island elements (Long, Blackledge, & Klose, 2013). The classic ZF-CXXC domain-containing protein, CFP1, binds unmethylated CpGs within CpG islands and recruits the SETD1 H3K4 methyltransferase complex, facilitating an enrichment of H3K4me3at nearby histones (Thompson, Fazzari, & Greally, 2010). In this way, the binding of CFP1, and other methyl-CpG readers, provides a mech-anism of directing histone modifications to specific regions of chromatin proximal to CpG islands. H3K4me3 is permissive for transcription (Long et al., 2013). Several additional ZF-CXXC domain-containing proteins include DNMT1, histone methyltransferases (MLL-1 and -2, KDM2 fam-ily), methyl-binding domain protein (MBD1), and Tet methylcytosine dioxygenases (TETs 1 and 3) all of which bind to DNA at CpGs and act to modify DNA or histones.
pathway involves hydroxylation of 5-mC to form 5-hydroxymethylcytosine (5-hmC) by members of the Tet methylcytosine dioxygenase enzyme family (TETs 1–3, see Section 2.3 below). TET proteins also further oxidize 5-hmC forming 5-formylcytosine (5-fC) and 5-carboxycytosine (5-caC), which are stable epigenetic marks that accumulate in brain. fC and 5-caC are specifically recognized by thymine deglycosylase (TDG) and are removed by base excision repair (BER) forming unmodified cytosine (Yu et al., 2012). The resultant TpG/mCpG or 5hmUpG/mCpG mis-matches are excised by DNA glycosylases (i.e., the methyl-CpG-binding domain protein 4 [MBD4] or thymine deglycosylase) (Hendrich, Hardeland, Ng, Jiricny, & Bird, 1999). Mechanisms that utilize TDG-mediated excision of 5-formylcytosine and 5-carboxycytosine have received the most experimental support (Shen, Song, He, & Zhang, 2014). The rap-idly inducible growth arrest and DNA-damage-inducible protein 45 family (GADD45), a small family of immediate early genes (Barreto et al., 2007; Ma et al., 2009; Matrisciano, Dong, Gavin, Nicoletti, & Guidotti, 2011), is thought to coordinate this process by selectively targeting specific 5-hmCs and recruiting deaminases and glycosylases to the corresponding genomic regions (Guo et al., 2011; Zhu, 2009).
Recently, a mechanism for protecting actively expressed genes from DNA methylation was described that links transcription, DNMT1, and extra coding RNA molecules (ecRNA; Di Ruscio et al., 2013). ecRNAs are ncRNAs that are widely expressed across the genome. While these are distinct from mRNAs and miRNAs, researchers are only beginning to understand how they regulate gene expression (see Peschansky & Wahlestedt, 2014for recent review). In addition to ecRNA protection from DNA methylation, there is evidence that RNA–DNA hybrids called “R-loops” also protect CpG islands from DNA methylation (Ginno, Lott, Christensen, Korf, & Che´din, 2012).
DNMT1 binds to the CEBPA ecRNA and prevents methylation of the gene. DNMT1 sequestration of the ecRNA activates transcription produc-ing CEBPA RNA (Di Ruscio et al., 2013). Moreover, genome-wide expression and methylation profiling of DNMT1-bound RNAs demon-strated that this protein–ecRNA association occurs across a large number of genes supporting a novel mechanism for blocking DNA methylation.
2.3. DNA hydroxymethylation
may represent an important neuronal- or cellular-specific epigenetic code (Szulwach & Jin, 2014). That is, the accumulation of 5-hmC is likely a crit-ical step in the activation of cell-specific enhancers during differentiation or lineage specification (Se´randour et al., 2012).
As noted above, proteins that contain CXXC domains bind to unmethylated CpGs and recruit ancillary chromatin modifiers. It has been known for some time that proteins containing methyl DNA-binding domains (MBDs) as part of their structure, bind to methylated CpGs. These include MBD2 and MECP2. A recent proteomics analysis shows that MECP2 binds to both 5-mC and 5-hmC, while MBD3 binds primarily to 5-hmC (Spruijt et al., 2013). Until recently, MECP2 was considered strictly as a global epigenetic repressor of gene expression. However, there is now a growing body of evidence supporting the idea that the function of
MECP2is to modulate neuronal transcription in both directions. MECP2 has a substantial role in modifying chromatin structure by binding to 5-hmC-enriched DNA regions within various gene domains, particularly in gene bodies where it correlates with those in the highest expression per-centiles (Melle´n, Ayata, Dewell, Kriaucionis, & Heintz, 2012). The increase of 5-hmC and the concomitant depletion of 5-mC from gene body regions produces a high 5-hmC/5-mC ratio which shows the highest correlation with active gene expression in the major cell types of the cerebellum (Purkinje cells, granule cells, and Bergman glia;Melle´n et al., 2012). Hence, recent data suggest that the phenotypic variability of RTT and other ASD-related conditions may be ASD-related to alterations in the 5-hmC/5-mC ratio and differential binding of MECP2 which could result from mutations or posttranslational modifications in a cell-, region-, or circuit-specific manner.
3. GENETIC DEFECTS WITH EPIGENETIC IMPLICATIONS
3.1. Methyl-CpG-binding protein 2 (MECP2)
More than 95% of RTT cases are caused by a mutation in theMECP2gene (Amir et al., 1999; Gonzales & Lasalle, 2010; Gra¨ff & Mansuy, 2009). RTT is an X-linked neurodevelopmental disorder that predominantly affects females and is characterized by autistic-like features, seizures, gait, ataxia, and stereotypical hand movements.MECP2is a member of the MBD family of proteins and is an essential epigenetic regulator of human brain develop-ment. MECP2 functions in coordination with multiple chromatin transcrip-tional repressors and is linked to the Sin3A repressor and HDACs (Della Ragione, Filosa, Scalabrı`, & D’Esposito, 2012; Nan et al., 1998). In the human neuronal SH-SY5Y cell line, the 63% of promoter-bound MECP2 is associated with actively transcribed genes (Yasui et al., 2007). Chromatin immunoprecipitation assays of Ntera2 (NT2) cells show that MECP2, DNMT1, DNMT3a, and HDAC2 bind to the same stretch of DNA in the glutamic acid decarboxylase 67 (GAD1) and Reelin (RELN) promoters (Kundakovic, Chen, Guidotti, & Grayson, 2009). Studies of MECP2
knock-out and over-expressing mice show that MECP2 positively regulates the expression of a wide range of genes by associating with the transcrip-tional activator CREB1 and binding to cAMP response element-binding (CREB) sites (Chahrour et al., 2008). In the last several years, studies support a role for MECP2 in (1) modulating RNA splicing (Young et al., 2005), (2) global alterations of chromatin condensation (Skene et al., 2010), (3) activat-ing retrotransposon transcription in neurons (Muotri et al., 2010), (4) pro-moting gene imprinting (Lasalle, 2007), and (5) regulating the expression of miRNAs important for brain development and plasticity (Klein et al., 2007; Urdinguio et al., 2010; Wu et al., 2010). Collectively, the evidence supports the concept that MeCP2 represents a complex and pleiotropic regulatory system that associates with 5-mC and 5-hmC and multiple ancillary proteins that serve to mediate downstream decisions regarding gene expression.
produces motor incoordination, while deletion in serotonergic neurons causes increased aggression (Samaco et al., 2009). Loss ofMECP2 in the amygdala impairs amygdala-dependent learning and memory (Adachi, Autry, Covington, & Monteggia, 2009), while loss in the hypothalamus affects feeding behaviors, aggression, and stress responses (Fyffe et al., 2008). Moreover, deletion of MECP2 in GABAergic neurons supports an important role for the corresponding protein in the function of inhibitory neurons (Chao et al., 2010). This study demonstrates that cortical wild-type GABAergic neurons express 50% more Mecp2 than non-GABAergic neurons. Mice with either a global loss of Mecp2in GABAergic neurons or a conditional loss in a subset of forebrain structures (striatum and cortex) develop autistic-like features including stereotyped behaviors, deficits in social behaviors, motor function, learning and memory, and sensorimotor gating (Chao et al., 2010).
The role of genetic and epigenetic factors was recently examined in a MZ twin pair discordant for RTT that shared the same de novomutation in exon 4 ofMeCP2(Miyake et al., 2013). Potential sources of discordance in RTT, in addition to non-shared environmental factors, include differen-tial patterns of X-chromosome inactivation. The exon 4 mutation in
MECP2in these twins was paternal in origin and occurred during spermato-genesis. Differential DNA methylation patterns located upstream of several genes relevant to brain function and skeletal tissue (includingBrain-type cre-atine kinase(CKB),Fyn proto-oncogene, andMohawk homeobox) were detected in skin fibroblasts. The corresponding mRNA levels also inversely correlate with DNA methylation levels (Miyake et al., 2013). CKB encodes a brain-selective isoform of creatine kinase which is important in brain energy homeostasis (Miyake et al., 2013) and to the function of GABAergic inter-neurons (Inoue, Yamada, Ueno, & Fukuda, 2006). No additional DNA dif-ferences, aside from methylation, were identified between these twins in SNPs, insertion–deletion polymorphisms (indels), CNVs, or patterns of X-chromosome inactivation that might account for the above findings.
Autistic-like features are characteristic of patients with MDS, including stereotypic hand movements, impaired speech development, or loss of speech after 4 years of age (Na, Nelson, Kavalali, & Monteggia, 2013). MDS patients also show stunted motor development, susceptibility to recur-rent respiratory infections, and anxiety (Ramocki, Tavyev, & Peters, 2010; Van Esch, 2012). While neurons isolated from MECP2-deficient mice show decreased excitation and increased inhibitory neurotransmission, MECP2
indicates thatMECP2gene dose bidirectionally affects excitatory transmis-sion (Na et al., 2013). In conclusion, monogenic defects inMECP2 expres-sion due to loss-of-function mutations (both SNPs and CNVs) present in RTT cases and gain-of-function mutations (MECP2duplication and trip-lication syndrome) are likely relevant to the altered epigenetic signature observed in ASDs.
3.2. DNA topoisomerase
Recent discoveries in chromatin biology have shown that the topology of DNA is complex and that local winding and unwinding of the DNA needs to occur to relax the torsional stress which accompanies nucleosome repositioning during transcription, as well as during DNA replication (Champoux, 2001). DNA topoisomerases 1 and 2 (TOP1 and TOP2) are vital to gene expression as they resolve the DNA supercoiling generated dur-ing transcription (Baranello, Levens, Gupta, & Kouzine, 2012; Capranico, Marinello, & Baranello, 2010; Pommier, 2013). Topoisomerases interact directly with RNA PolII and enable transcription elongation over long stretches of DNA (Capranico et al., 2010; Rahl et al., 2010; Wu, Shyy, Wang, & Liu, 1998). In a recent study (King et al., 2013), topoisomerase was shown to facilitate the expression of long genes, >200 kb, many of
which are associated with synaptic function and ASD. An impressive per-centage of long genes (27%) are also known ASD risk genes. By pharmaco-logically inhibiting TOP1 or knocking-down the expression of TOP1 or
TOP2b in neurons, the expression of long primary transcripts in mouse and human neurons are reduced (King et al., 2013). The TOP1 inhibitor, topotecan, mediates transcriptional silencing of theUBE3Aantisense tran-script (UBE3A-ATS) which is required for silencing the paternal allele of
Ube3a in mouse cortical neurons (Huang et al., 2011). Independently, topotecan was shown to stabilize the formation of RNA:DNA hybrids (R-loops) at G-skewed repeat elements within paternalSnord116, a small nuclear ncRNA molecule inhibiting expression of Ube3a-ATS (Powell et al., 2013). Topotecan has a similar effect on transcription of the human
UBE3Alocus (King et al., 2013). In the imprinting disorder AS, in which
UBE3A deficiency is mediated by mutations in the maternal allele, topotecan appears to be useful in activating expression of the paternal
3.3. Chromodomain helicase DNA-binding protein 8
The chromodomain helicase DNA-binding proteins are ATP-dependent chromatin-remodeling proteins that regulate transcription by alter the posi-tioning of nucleosomes over the DNA. Chromatin regulators contribute to both dynamic changes in gene expression and heritable states of gene expres-sion as required during brain development (Ronan, Wu, & Crabtree, 2013). Chromodomain helicase DNA-binding protein 8 (CHD8) bindsβ-catenin (CTNNB1) and negatively regulates WNT signaling which plays a critical role in early vertebrate development and morphogenesis. Mice lackingChd8
exhibit early embryonic death thought to the result of widespread p-53-induced apoptosis (Nishiyama et al., 2009). It was recently shown, by ChIP-on-chip analysis, that CHD8 binds to upward of 2000 transcrip-tionally active promoters in transformed cell lines (Subtil-Rodrı´guez et al., 2013). Interestingly, the targets of CHD8 binding are also targets of the E2F family of transcription factors that regulate genes related to the cell cycle and proliferation (Subtil-Rodrı´guez et al., 2013). The same study showed that CHD8 binds to promoters that are enriched in histone marks (H3K4me2 and H3K4me3) positively associated with transcription. Based on these and other studies, it appears that CHD8 acts as a negative transcrip-tional regulator in repressing p53 and CTNNB1 gene expression while positively modulating expression of large numbers of genes containing E2F-binding sites. Similar to MECP2 and TOP-1 and -2, CHD8 is involved in regulating large numbers of genes in different families at different stages during brain development.
from alterations in local chromatin conformation induced by these and other chromatin modifiers.
Additional large-scale resequencing studies identified several genes, in addition to CHD8, including cadherin-associated protein,β1 (CTNNB1), and dual specificity tyrosine-phosphorylation-regulated kinase 1A in multiple patients (reviewed in Krumm, O’Roak, Shendure, & Eichler, 2014). Protein-interacting networks built from mutations associated with ASD and intellectual disabilities indicate the presence of three large interconnected networks which include genes whose protein products function in (1) chromatin modification (primary hub gene¼CHD8), (2) WNT/CTNNB1 signaling pathway (primary hub¼CTNNB1), and (3) synaptic function (multiple hubs) (Krumm et al., 2014). As noted above, the downstream targets of the CHD proteins are only now being identified (Subtil-Rodrı´guez et al., 2013). As more and morede novomutations in ASD and related disorders are identified, genes that are epigenetically coupled to large downstream gene networks, such asCHD8(andMECP2,TOP1, and
2), are likely to be identified and replicated in additional studies. Moreover, deciphering the function of the CHD proteins during brain development is still largely under explored.
4. EPIGENETIC DYSREGULATION OF ASD CANDIDATE GENES
4.1. GABAergic genes
A role for the GABAergic inhibitory system in the pathophysiology of ASD has been consistently reported based on the results of several postmortem human brain studies. These studies include reports of (1) reduction in the number of GABAergic Purkinje cells in cerebellar cortex (Bailey et al., 1998; Bauman & Kemper, 1985; Whitney, Kemper, Bauman, & Blatt, 2004), (2) reduction by50% of the GABA-synthesizing enzymes glutamic acid decarboxylase 65 (GAD2) in cerebellum and GAD1 in parietal cortex of ASD patients (Fatemi et al., 2012; Fatemi, Halt, et al., 2002), (3) 40% down-regulation of GAD1 mRNA levels in cerebellar GABAergic Purkinje cells of ASD patients (Yip, Soghomonian, & Blatt, 2007), (4) reduction of GABAA receptor binding in the hippocampus and anterior and posterior
(BA-40), (7) reduction of GABRA1 and GABRB3 in cerebellum, and (8) reduction of GABRA1 receptor subunit in the superior frontal cortices (BA-9) of ASD subjects (Fatemi, Folsom, Reutiman, & Thuras, 2009). It has been hypothesized that the dysfunction of GABAA receptor subunits is
most likely responsible for the observed inhibitory signaling deficits which could explain the high comorbidity of RTT and ASD with seizures (80% in the RTT and 10–25% in ASD). In addition, 15–30% of children with epilepsy have ASD (Berg, Plioplys, & Tuchman, 2011; Tuchman & Rapin, 2002).
4.2. GAD67 (GAD1)
Glutamate decarboxylase is the enzyme that catalyzes the decarboxylation of glutamate to form GABA. GAD67 and GAD65 were named based on the molecular size of the corresponding protein on Western blots (Kaufman, Houser, & Tobin, 1991), and this nomenclature has been replaced with the gene symbols GAD1 and GAD2, respectively. GAD67 and 65 are the protein products of different genes located on separate chromosomes and have distinct intracellular locations and distinct cofactor/substrate requirements (Erlander, Tillakaratne, Feldblum, Patel, & Tobin, 1991). Studies from our laboratories show that the GAD1 promoter is GC rich (Chen, Dong, & Grayson, 2011) and its regulation negatively correlates with promoter proximal hypermethylation in SZ and bipolar (BP) subjects (Grayson et al., 2005; Guidotti et al., 2000; Ruzicka et al., 2007; Veldic, Guidotti, Maloku, Davis, & Costa, 2005). GAD1 expression in ASD is also likely regulated by epigenetic mechanisms including DNMT-mediated DNA hypermethylation and DNA demethylation involving the initial hydroxylation of 5-mC to form 5-hmC by members of the TET protein family (Dong, Gavin, Chen, & Davis, 2012). Both 5-hmC and TET1 are highly expressed in human cerebellum. Moreover, several studies have reported that Purkinje cell loss is one of the more consistent neuropatholog-ical findings in the postmortem cerebellum of ASD subjects (Bailey et al., 1998; Bauman & Kemper, 1985; Whitney et al., 2004).
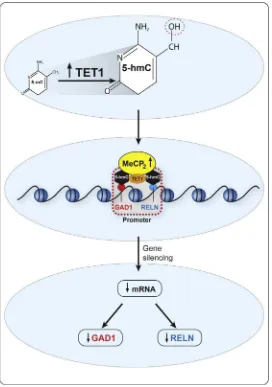
TET1 mRNAs and also the binding of MECP2, DNMT1, and TET1 to pro-moter and gene body regions ofGAD1and2were examined. The results show an upregulation of TET1 mRNA and increased TET1 binding to theGAD1promoter in ASD versus CON. The increased TET1 is associated with an enrichment of 5-hmC at theGAD1promoter region (Fig. 6.2). In contrast, the levels of 5-mC at the promoter and gene body ofGAD1were unchanged. There were no significant differences in the levels of DNMT1 mRNA and protein binding to any GAD1 promoter regions. The increased levels of 5-hmC at the promoter in ASD may facilitate the binding of MECP2. This could reflect an increased binding affinity of a posttranslationally modified form of MECP2, since the protein levels are not different in ASD versus CON (Zhubi et al., 2014). In ASD, MECP2 likely acts to repress
GAD1 as MECP2 binding negatively correlates with the levels of GAD1
mRNA. This repression shows some selectivity, since the levels of mRNA, MECP2 binding, and 5-hmC enrichment at the GAD2promoter do not change in cerebellum of ASD versus CON subjects (Zhubi et al., 2014).
4.3. Reelin
RELN is a large extracellular matrix glycoprotein expressed in corticolimbic GABAergic neurons (Kadriu, Guidotti, Chen, & Grayson, 2012; Ruzicka et al., 2007; Veldic et al., 2005, 2007). RELN that plays a pivotal role in neuronal migration and cortical lamination during embryonic development (D’Arcangelo & Curran, 1998). In addition, RELN also significantly impacts synaptic strength and connectivity in the adult brain (Costa et al., 2001; Levenson, Qiu, & Weeber, 2008). Abnormal RELN levels associated with decreased dendritic spine densities have been identified in several psy-chiatric conditions including ASD, SZ, BP disorder, Alzheimer disease, etc. Several studies using postmortem tissue from ASD subjects have reported significant reductions in RELN protein (180-kDa protein fragment) in cerebella of subjects with ASD (Fatemi, Kroll, & Stary, 2001), and a reduc-tion in RELN protein levels (410-, 310-, and 180-kDa protein fragments) in superior frontal cortex, parietal cortex, and cerebella of ASD subjects (Fatemi, 2005). Similar studies have shown a significant reduction in
(Grayson & Guidotti, 2013; Guidotti et al., 2000; Ruzicka et al., 2007; Veldic et al., 2005). Moreover, recent studies indicate that sex hormones might play a role in the methylation of the RELNpromoter, particularly in the pubertal and postpubertal periods, which coincide with a worsening of autistic behaviors and the onset of SZ (Lintas & Persico, 2010). In our study of postmortem cerebellar samples, we observed that decreased RELN mRNA level in the cerebellum of ASD was associated with increased bind-ing of MECP2, which coincides with an enrichment of 5-hmC at theRELN
promoter. This methylcytosine modification, catalyzed by TET1, is upregulated in ASD postmortem brain (Zhubi et al., 2014). As shown in
Fig. 6.2, increased 5-hmC at theRELNandGAD1promoters is coincident with decreased transcription and decreased levels of the corresponding pro-teins. It is currently unclear why the increase in 5-hmC correlates with reduced expression in psychiatric disorders.
4.4. GABA β3
15q11.2-13.3 is a genomic region prone to chromosomal rearrangements of various sizes that contains three distinct ASD susceptibility and CNV loci that vary in their genomic boundaries (Menashe, Larsen, & Banerjee-Basu, 2013). This is a complex locus that includes several imprinted genes, including
GABRB3, GABRA5, and GABRG3. Chromatin immunoprecipitation studies show that MECP2 binds to methylated sites in the first intron of
GABRB3(Hogart, Nagarajan, Patzel, Yasui, & Lasalle, 2007). The expres-sion and binding of the sequence-specific transcription factor Specificity Pro-tein 1 (Sp1) in various brain regions of postmortem ASD subjects showed increased binding to DNA that negatively correlated with the levels of expression of several ASD candidate genes includingGABRB3andRELN
4.5. Oxytocin receptor (OXTR)
OXT is a peptide hormone important for social cognition and behaviors, and facilitates affiliative bonding in humans and animals ( Meyer-Lindenberg, Domes, Kirsch, & Heinrichs, 2011). OXT mediates its effects through interaction with the OXT receptor (OXTR). An increasing num-ber of studies support a role for the involvement of the OXTR in the path-ophysiology of ASD (Andari et al., 2010; Guastella et al., 2010; Heinrichs, Von Dawans, & Domes, 2009). One plausible mechanism for the down-regulation ofOXTRin ASD patients is an increased methylation of a region spanning the 30portion of the first exon and 50part of the first intron which
suppresses transcription (Kusui et al., 2001). Increased OXTR promoter methylation in DNA of peripheral blood mononuclear cells was demon-strated in 20 individuals with ASD, as compared with the same number of CON subjects (Gregory et al., 2009). A similar finding was reported using DNA from temporal cortices of ASD subjects which show an association between increased methylation and decreased mRNA expression (Gregory et al., 2009). In a more recent study, a genome-wide analysis examined epigenetic differences in the methylation profiles of whole-blood DNA in a cohort of MZ twins discordant for ASD (Wong et al., 2013). These results support previous reports (Dempster et al., 2011; Rakyan et al., 2011) of DNA methylation profiles with phenotypic differences between MZ twins discordant for ASD and ASD-related traits (matched for genotype, age, sex, and maternal environment). Interestingly, there is a significant correlation between DNA methylation profiles and ASD symp-tom scores suggesting a relationship between ASD phenotype severity and epigenetic variation at specific gene promoters (Wong et al., 2013).
4.6. Brain-derived neutrophic factor (BDNF)
decreased in children diagnosed with ASD later in life as compared with matched controls (Abdallah et al., 2013). In addition, NT-4 and TGF-β mRNA levels tended to decrease, although the decreases did not reach statistical significance (Abdallah et al., 2013). The expression ofBDNF is altered in several psychiatric disorders (major depressive (MD) disorder, BP, SZ, addiction, ASD, Alzheimer’s, Parkinson’s, and Huntington disease, see Autry & Monteggia, 2012).
Numerous studies suggest that adverse social interactions, trauma, and environmental factors during early development alter brain levels of BDNF likely through epigenetic mechanisms including DNA methylation, histone modifications, and miRNA/ncRNA expression (Ikegame et al., 2013; Roth & Sweatt, 2011). The differential sensitivities of the mouse and rat promoters to epigenetic regulation have been described (Aid, Kazantseva, Piirsoo, Palm, & Timmusk, 2007).MECP2binds and regulates transcription ofBDNFin cooperation with REST1 (RE1-silencing factor) and other fac-tors (Karpova, 2014). This suggests the possibility that both factors interact negatively to regulateBDNFor that these repressors cooperate at different
BDNF promoters. In this context, several studies have suggested potential environmental interventions which can restore BDNF levels by modulating epigenetic mechanisms at the corresponding promoters. One example is that music and physical exercise may modulate the restoration of BDNF-dependent synaptic plasticity (Angelucci, Ricci, Padua, Sabino, & Tonali, 2007). Moreover, improvements in cognition are dependent on BDNF-mediated neurogenesis and synapse formation (Lee et al., 2006).
4.7. Ubiquitin–protein ligase E3A (UBE3A)
UBE3A is a “Homologous to the E6-AP Carboxyl Terminus” domain E3 ubiquitin ligase specialized in protein degradation. As indicated above (see
communication defects, microcephaly, seizures, and autistic features. Genetic analyses of the 15q11-q13 region reveal significant linkage disequi-librium among patients with ASD (Cook et al., 1998; Nurmi et al., 2001), while duplication of this chromosomal region on the maternally derived chromosome is associated with ASD (Cook et al., 1997; Moreno-De-Luca et al., 2013). Mice with three copies ofUbe3a exhibit behaviors that are considered typical of the ASD phenotype (Smith et al., 2011). That is, these mice prefer less social interactions (fewer vocalizations in the pres-ence of a second mouse of the same sex) and increased repetitive behaviors (increased self-grooming) (Smith et al., 2011).
Implications of the epigenetic regulation ofUBE3A expression have been recently described (Flashner, Russo, Boileau, Leong, & Gallicano, 2013; Mabb, Judson, Zylka, & Philpot, 2011). Changes in CpG island methylation proximal to theUBE3A CpG island correlate with reduced mRNA levels in a small number of AS patients (Jiang et al., 2004). A defi-ciency inMECP2results in a significant reduction of UBE3A and GABA β3 mRNA expression in mouse brain without also disrupting allele-specific expression (Samaco, Hogart, & Lasalle, 2005). Furthermore, sig-nificant reductions inUBE3AandGABRB3mRNA levels are observed in human postmortem AS, RTT, and ASD subjects, implicating UBE3A
in these disorders (Jiang et al., 2004). A recent study further elaborated on the potential mechanism of chromatin R-loop formation and the activa-tion of UBE3A expression (Powell et al., 2013). They showed that the topoisomerase inhibitor (topotecan) and possible AS drug act to stabilize RNA:DNA hybrids (so-called R-loops) within the paternal Snord116 gene which increases chromatin decondensation and reduces Ube3a-ATS expression with concomittant increased paternal Ube3a expression (Powell et al., 2013).
4.8. Engrailed-2 (EN-2)
and delayed maturation and migration of germinal layers are observed (Baader, Sanlioglu, Berrebi, Parker-Thornburg, & Oberdick, 1998; Holst et al., 2008; Jankowski et al., 2004). WhenEN2is overexpressed during late fetal and early postnatal development, it results in a deficit of dendritogenesis and alterations in the patterns of afferent innervation (Cheng et al., 2010; Logan et al., 1992; Sillitoe, Vogel, & Joyner, 2010). Postmortem studies of human ASD cerebellum consistently report a reduction of Purkinje cell numbers. The failure ofEN2to downregulate normally during the critical perinatal period (which coincides with Purkinje cell maturation) may impair cognitive functioning in ASD (Fatemi et al., 2012; O’Halloran, Kinsella, & Storey, 2012). A recent study conducted on a cohort of ASD cerebellar sam-ples measured the epigenetic profile of EN2, which included measures of global DNA methylation, EN2promoter methylation, andEN-2mRNA and protein levels (James, Shpyleva, Melnyk, Pavliv, & Pogribny, 2013). In addition, they carried out measurements of the status of H3K27me3 (repressed) and H3K4me3 (activated) using chromatin immunoprecipita-tion. The data showed thatEN2promoter hypermethylation paradoxically correlates with a significant increase in EN2 expression and protein levels (James et al., 2013). In contrast, the levels of H3K27me3 deceased, while H3K4me3 increased in ASD. The authors conclude that failure of EN2
to downregulate during Purkinje cell maturation (during late prenatal and early postnatal developments) likely contributes to the cerebellar dysfunc-tion observed in ASD (James et al., 2013).
4.9. SH3 and multiple ankyrin repeat domains (SHANK3)
SHANK3 is a multifunctional synaptic protein associated with ASD. This protein is composed of several domains that define the other proteins with which SHANK3 interacts and mediate it’s functions at the postsynaptic den-sities of excitatory synapses (Grabrucker et al., 2011; Jiang & Ehlers, 2013). Moreover, microdeletions in chromosome 22q13.3 syndrome, as occur in Phelan-McDermid syndrome, are a condition in which SHANK3 plays a crucial role and is associated with autistic-like features (Phelan & McDermid, 2012).
islands (CGI-1 to CGI-5) ofSHANK3in DNA from postmortem brains of ASD and CON subjects (Zhu et al., 2013). The data show increased levels of methylation in CpG islands 1-2 and -4 in15% of ASD subjects (cerebel-lum and cortex). Moreover, the methylation significantly impacts expression and alternative splicing of theSHANK3isoforms measured in those tissues (Zhu et al., 2013). To validate these results, human cell culturesin vitrowere treated with the DNA methylation inhibitor 5-azacytidine which reversed the CGI methylation status and ultimately altered SHANK3 isoform-specific expression. This study, and others, indicate that there is a strong con-nection between altered DNA methylation levels and gene expression in ASD which warrants additional investigation.
5. ENVIRONMENTAL MODEL OF AUTISM
postmortem subjects. Furthermore, mitochondrial-induced oxidative stress also disrupts epigenetic mechanisms in the brain of ASD (Legido, Jethva, & Goldenthal, 2013).
6. CONCLUSIONS
phenotypic stratification that utilizes the full spectrum of ASD-associated behaviors are likely to provide better insight into how susceptibility genes interact with biological factors in the development of ASD (Hu & Lai, 2013). The Human Phenotype Ontology (HPO) project has assembled a comprehensive set of phenotypic abnormalities accompanying human diseases in a searchable database to allow crossreferencing of symptoms with genetic diseases (K€ohler et al., 2014). Databases like HPO and others, such as OMIM, Orphanet, and phenoDB, will allow for the integration of genome sequence data with phenotypic abnormalities, which will eventually allow investigators and physicians to focus on those genes relevant to the specific patient under examination. While it is still too early to predict, this will likely lead the way to personalized therapy that is centered on the treatment of specific symptoms rather than a more generic hit-or-miss approach.
ACKNOWLEDGMENTS
This work was supported by a Lever Award from the Chicago Biomedical Consortium and NIH grants 1P50 MH094267 (E. H. C.), 1P50 HD055751 (E. H. C.), and 5 R01 MH093348-03 (A. G.). The authors would like to thank Myhidin Shehu, M.D., for help in preparing the figures.
REFERENCES
Abdallah, M. W., Mortensen, E. L., Greaves-Lord, K., Larsen, N., Bonefeld-Jørgensen, E. C., Nørgaard-Pedersen, B., et al. (2013). Neonatal levels of neurotrophic factors and risk of autism spectrum disorders.Acta Psychiatrica Scandinavica,128, 61–69.
Abrahams, B. S., & Geschwind, D. H. (2008). Advances in autism genetics: On the threshold of a new neurobiology.Nature Reviews. Genetics,9, 341–355.
Adachi, M., Autry, A. E., Covington, H. E., 3rd., & Monteggia, L. M. (2009). MeCP2-mediated transcription repression in the basolateral amygdala may underlie heightened anxiety in a mouse model of Rett syndrome.Journal of Neuroscience, 29, 4218–4227.
Aid, T., Kazantseva, A., Piirsoo, M., Palm, K., & Timmusk, T. (2007). Mouse and rat BDNF gene structure and expression revisited. Journal of Neuroscience Research, 85, 525–535.
Albrecht, U., Sutcliffe, J. S., Cattanach, B. M., Beechey, C. V., Armstrong, D., Eichele, G., et al. (1997). Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons.Nature Genetics,17, 75–78.
Amir, R. E., Van Den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., & Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2.Nature Genetics,23, 185–188.
Andari, E., Duhamel, J. R., Zalla, T., Herbrecht, E., Leboyer, M., & Sirigu, A. (2010). Pro-moting social behavior with oxytocin in high-functioning autism spectrum disorders.
Proceedings of the National Academy of Sciences of the United States of America, 107, 4389–4394.
Angelucci, F., Ricci, E., Padua, L., Sabino, A., & Tonali, P. A. (2007). Music exposure dif-ferentially alters the levels of brain-derived neurotrophic factor and nerve growth factor in the mouse hypothalamus.Neuroscience Letters,429, 152–155.
Autry, A. E., & Monteggia, L. M. (2012). Brain-derived neurotrophic factor and neuropsy-chiatric disorders.Pharmacological Reviews,64, 238–258.
Baader, S. L., Sanlioglu, S., Berrebi, A. S., Parker-Thornburg, J., & Oberdick, J. (1998). Ectopic overexpression of engrailed-2 in cerebellar Purkinje cells causes restricted cell loss and retarded external germinal layer development at lobule junctions.Journal of Neu-roscience,18, 1763–1773.
Bailey, A., Le Couteur, A., Gottesman, I., Bolton, P., Simonoff, E., Yuzda, E., et al. (1995). Autism as a strongly genetic disorder: Evidence from a British twin study.Psychological Medicine,25, 63–77.
Bailey, A., Luthert, P., Dean, A., Harding, B., Janota, I., Montgomery, M., et al. (1998). A clinicopathological study of autism.Brain,121, 889–905.
Bambini-Junior, V., Rodrigues, L., Behr, G. A., Moreira, J. C., Riesgo, R., & Gottfried, C. (2011). Animal model of autism induced by prenatal exposure to valproate: Behavioral changes and liver parameters.Brain Research,23, 8–16.
Bannister, A. J., & Kouzarides, T. (2011). Regulation of chromatin by histone modifications.
Cell Research,21, 381–395.
Baranello, L., Levens, D., Gupta, A., & Kouzine, F. (2012). The importance of being supercoiled: How DNA mechanics regulate dynamic processes.Biochimica et Biophysica Acta,1819, 632–638.
Baron-Cohen, S., Scott, F. J., Allison, C., Williams, J., Bolton, P., Matthews, F. E., et al. (2009). Prevalence of autism-spectrum conditions: UK school-based population study.
British Journal of Psychiatry,194, 500–509.
Barreto, G., Scha¨fer, A., Marhold, J., Stach, D., Swaminathan, S. K., Handa, V., et al. (2007). Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation.
Nature,445, 671–675.
Batsukh, T., Pieper, L., Koszucka, A. M., Von Velsen, N., Hoyer-Fender, S., Elbracht, M., et al. (2010). CHD8 interacts with CHD7, a protein which is mutated in CHARGE syndrome.Human Molecular Genetics,19, 2858–2866.
Bauman, M., & Kemper, T. L. (1985). Histoanatomic observations of the brain in early infan-tile autism.Neurology,35, 866–874.
Ben-David, E., & Shifman, S. (2013). Combined analysis of exome sequencing points toward a major role for transcription regulation during brain development in autism.Molecular Psychiatry,18, 1054–1056.
Berg, A. T., Plioplys, S., & Tuchman, R. (2011). Risk and correlates of autism spectrum disorder in children with epilepsy: A community-based study.Journal of Child Neurology,
26, 540–547.
Beri, S., Tonna, N., Menozzi, G., Bonaglia, M. C., Sala, C., & Giorda, R. (2007). DNA methylation regulates tissue-specific expression of Shank3. Journal of Neurochemistry,
101, 1380–1391.
Berry, R. J. (2013). Maternal prenatal folic acid supplementation is associated with a reduc-tion in development of autistic disorder.Journal of Pediatrics,163, 303–304.
Bestor, T. H. (1990). DNA methylation: Evolution of a bacterial immune function into a regulator of gene expression and genome structure in higher eukaryotes.Philosophical Transactions of the Royal Society of London B,326, 179–187.
Bhutani, N., Burns, D. M., & Blau, H. M. (2011). DNA demethylation dynamics.Cell,146, 866–872.
Bittigau, P., Sifringer, M., Genz, K., Reith, E., Pospischil, D., Govindarajalu, S., et al. (2002). Antiepileptic drugs and apoptotic neurodegeneration in the developing brain.
Proceedings of the National Academy of Sciences of the United States of America, 99, 15089–15094.
Blatt, G. J., Fitzgerald, C. M., Guptill, J. T., Booker, A. B., Kemper, T. L., & Bauman, M. L. (2001). Density and distribution of hippocampal neurotransmitter receptors in autism: An autoradiographic study.Journal of Autism and Developmental Disorders,31, 537–543. Bromley, R. L., Mawer, G. E., Briggs, M., Cheyne, C., Clayton-Smith, J., Garcia-Finana, M., et al. (2013). The prevalence of neurodevelopmental disorders in children prenatally exposed to antiepileptic drugs. Journal of Neurology, Neurosurgery, and Psychiatry, 84, 637–643.
Bromley, R. L., Mawer, G., Clayton-Smith, J., Baker, G. A., & Liverpool and Manchester Neurodevelopment Group. (2008). Autism spectrum disorders following in utero expo-sure to antiepileptic drugs.Neurology,71, 1923–1924.
Capranico, G., Marinello, J., & Baranello, L. (2010). Dissecting the transcriptional functions of human DNA topoisomerase I by selective inhibitors: Implications for physiological and therapeutic modulation of enzyme activity. Biochimica et Biophysica Acta, 1806, 240–250.
Chahrour, M., Jung, S. Y., Shaw, C., Zhou, X., Wong, S. T., Qin, J., et al. (2008). MeCP2, a key contributor to neurological disease, activates and represses transcription.Science,320, 1224–1229.
Chakravarthy, S., Park, Y. J., Chodaparambil, J., Edayathumangalam, R. S., & Luger, K. (2005). Structure and dynamic properties of nucleosome core particles.FEBS Letters,
579, 895–898.
Champoux, J. J. (2001). DNA topoisomerases: Structure, function, and mechanism.Annual Review of Biochemistry,70, 369–413.
Chandler, C. H., Chari, S., & Dworkin, I. (2013). Does your gene need a background check? How genetic background impacts the analysis of mutations, genes, and evolution.Trends in Genetics,29, 358–366.
Chao, H. T., Chen, H., Samaco, R. C., Xue, M., Chahrour, M., & Yoo, J. (2010). Dys-function in GABA signalling mediates autism-like stereotypies and Rett syndrome phe-notypes.Nature,468, 263–269.
Chao, H. T., Zoghbi, H. Y., & Rosenmund, C. (2007). MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number.Neuron,6, 58–65.
Chaste, P., & Leboyer, M. (2012). Autism risk factors: Genes, environment, and gene-environment interactions.Dialogues in Clinical Neuroscience,14, 281–292.
Chen, Y., Dong, E., & Grayson, D. R. (2011). Analysis of the GAD1 promoter: Trans-acting factors and DNA methylation converge on the 50untranslated region.Neuropharmacology, 60, 1075–1087.
Chen, Z. X., & Riggs, A. D. (2011). DNA methylation and demethylation in mammals.
Journal of Biological Chemistry,286, 18347–18353.
Cheng, Y., Sudarov, A., Szulc, K. U., Sgaier, S. K., Stephen, D., Turnbull, D. H., et al. (2010). The engrailed homeobox genes determine the different foliation patterns in the vermis and hemispheres of the mammalian cerebellum.Development,137, 519–529. Chess, S. (1971). Autism in children with congenital rubella.Journal of Autism and Childhood
Schizophrenia,1, 33–47.
Ching, T. T., Maunakea, A. K., Jun, P., Hong, C., Zardo, G., & Pinkel, D. (2005). Epigenome analyses using BAC microarrays identify evolutionary conservation of tissue-specific methylation of SHANK3.Nature Genetics,37, 645–651.
Christensen, J., Grønborg, T. K., Sørensen, M. J., Schendel, D., Parner, E. T., Pedersen, L. H., et al. (2013). Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism.JAMA,309, 1696–1703.
Clayton-Smith, J., & Donnai, D. (1995). Fetal valproate syndrome.Journal of Medical Genetics,
32, 724–747.
Cook, E. H., Jr., Courchesne, R. Y., Cox, N. J., Lord, C., Gonen, D., & Guter, S. J. (1998). Linkage-disequilibrium mapping of autistic disorder, with 15q11-13 markers.American Journal of Human Genetics,62, 1077–1083.
Cook, E. H., Jr., Lindgren, V., Leventhal, B. L., Courchesne, R., Lincoln, A., Shulman, C., et al. (1997). Autism or atypical autism in maternally but not paternally derived proximal 15q duplication.American Journal of Human Genetics,60, 928–934.
Cook, E. H., Jr., & Scherer, S. W. (2008). Copy-number variations associated with neuro-psychiatric conditions.Nature,455, 919–923.
Costa, E., Davis, J., Grayson, D. R., Guidotti, A., Pappas, G. D., & Pesold, C. (2001). Den-dritic spine hypoplasticity and downregulation of reelin and GABAergic tone in schizo-phrenia vulnerability.Neurobiology of Disease,8, 723–742.
Czyz, W., Morahan, J. M., Ebers, G. C., & Ramagopalan, S. V. (2012). Genetic, environ-mental and stochastic factors in monozygotic twin discordance with a focus on epigenetic differences.BMC Medicine,10, 93.
D’Arcangelo, G., & Curran, T. (1998). Reeler: New tales on an old mutant mouse.Bioessays,
De Lacy, N., & King, B. H. (2013). Revisiting the relationship between autism and schizophre-nia: Toward an integrated neurobiology.Annual Review of Clinical Psychology,9, 555–587. Della Ragione, F., Filosa, S., Scalabrı`, F., & D’Esposito, M. (2012). MeCP2 as a
genome-wide modulator: The renewal of an old story.Frontiers in Genetics,11, 181.
Dempster, E., Pidsley, R., Schalkwyk, L., Owens, S., Georgiades, A., Kane, F., et al. (2011). Disease-associated epigenetic changes in monozygotic twins discordant for schizophre-nia and bipolar disorder.Human Molecular Genetics,20, 4786–4796.
Deth, R. C. (2013). Autism: A redox/methylation disorder.Global Advances in Health and Medicine,2, 68–73.
Di Ruscio, A., Ebralidze, A. K., Benoukraf, T., Amabile, G., Goff, L. A., Terragni, J., et al. (2013). DNMT1-interacting RNAs block gene-specific DNA methylation.Nature,503, 371–376.
Dong, E., Gavin, D. P., Chen, Y., & Davis, J. (2012). Upregulation of TET1 and down-regulation of APOBEC3A and APOBEC3C in the parietal cortex of psychotic patients.
Translational Psychiatry,2, e159.
Dufour-Rainfray, D., Vourc’h, P., Tourlet, S., Guilloteau, D., Chalon, S., & Andres, C. R. (2011). Fetal exposure to teratogens: Evidence of genes involved in autism.Neuroscience and Biobehavioral Reviews,35, 1254–1265.
Eapen, V., Crncˇec, R., & Walter, A. (2013). Exploring links between genotypes, pheno-types, and clinical predictors of response to early intensive behavioral intervention in autism spectrum disorder.Frontiers in Human Neuroscience,7, 567.
Erlander, M. G., Tillakaratne, N. J., Feldblum, S., Patel, N., & Tobin, A. J. (1991). Two genes encode distinct glutamate decarboxylases.Neuron,7, 91–100.
Fatemi, S. H. (2005). Reelin glycoprotein: Structure, biology and roles in health and disease.
Molecular Psychiatry,10, 251–257.
Fatemi, S. H., Aldinger, K. A., Ashwood, P., Bauman, M. L., Blaha, C. D., Blatt, G. J., et al. (2012). Consensus paper: Pathological role of the cerebellum in autism.Cerebellum,11, 777–807.
Fatemi, S. H., Folsom, T. D., Reutiman, T. J., & Thuras, P. D. (2009). Expression of GABA(B) receptors is altered in brains of subjects with autism.Cerebellum,8, 64–69. Fatemi, S. H., Halt, A. R., Stary, J. M., Kanodia, R., Schulz, S. C., & Realmuto, G. R.
(2002). Glutamic acid decarboxylase 65 and 67 kDa proteins are reduced in autistic pari-etal and cerebellar cortices.Biological Psychiatry,52, 805–810.
Fatemi, S. H., Kroll, J. L., & Stary, J. M. (2001). Altered levels of Reelin and its isoforms in schizophrenia and mood disorders.Neuroreport,12, 3209–3215.
Fatemi, S. H., Stary, J. M., & Egan, E. A. (2002). Reduced blood levels of reelin as a vul-nerability factor in pathophysiology of autistic disorder.Cellular and Molecular Neurobiol-ogy,22, 139–152.
Favre, M. R., Barkat, T. R., Lamendola, D., Khazen, G., Markram, H., & Markram, K. (2013). General developmental health in the VPA-rat model of autism.Frontiers in Behav-ioral Neuroscience,7, 88.
Fenouil, R., Cauchy, P., Koch, F., Descostes, N., Cabeza, J. Z., Innocenti, C., et al. (2012). CpG islands and GC content dictate nucleosome depletion in a transcription-independent manner at mammalian promoters.Genome Research,22, 2399–2408. Flashner, B. M., Russo, M. E., Boileau, J. E., Leong, D. W., & Gallicano, G. I. (2013).
Epi-genetic factors and autism spectrum disorders.Neuromolecular Medicine,15, 339–350. Fyffe, S. L., Neul, J. L., Samaco, R. C., Chao, H. T., Ben-Shachar, S., & Moretti, P. (2008).
Deletion of Mecp2 in Sim1-expressing neurons reveals a critical role for MeCP2 in feeding behavior, aggression, and the response to stress.Neuron,59, 947–958. Gardener, H., Spiegelman, D., & Buka, S. L. (2011). Perinatal and neonatal risk factors for
Gardiner-Garden, M., & Frommer, M. (1987). CpG islands in vertebrate genomes.Journal of Molecular Biology,196, 261–282.
Ginno, P. A., Lott, P. L., Christensen, H. C., Korf, I., & Che´din, F. (2012). R-loop forma-tion is a distinctive characteristic of unmethylated human CpG island promoters. Molec-ular Cell,45, 814–825.
Goll, M. G., & Bestor, T. H. (2005). Eukaryotic cytosine methyltransferases.Annual Review of Biochemistry,74, 481–514.
Gonzales, M. L., & Lasalle, J. M. (2010). The role of MeCP2 in brain development and neu-rodevelopmental disorders.Current Psychiatry Reports,12, 127–134.
Grabrucker, A. M. (2013). Environmental factors in autism.Frontiers in Psychiatry,3, 118. Grabrucker, A. M., Knight, M. J., Proepper, C., Bockmann, J., Joubert, M., Rowan, M.,
et al. (2011). Concerted action of zinc and ProSAP/Shank in synaptogenesis and synapse maturation.EMBO Journal,30, 569–581.
Gra¨ff, J., Kim, D., Dobbin, M. M., & Tsai, L. H. (2011). Epigenetic regulation of gene expression in physiological and pathological brain processes.Physiological Reviews,91, 603–649.
Gra¨ff, J., & Mansuy, I. M. (2009). Epigenetic dysregulation in cognitive disorders.European Journal of Neuroscience,30, 1–8.
Grayson, D. R., & Guidotti, A. (2013). The dynamics of DNA methylation in schizophrenia and related psychiatric disorders.Neuropsychopharmacology,38, 138–166.
Grayson, D. R., Jia, X., Chen, Y., Sharma, R. P., Mitchell, C. P., Guidotti, A., et al. (2005). Reelin promoter hypermethylation in schizophrenia.Proceedings of the National Academy of Sciences of the United States of America,102, 9341–9346.
Gregory, S. G., Connelly, J. J., Towers, A. J., Johnson, J., Biscocho, D., Markunas, C. A., et al. (2009). Genomic and epigenetic evidence for oxytocin receptor deficiency in autism.BMC Medicine,7, 62.
Guastella, A. J., Einfeld, S. L., Gray, K. M., Rinehart, N. J., Tonge, B. J., Lambert, T. J., et al. (2010). Intranasal oxytocin improves emotion recognition for youth with autism spec-trum disorders.Biological Psychiatry,67, 692–694.
Guidotti, A., Auta, J., Davis, J. M., Di-Giorgi-Gerevini, V., Dwivedi, Y., Grayson, D. R., et al. (2000). Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: A postmortem brain study.Archives of General Psy-chiatry,57, 1061–1069.
Guidotti, A., Dong, E., Kundakovic, M., Satta, R., Grayson, D. R., & Costa, E. (2009). Characterization of the action of antipsychotic subtypes on valproate-induced chromatin remodeling.Trends in Pharmacological Sciences,30, 55–60.
Guilmatre, A., Dubourg, C., Mosca, A. L., Legallic, S., Goldenberg, A., Drouin-Garraud, V., et al. (2009). Recurrent rearrangements in synaptic and neurodevelopmental genes and shared biologic pathways in schizophrenia, autism, and mental retardation.Archives of General Psychiatry,66, 947–956.
Guo, J. U., Ma, D. K., Mo, H., Ball, M. P., Jang, M. H., Bonaguidi, M. A., et al. (2011). Neuronal activity modifies the DNA methylation landscape in the adult brain.Nature Neuroscience,14, 1345–1351.
Guo, J. U., Su, Y., Shin, J. H., Shin, J., Li, H., Xie, B., et al. (2014). Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain.Nature Neurosci-ence,17, 215–222.
Hallmayer, J., Cleveland, S., Torres, A., Phillips, J., Cohen, B., Torigoe, T., et al. (2011). Genetic heritability and shared environmental factors among twin pairs with autism.
Archives of General Psychiatry,68, 1095–1102.
Hendrich, B., Hardeland, U., Ng, H. H., Jiricny, J., & Bird, A. (1999). The thymine gly-cosylase MBD4 can bind to the product of deamination at methylated CpG sites.
Nature,401, 301–304.
Hoerder-Suabedissen, A., Oeschger, F. M., Krishnan, M. L., Belgard, T. G., Wang, W. Z., Lee, S., et al. (2013). Expression profiling of mouse subplate reveals a dynamic gene net-work and disease association with autism and schizophrenia.Proceedings of the National Academy of Sciences of the United States of America,110, 3555–3560.
Hogart, A., Nagarajan, R. P., Patzel, K. A., Yasui, D. H., & Lasalle, J. M. (2007). 15q11-13 GABAA receptor genes are normally biallelically expressed in brain yet are subject to epigenetic dysregulation in autism-spectrum disorders. Human Molecular Genetics,16, 691–703.
Holst, M. I., Maercker, C., Pintea, B., Masseroli, M., Liebig, C., Jankowski, J., et al. (2008). Engrailed-2 regulates genes related to vesicle formation and transport in cerebellar Purkinje cells.Molecular and Cellular Neurosciences,38, 495–504.
Houston, I., Peter, C. J., Mitchell, A., Straubhaar, J., Rogaev, E., & Akbarian, S. (2013). Epigenetics in the human brain.Neuropsychopharmacology,38(1), 183–197.
Hu, V. W., & Lai, Y. (2013). Developing a predictive gene classifier for autism spectrum disorders based upon differential gene expression profiles of phenotypic subgroups.North American Journal of Medicine & Science (Boston),6, 107–116.
Huang, H. S., Allen, J. A., Mabb, A. M., King, I. F., Miriyala, J., Taylor-Blake, B., et al. (2011). Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons.
Nature,481, 185–189.
Huguet, G., Ey, E., & Bourgeron, T. (2013). The genetic landscapes of autism spectrum dis-orders.Annual Review of Genomics and Human Genetics,14, 191–213.
Ikegame, T., Bundo, M., Murata, Y., Kasai, K., Kato, T., & Iwamoto, K. (2013). DNA methylation of the BDNF gene and its relevance to psychiatric disorders.Journal of Human Genetics,58, 434–438.
Illingworth, R. S., & Bird, A. P. (2009). CpG islands—‘A rough guide’.FEBS Letters,583, 1713–1720.
Ingram, J. L., Peckham, S. M., Tisdale, B., & Rodier, P. M. (2000). Prenatal exposure of rats to valproic acid reproduces the cerebellar anomalies associated with autism. Neu-rotoxicology and Teratology,22, 319–324.
Inoue, K., Yamada, J., Ueno, S., & Fukuda, A. (2006). Brain-type creatine kinase activates neuron-specific K + –Cl co-transporter KCC2.Journal of Neurochemistry,96, 598–608. Ito, S., Shen, L., Dai, Q., Wu, S. C., Collins, L. B., Swenberg, J. A., et al. (2011). Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine.Science,333, 1300–1303.
James, S. J., Shpyleva, S., Melnyk, S., Pavliv, O., & Pogribny, I. P. (2013). Complex epige-netic regulation of engrailed-2 (EN-2) homeobox gene in the autism cerebellum. Trans-lational Psychiatry,3, e232.
Jankowski, J., Holst, M. I., Liebig, C., Oberdick, J., & Baader, S. L. (2004). Engrailed-2 neg-atively regulates the onset of perinatal Purkinje cell differentiation.The Journal of Com-parative Neurology,472, 87–99.
Jenuwein, T., & Allis, C. D. (2001). Translating the histone code.Science,293, 1074–1080. Jiang, Y. H., & Ehlers, M. D. (2013). Modeling autism by SHANK gene mutations in mice.
Neuron,78, 8–27.
Jiang, Y. H., Sahoo, T., Michaelis, R. C., Bercovich, D., Bressler, J., & Kashork, C. D. (2004). A mixed epigenetic/genetic model for oligogenic inheritance of autism with a limited role for UBE3A.American Journal of Medical Genetics,131, 1–10.
Kar, S., Deb, M., Sengupta, D., Shilpi, A., Parbin, S., Torrisani, J., et al. (2012). An insight into the various regulatory mechanisms modulating human DNA methyltransferase 1 stability and function.Epigenetics,7, 994–1007.
Karlic´, R., Chung, H. R., Lasserre, J., Vlahovicek, K., & Vingron, M. (2010). Histone mod-ification levels are predictive for gene expression.Proceedings of the National Academy of Sciences of the United States of America,107, 2926–2931.
Karpova, N. N. (2014). Role of BDNF epigenetics in activity-dependent neuronal plasticity.
Neuropharmacology,76(Pt C), 709–718.
Kaufman, D. L., Houser, C. R., & Tobin, A. J. (1991). Two forms of the gamma-aminobutyric acid synthetic enzyme glutamate decarboxylase have distinct intraneuronal distributions and cofactor interactions.Journal of Neurochemistry,56, 720–723.
Kemper, T. L., & Bauman, M. (1998). Neuropathology of infantile autism.Journal of Neu-ropathology and Experimental Neurology,157, 645–652.
King, I. F., Yandava, C. N., Mabb, A. M., Hsiao, J. S., Huang, H. S., Pearson, B. L., et al. (2013). Topoisomerases facilitate transcription of long genes linked to autism.Nature,
501, 58–62.
Klei, L., Sanders, S. J., Murtha, M. T., Hus, V., Lowe, J. K., & Willsey, A. J. (2012). Com-mon genetic variants, acting additively, are a major source of risk for autism.Molecular Autism,3, 9.
Klein, M. E., Lioy, D. T., Ma, L., Impey, S., Mandel, G., & Goodman, R. H. (2007). Homeostatic regulation of MeCP2 expression by a CREB-induced microRNA.Nature Neuroscience,10, 1513–1514.
Kohler, S., Doelken, S. C., Mungall, C. J., Bauer, S., Firth, H. V., & Bailleul-Forestier, I.€
(2014). The Human Phenotype Ontology project: Linking molecular biology and dis-ease through phenotype data.Nucleic Acids Research,42, D966–D974.
Kohli, R. M., & Zhang, Y. (2013). TET enzymes, TDG and the dynamics of DNA demeth-ylation.Nature,502, 472–479.
Kouzarides, T. (2007). Chromatin modifications and their function.Cell,128, 693–705. Kozlenkov, A., Roussos, P., Timashpolsky, A., Barbu, M., Rudchenko, S., Bibikova, M.,
et al. (2014). Differences in DNA methylation between human neuronal and glial cells are concentrated in enhancers and non-CpG sites. Nucleic Acids Research, 42, 109–127.
Kriaucionis, S., & Heintz, N. (2009). The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain.Science,324, 929–930.
Krumm, N., O’Roak, B. J., Shendure, J., & Eichler, E. E. (2014). A de novo convergence of autism genetics and molecular neuroscience.Trends in Neurosciences,37, 95–105. Kundakovic, M., Chen, Y., Guidotti, A., & Grayson, D. R. (2009). The reelin and GAD67
promoters are activated by epigenetic drugs that facilitate the disruption of local repressor complexes.Molecular Pharmacology,75, 342–354.
Kusui, C., Kimura, T., Ogita, K., Nakamura, H., Matsumura, Y., Koyama, M., et al. (2001). DNA methylation of the human oxytocin receptor gene promoter regulates tissue-specific gene suppression. Biochemical and Biophysical Research Communications, 289, 681–686.
Lasalle, J. M. (2007). The Odyssey of MeCP2 and parental imprinting.Epigenetics,2, 5–10. Lasalle, J. M. (2013). Autism genes keep turning up chromatin.OA Autism,1, 14. Lasalle, J. M., Goldstine, J., Balmer, D., & Greco, C. M. (2001). Quantitative localization of
heterogeneous methyl-CpG-binding protein 2 (MeCP2) expression phenotypes in nor-mal and Rett syndrome brain by laser scanning cytometry.Human Molecular Genetics,10, 1729–1740.