BAB 1 PENDAHULUAN
Sindroma Treacher Collins adalah kelainan genetik yang ditandai dengan
adanya kelainan bentuk kraniofasial.1 Gambaran yang sangat umum dari Sindroma
Treacher Collins yaitu kelopak mata yang miring ke bawah (89% dari kasus),
hipoplasia tulang zigoma (81 % dari kasus) dan mandibula (78% dari kasus).2
Para peneliti memperkirakan bahwa Sindroma Treacher Collins dialami
sekitar 1 dari setiap 50.000 kelahiran hidup. Sekitar 40% dari kasus Sindroma
Treacher Collins diturunkan oleh orang tua, sementara 60% dari kasus Sindroma
Treacher Collins timbul sebagai mutasi de novo.2,3,4,5,6 Prevalensi jenis kelamin pria
dan wanita yang terkena Sindroma Treacher Collins adalah sama.5,6
Sindrom ini diberi nama Sindroma Treacher Collins setelah dokter spesialis
mata terkemuka Inggris Edward Treacher Collins (1862-1932), yang menggambarkan
ciri-ciri penting sindrom ini dalam sebuah makalah pada tahun 1900.1,3,4,6 Di benua
Eropa, nama yang lebih umum untuk kondisi ini adalah Sindroma
Franceschetti-Zwahlen-Klein, didasarkan pada studi yang ekstensif terhadap disostosis
mandibulofasial yang diterbitkan oleh dokter spesialis mata Franceschetti dari Swiss
dan dokter spesialis genetika Klein (1949).3,4,6 Sedangkan Van der Meulen
menggolongkan kondisi ini sebagai displasia zigoauromandibular.3
Salah satu penyebab terjadinya Sindroma Treacher Collins adalah adanya
mutasi gen TCOF1, pada kromosom 5q32-q33.1. Gen ini mengkode protein yang
tertentu dalam perkembangan embrio, terutama pada struktur kepala dan wajah.
Kelainan ini diturunkan dalam pola autosomal dominan.1,2,3,5,7,8 Apabila terdapat
riwayat keluarga, diagnosa dapat ditegakkan dengan mudah berdasarkan evaluasi
klinis dan dapat dikonfirmasikan dengan studi genetik.6,9 Untuk mendiagnosa bayi
dalam kandungan dapat digunakan alat ultrasonografi.2
Untuk mencegah anomali sindroma ini, telah dilakukan percobaan pada model
hewan dengan menghambat fungsi p53. Namun, p53 berperan penting dalam fungsi
seluler dan penekanan fungsi p53 secara total sangat beresiko. Karena itu, dilakukan
penghambatan p53 secara genetik dan khemis untuk menekan apoptosis
neuroepithelial yang dikaitkan dengan Sindroma Treacher Collins sehingga dapat
mencegah patogenesis anomali kraniofasial dari sindroma tersebut.2,10
Penatalaksanaan pasien dengan Sindroma Treacher Collins memerlukan
perawatan multidisiplin dari sejak ia lahir dan sepanjang hidupnya untuk
meminimalkan atau mengkoreksi masalah fungsional dan cacat kraniofasial.2,3
Perawatan emergensi umumnya ditujukan untuk mengatasi masalah pernafasan yang
mungkin diakibatkan mikrognasia dan obstruksi hipofaring oleh lidah pada bayi baru
lahir dengan Sindroma Treacher Collins. Selain itu, apabila bayi mengalami kesulitan
menelan, perlu dilakukan pemberian makan dengan gavage atau tube gastrostomi.
Sedangkan perawatan definitif dilakukan setelah pasien selesai masa
pertumbuhannya. Keseluruhan rencana perawatan Sindroma Treacher Collins harus
berorientasi sesuai masalah, namun cukup fleksibel untuk memenuhi keinginan serta
Didalam skripsi ini penulis akan membahas mengenai Sindroma Treacher
Collins sendiri serta cara penatalaksanaan pasien Sindroma Treacher Collins dengan
BAB 1 PENDAHULUAN
Sindroma Treacher Collins adalah kelainan genetik yang ditandai dengan
adanya kelainan bentuk kraniofasial.1 Gambaran yang sangat umum dari Sindroma
Treacher Collins yaitu kelopak mata yang miring ke bawah (89% dari kasus),
hipoplasia tulang zigoma (81 % dari kasus) dan mandibula (78% dari kasus).2
Para peneliti memperkirakan bahwa Sindroma Treacher Collins dialami
sekitar 1 dari setiap 50.000 kelahiran hidup. Sekitar 40% dari kasus Sindroma
Treacher Collins diturunkan oleh orang tua, sementara 60% dari kasus Sindroma
Treacher Collins timbul sebagai mutasi de novo.2,3,4,5,6 Prevalensi jenis kelamin pria
dan wanita yang terkena Sindroma Treacher Collins adalah sama.5,6
Sindrom ini diberi nama Sindroma Treacher Collins setelah dokter spesialis
mata terkemuka Inggris Edward Treacher Collins (1862-1932), yang menggambarkan
ciri-ciri penting sindrom ini dalam sebuah makalah pada tahun 1900.1,3,4,6 Di benua
Eropa, nama yang lebih umum untuk kondisi ini adalah Sindroma
Franceschetti-Zwahlen-Klein, didasarkan pada studi yang ekstensif terhadap disostosis
mandibulofasial yang diterbitkan oleh dokter spesialis mata Franceschetti dari Swiss
dan dokter spesialis genetika Klein (1949).3,4,6 Sedangkan Van der Meulen
menggolongkan kondisi ini sebagai displasia zigoauromandibular.3
Salah satu penyebab terjadinya Sindroma Treacher Collins adalah adanya
mutasi gen TCOF1, pada kromosom 5q32-q33.1. Gen ini mengkode protein yang
tertentu dalam perkembangan embrio, terutama pada struktur kepala dan wajah.
Kelainan ini diturunkan dalam pola autosomal dominan.1,2,3,5,7,8 Apabila terdapat
riwayat keluarga, diagnosa dapat ditegakkan dengan mudah berdasarkan evaluasi
klinis dan dapat dikonfirmasikan dengan studi genetik.6,9 Untuk mendiagnosa bayi
dalam kandungan dapat digunakan alat ultrasonografi.2
Untuk mencegah anomali sindroma ini, telah dilakukan percobaan pada model
hewan dengan menghambat fungsi p53. Namun, p53 berperan penting dalam fungsi
seluler dan penekanan fungsi p53 secara total sangat beresiko. Karena itu, dilakukan
penghambatan p53 secara genetik dan khemis untuk menekan apoptosis
neuroepithelial yang dikaitkan dengan Sindroma Treacher Collins sehingga dapat
mencegah patogenesis anomali kraniofasial dari sindroma tersebut.2,10
Penatalaksanaan pasien dengan Sindroma Treacher Collins memerlukan
perawatan multidisiplin dari sejak ia lahir dan sepanjang hidupnya untuk
meminimalkan atau mengkoreksi masalah fungsional dan cacat kraniofasial.2,3
Perawatan emergensi umumnya ditujukan untuk mengatasi masalah pernafasan yang
mungkin diakibatkan mikrognasia dan obstruksi hipofaring oleh lidah pada bayi baru
lahir dengan Sindroma Treacher Collins. Selain itu, apabila bayi mengalami kesulitan
menelan, perlu dilakukan pemberian makan dengan gavage atau tube gastrostomi.
Sedangkan perawatan definitif dilakukan setelah pasien selesai masa
pertumbuhannya. Keseluruhan rencana perawatan Sindroma Treacher Collins harus
berorientasi sesuai masalah, namun cukup fleksibel untuk memenuhi keinginan serta
Didalam skripsi ini penulis akan membahas mengenai Sindroma Treacher
Collins sendiri serta cara penatalaksanaan pasien Sindroma Treacher Collins dengan
BAB 2
SINDROMA TREACHER COLLINS
Sindroma Treacher Collins, yang dikenal sebagai disostosis mandibulofasial
dan Sindroma Franceschetti-Zwahlen-Klein, merupakan kelainan genetik yang
diturunkan secara autosomal dominan dan biasanya terjadi secara bilateral.
Karakterisitik dari Sindroma Treacher Collins meliputi hipoplasia tulang wajah,
terutama mandibula dan tulang zigoma, celah palatum, fisur palpebra yang miring ke
bawah dengan koloboma pada kelopak mata bawah dan kelainan bentuk telinga
bagian luar.1,2,3,4 Gejala yang ditimbulkan bervariasi dari ringan sampai parah. Pada
pasien dengan dismorfologi kraniofasial yang parah dapat terdeteksi sebelum
kelahiran dengan USG, sedangkan pasien dengan dismorfologi kraniofasial yang
ringan, mungkin terdiagnosis pada saat lahir.3,6
2.1 Definisi
Sindroma Treacher Collins adalah kelainan yang diturunkan secara autosomal
dominan yang timbul akibat penyimpangan dalam perkembangan struktur wajah
selama morfogenesis histodiferensiasi antara 20 hari dan minggu ke-12 IU.9,11
Walaupun pertama sekali dilaporkan oleh Thompson (1846), sindroma ini dikenal
masyarakat karena Berry dan terutama Treacher Collins (1900), dokter mata Inggris,
melaporkan 2 kasus dan mendeskripsikan komponen penting sindroma ini.
dan mengemukakan istilah “disostosis mandibulofasial” yang dikenal dalam
literatur.3,11
2.2 Etiologi
Sindroma Treacher Collins merupakan gangguan perkembangan kraniofasial
yang disebabkan kelainan genetik. Kelainan genetik ini dapat terjadi karena
diturunkan oleh orang tua ataupun mutasi baru.3,12
Pertama, terjadinya Sindroma Treacher Collins sebagai hasil dari mutasi de
novo (60% dari kasus).2,3,4,10 Ini berarti bahwa kedua orang tua pasien menurunkan gen yang normal kepada anaknya dan terjadinya mutasi akibat perubahan salah satu
gen.12,13
Kedua, jika salah satu dari orang tua menderita Sindroma Treacher Collins
maka dapat diasumsikan bahwa penyebab terjadinya sindroma ini diperoleh dari gen
orang tua yang diturunkan secara autosomal dominan kepada anaknya (40 % dari
kasus).2,3,4,10,11 Terdapat probabilitas 50% bagi anak untuk menderita Sindroma
Treacher Collins apabila salah satu dari orang tua memiliki gen abnormal pada
kromosom autosomal.5,7,12,13 Namun, dapat terjadi hambatan secara klinis untuk
mengetahui apakah orang tua pasien menderita sindroma ini. Karena orang tua pasien
Gambar 1. Kelainan yang diturunkan secara autosomal dominan,T mewakili gen dominan, yang menyebabkan terjadinya Sindroma Treacher Collins; t mewakili gen resesif yang merupakan gen normal14
2.3 Patogenesis
Terjadinya Sindroma Treacher Collins disebabkan karena adanya mutasi dari
gen TCOF1. Gen TCOF1 terpeta dalam kromosom band 5q31.3-33.3. Gen ini
mengkode protein treacle, yang diperlukan dalam perkembangan kraniofasial yang
normal.2,6 Mutasi tunggal pada gen ini mengakibatkan terminasi prematur dari produk
protein (The Treacher Collins Syndrome Collaborative Group, 1996; Wise, 1997).6
Dixon (1996) meninjau gambaran klinis dan molekular Sindroma Treacher
Collins, dari total 20 mutasi gen TCOF1, 2 diantaranya merupakan mutasi nonsense,
5 terjadi insersi, 11 terjadi delesi, dan 2 terjadi mutasi penyambungan. Keseluruhan
haploinsufisiensi dimana hal ini sebagai mekanisme molekular yang mendasari
terjadinya sindroma ini. Menurut Dixon, selama perkembangan embrio, treacle
dinyatakan berada pada level puncak dalam lengkung brakhial pertama dan kedua.6,9
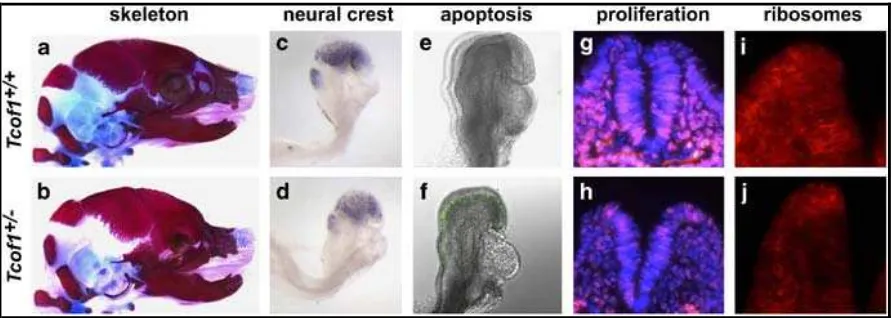
Gambar 2. Dasar perkembangan anomali kraniofasial Sindroma Treacher Collins2 2A.Pewarnaan Skeletal pada embrio tipe liar, 2B. Pewarnaan Skeletal pada embrio yang terkena STC menunjukkan keparahan hipoplasia frontonasal, 2C. Terjadi migrasi neural-crest pada embrio tipe liar, 2D. Terjadi pengurangan populasi sel neural-neural-crest yang parah pada embrio yang terkena STC, 2E. Pewarnaan untuk melihat apoptosis pada embrio tipe liar menunjukkan rendahnya level kematian sel endogen, 2F. Pewarnaan untuk melihat apoptosis pada embrio yang terkena STC menunjukkan peningkatan tingkat kematian sel , 2G. Pewarnaan untuk melihat proliferasi embrio tipe liar, 2H. Pewarnaan untuk melihat proliferasi yang terkena STC menunjukkan penurunan proliferasi sel, 2I. Pewarnaan untuk melihat ribosom pada embrio tipe liar, 2J. Pewarnaan untuk melihat ribosom pada embrio yang terkena STC menunjukkan kekurangan biogenesis ribosom
Sel neural crest adalah populasi sel yang multipoten, stem dan progenitor,
dibentuk dalam ektoderm neural pada batas dengan ektoderm non-neural sepanjang
sumbu tubuh selama awal embriogenesis. Sel neural crest menjalani sebuah transisi
ephitelial-mesenkimal dan di region kranial, sel-sel ini dideliminasi dari ektoderm
kraniofasial diperkirakan disebabkan oleh defek pada pembentukan, proliferasi,
migrasi dan atau diferensiasi dari sel neural crest kranial (Gambar 2).2,10
Treacle merupakan protein yang sangat sederhana yang dikode oleh TCOF1 dan berperan dalam biogenesis ribosom serta mengatur kelangsungan hidup
neuroepithelial dan proliferasi sel neural crest. Haploinsufisiensi TCOF1 mengurangi
biogenesis ribosom yang diukur dengan produksi 28s subunit dalam neuroepithelial
dan sel neural crest, dimana defisiensi biogenesis ribosom berhubungan dengan
kurangnya proliferasi dalam sel neural crest dan sel neuroepithelial yang diamati
pada mutan TCOF1. Akibat kekurangan biogenesis ribosom yang tidak dapat
mengimbangi kebutuhan seluler dan metabolik dari populasi sel yang berproliferasi
tinggi, menyebabkan terjadinya aktivasi p53. Stabilisasi p53 mengaktifkan banyak
gen efektor proapoptotik, seperti Ccng1, Trp53inp1, Noxa, Perp dan Wig1, dalam
neuroepithelium, yang secara kolektif bertanggungjawab terhadap tingginya tingkat
kematian jaringa n tertentu yang diamati dalam patogenesis Sindroma Treacher
Collins.2,10
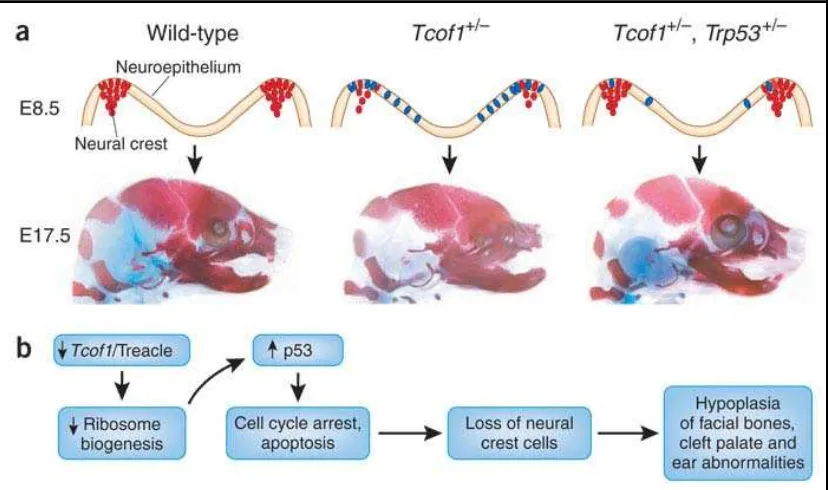
Korelasi langsung antara stabilisasi nuklear protein p53, aktivasi transkripsi
gen p53 dependent, dan induksi apoptosis neuroepithelial menunjukkan defisiensi
yang diamati dalam migrasi sel neural crest pada Sindroma Treacher Collins. Dari
percobaan yang dilakukan, diketahui penghambatan p53 secara genetik berhasil
menghambat apoptosis neuroepithelial yang terjadi tanpa mengubah biogenesis
ribosom yaitu dengan menghambat p53 secara genetik dan khemis, aktivitas Ccng1
hipoplasia kraniofasial dan menghasilkan perkembangan kraniofasial yang normal
setelah dilahirkan.2
Gambar 3. A. Gambar kiri menunjukkan perkembangan neuroepithelium yang normal, gambar tengah menunjukkan apoptosis neuroepithelium dan pengurangan pembentukan dan migrasi sel neural crest, gambar kanan menunjukkan penghambatan p53 dan migrasi sel neural crest yang normal, B. Diagram alir yang menunjukkan patogenesis terjadinya Sindroma Treacher Collins.2
2.4 Manifestasi Klinis
Wajah yang khas dari Sindroma Treacher Collins biasanya membuat diagnosa
lebih mudah. Terdapat fisur palpebra yang miring ke bawah (antimongoloid), tulang
malar yang tidak berkembang atau bahkan absen, mandibula yang retrusif dan
2.4.1 Tulang Tengkorak
Abnormalitas yang terjadi umumnya secara bilateral dan simetris.6 Panjang
kranial anterior lebih pendek dengan kompensasi peningkatan panjang kranial
posterior, dan panjang keseluruhan tulang tengkorak normal atau berkurang
dibandingkan dengan kontrol. Prosesus mastoideus sering tidak memiliki rongga
(unpneumatized) dan mungkin skrelotik.3,9,18 Basis kranial secara progresif
mengalami pembengkokan (kyphotic) dan kalvaria umumnya normal. Sudut basis
kranial tinggi, menyebabkan penyempitan anteroposterior dari ruang faring.3,6
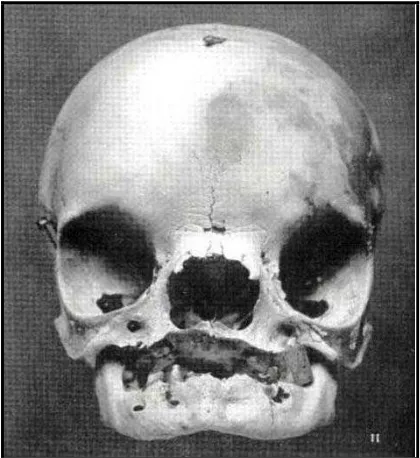
Gambar 4. Tulang tengkorak dengan defek kraniofasial pada pasien Sindroma Treacher Collins20
Derajat ketidak berkembangan atau absennya tulang malar bervariasi dan
cenderung cukup simetris. Hipoplasia tulang malar menyebabkan orbit menjadi
malar dan arkus zigoma yang tidak berfusi, sinus paranasal juga mengalami
hipoplasia.3,6,18,19
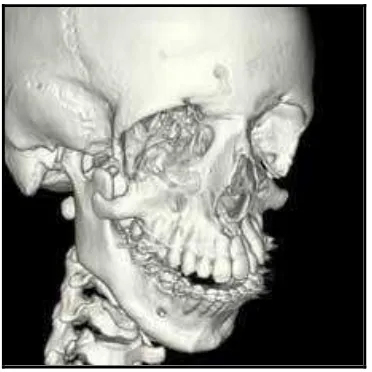
Gambar 5. CT Scan pada pasien Sindroma Treacher Collins tanpa tulang pipi20
2.4.2 Hidung dan Jaringan Lunak Wajah
Sudut frontonasal yang tinggi dan hipoplastik tulang malar dan ridge
supraorbital membuat hidung tampak lebih menonjol. Lebar dasar hidung meningkat,
tetapi hidung biasanya berukuran normal atau optimal.3,5,6 Sekitar 25% dari kasus,
kulit berambut meluas turun ke pipi dari regio temporal.3,21,22,23 Kulit yang melapisi
tulang malar yang hipoplastik sering tipis dengan jaringan subkutan yang minimal.3
2.4.3 Mata
Hampir seluruh pasien dilaporkan memiliki beberapa masalah okular dan
adneksal.3 Pasien Sindroma Treacher Collins sering memiliki mata yang miring ke
yang disebut koloboma.11,18,24,25 Kemiringan antimongoloid pada celah palpebra dan
ligamen lateral kanthus yang terletak inferior dan rudimenter hampir seragam.
Koloboma sejati terjadi pada 25% kasus, dan koloboma pseudo muncul sekitar 50%
kasus, dimana cenderung berada di sepertiga luar dari kelopak mata bawah. Yang
umum terjadi adalah gangguan refraktif, dan jarang terjadi ambliopia.3
Gambar 6A. Mata yang miring ke bawah pada Sindroma Treacher Collins26 6B. Adanya koloboma pada kelopak mata bawah27
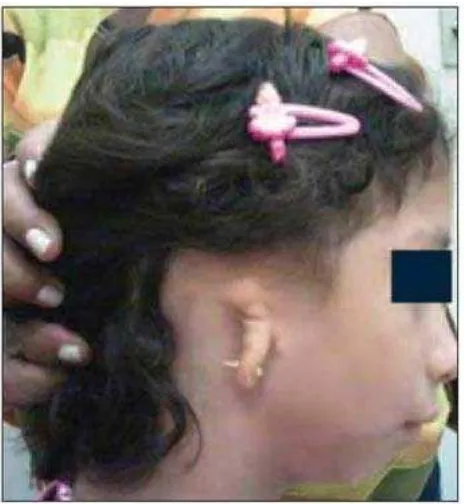
2.4.4 Telinga
Pada telinga luar (pinna) penderita Sindroma Treacher Collins dapat terjadi
kelainan yang berbeda dalam bentuk, ukuran dan posisi.1,3,26 Pinna juga mungkin
relatif normal dalam bentuk dan ukuran, diikuti kehadiran ada penonjolan kecil (tag)
telinga dengan satu atau lebih.3 Dengan derajat kecacatan apapun, pinna cenderung
terletak lebih anterior dan inferior dan tampak kusut.3,23
Cacat meatus auditorius eksternal berkisar dari menjadi paten hingga oklusi
tulang yang komplit. Pada sebagian besar kasus, sangat sedikit terjadi stenosis
jaringan lunak. 3
Telinga tengah hampir selalu hipoplastik dengan sebagian besar
mempengaruhi rongga timpani bagian atas di atas membran timpani (attic).2,3 Rantai osikular sering abnormal dan keparahannya bervariasi, rantai sering berpindah secara
anterolateral. Telinga dalam biasanya normal dengan beberapa pemendekan dan
perpindahan anterior dari nervus fasialis yang desending.3,5,13,23
Gambar 7. Malformasi bentuk telinga luar21
2.4.5 Mandibula
Komponen mandibula sering mengalami hipoplasia, dengan bentuk cekung
pada permukaan bawah bodi mandibula. Terjadi hipoplastik mandibula pada ramus
yang retrusi.1,3,11,24,28 Notch antegonial yang ditandai muncul dengan sudut gonial yang tumpul dan kecenderungan dagu untuk berputar inferior.3,5,12 Ini memberikan
mandibula tampak dibengkokkan. Pola pembengkokan ini berbeda dari yang tampak
pada kondisi lain seperti penyakit Still’s, dan beberapa perbedaan telah diukur. Selain
itu juga dijumpai abnormalitas perlekatan muskular dari pterigomasseter.3
2.4.6 Maksila
Maksila cenderung memiliki palatum yang melengkung tinggi atau celah
palatum (30% kasus).3,11,21 Tinggi gigi posterior menjadi rendah dengan tingkat
hiperproyeksi maksila. Pada beberapa kasus, hiperproyeksi tampak sangat nyata, dan
menjadi sulit untuk memastikan apakah abnormalitas terletak pada maksila atau basis
cranial.3
Choanal atresia mungkin terjadi, dimana keberadaannya menyebabkan masalah jalan nafas, terutama pada periode neonatal. Hubungan skeletal rahang
biasanya Klas II dengan gigitan terbuka anterior.3,4,23,24 Gigi biasanya berkembang
dengan normal dan mempunyai ukuran normal. Namun, mandibula yang mempunyai
lengkung sempit dan berbentuk abnormal sering mengakibatkan gigi berjejal yang
parah.3
Derajat malformasi yang terlihat sewaktu bayi dilahirkan dengan Sindroma
Treacher Collins diyakinkan relatif stabil dan tidak progresif seiring usia. Robert dkk
(1975) meninjau berturut-turut radiografi sefalometri dari pasien Sindroma Treacher
Collins dan mendokumentasikan stabilitas batas inferior mandibula dari waktu ke
waktu. Garner (1967) melaporkan penemuannya terhadap analisis sefalometri 3
pasien dengan variasi umur yang dikonfirmasikan menderita Sindroma Treacher
Collins. Dia mendokumentasikan deformasi yang relatif stabil yang diobservasi pada
umur yang bervariasi. Tidak ada bukti pasti yang mengkonfirmasi secara signifikan
bahwa dismorfologi Sindroma Treacher Collins makin memburuk dengan
pertumbuhan wajah.5
Diagnosa Sindroma Treacher Collins ini dibuat berdasarkan gambaran klinis
karena karakteritiknya yang khas dengan mata yang miring ke bawah, abnormalitas
telinga dan rahang bawah yang kecil dan pemeriksaan radiografi seperti
Computerized tomography (CTscan) dapat digunakan untuk menentukan derajat ketidakberkembangan struktur tulang wajah.30
Ada banyak sindroma yang penampilan wajahnya menyerupai Sindroma
Treacher Collins. Pemeriksaan fisik yang lengkap terhadap sistem tubuh yang lain
dapat membantu menegakkan diagnosis Sindroma Treacher Collins. Sindroma
Treacher Collins dapat dibedakan dari Sindroma Nager dan Sindroma Miller jika
tidak ada abnormalitas pada tangan atau lengan. Dengan keterlibatan wajahnya
bilateral (mengenai kedua sisi wajah) dan spinal column normal, Sindroma Treacher
Collins dapat dibedakan dari kondisi Oculoauriculovertebral (OAV) seperti
Sindroma Goldenhar.30
Jika terdapat beberapa orang dalam sebuah keluarga mengalami Sindroma
Treacher Collins, studi hubungan (linkage) genetik dapat dilakukan. Studi hubungan
ini memerlukan sampel darah dari berbagai anggota keluarga, yang terkena dan yang
tidak terkena.30 Ditandai pada gen TCOF1 dan dianalisa serta dibandingkan untuk
menentukan terjemahan gen yang dibagi kepada anggota keluarga yang terkena. Gen
yang menyebabkan terjadinya Sindroma Treacher Collins ada pada semua anggota
keluarga yang terkena, dan absen pada semua anggota yang tidak terkena. Studi
hubungan dapat dilakukan pada bayi belum lahir untuk menentukan apakah bayi
mewarisi gen yang menyebabkan sindroma tersebut. Ultrasonografi prenatal dapat
laporan tentang diagnosa prenatal dengan ultrasonografi saja, bayi dengan manifestasi
ringan dapat terlihat normal. Deteksi tergantung pada keterampilan dokter yang
melakukan ultrasonografi dan pengalamannya terhadap gambaran Sindroma Treacher
BAB 3
PENATALAKSANAAN HIPOPLASIA MANDIBULA DAN ZIGOMA PADA PASIEN SINDROMA TREACHER COLLINS
Karena terdapat banyak anomali kraniofasial, penatalaksanaan pasien
Sindroma Treacher Collins serta orang tua pasien harus memiliki tujuan yang spesifik
dan bertujuan untuk memaksimalkan potensi anak. Keseluruhan rencana perawatan
Sindroma Treacher Collins harus berorientasi sesuai masalah, namun cukup fleksibel
untuk memenuhi keinginan serta kebutuhan pasien dan orang tua pasien. Perawatan
harus disesuaikan dengan kebutuhan spesifik setiap individu, dimana perawatan
terhadap kondisi ini merupakan perawatan terhadap simptom yang muncul, bersifat
jangka panjang dan memerlukan pendekatan yang multidisiplin.3,6 Karena hipoplasia
mandibula dan zigoma merupakan kasus yang sering dijumpai pada Sindroma
Treacher Collins, maka akan dibahas penatalaksanaannya berikut ini.
3.1 Perawatan Emergensi
Pada bayi yang baru lahir dengan Sindroma Treacher Collins, perlu segera
diperhatikan jalan nafas dan kemampuan menelan.3,6,12 Hambatan jalan nafas dapat
terjadi akibat dari dua faktor. Pertama adalah hipoplasia maksila, yang cenderung
mengkonstriksikan jalan lintasan nasal dan menyebabkan derajat penyempitan koana
(choanal stenosis). Kedua adalah mandibula yang mikrognasia dan lidah yang
keparahan deformasi, kesulitan jalan nafas dapat timbul dan diperlukan posisi bayi
yang khusus dan rawat inap di rumah sakit dengan monitor denyut oksimeter.3,6,23
Pada bayi dengan manifestasi parah dimana inadekuat jalan nafas merupakan
gambaran yang menonjol setelah dilahirkan, maka dilakukan trakeostomi. Alternatif
lain jika anak tidak dapat memperoleh oksigen secara adekuat yaitu dengan
memanjangkan mandibula sehingga lidah dan struktur dasar mulut dapat diposisikan
anterior .3,6,7,23
Tingkat malformasi muskuloskeletal pada Sindroma Treacher Collins juga
mengakibatkan kesulitan menelan cairan dengan efektif dan memperoleh nutrisi yang
adekuat.Diperlukan pemberian makan dengan bantuan gavage, atau yang ekstrem
dengan penempatan tube gastrostomi untuk memastikan asupan jumlah kalori dan
hidrasi yang adekuat. 3,12,13
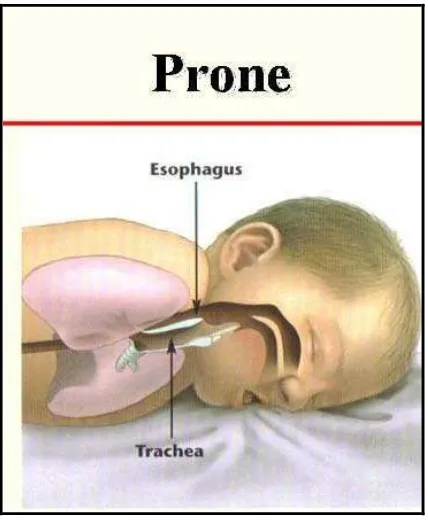
3.1.1 Penatalaksanaan jalan nafas
Obstruksi yang ringan umumnya dapat ditangani dengan cara yang sangat
konservatif yaitu dengan perubahan posisi. Obstruksi ini disebabkan mandibula yang
kecil dan ukuran lidah yang normal. Dengan meletakkan bayi dalam posisi prone,
yakni wajah menghadap ke bawah, gravitasi menarik lidah ke depan dan
Gambar 9. Bayi dengan posisi prone32
Penatalaksanaan obstruksi jalan nafas yang sedang sampai parah dapat dilakukan
dengan trakeostomi dan glosopeksi. Diindikasikan ketika kesulitan bernafas tetap
terjadi walaupun telah dilakukan perubahan posisi.31
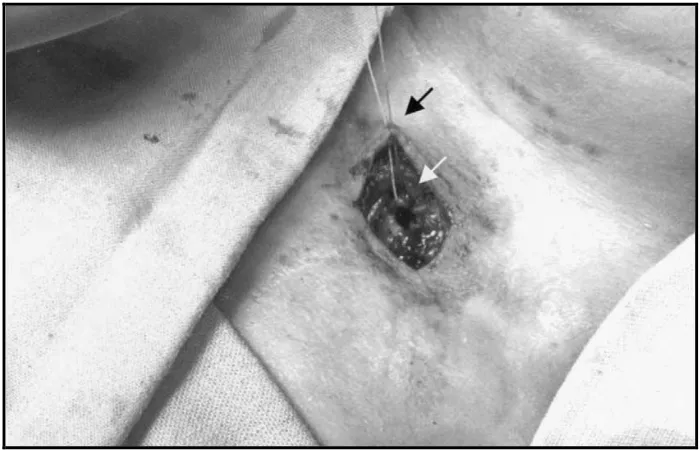
3.1.1.1 Trakeostomi
Trakeostomi masih merupakan penatalaksanaan standar dalam menangani
sumbatan jalan nafas yang parah.Tube trakeostomi secara efektif melewati obstruksi
di faring dan hipofaring. Ketika obstruksi jalan nafas bayi telah diselesaikan, tube
trakeostomi dapat disingkirkan. Sayangnya, tube trakeostomi memerlukan
pemantauan yang ketat. Jika tube ini terjadi penyumbatan atau lepas, pasien dapat
Trakeostomi neonatal berhubungan secara signifikan terhadap morbilitas dan
mortalitas, termasuk terhambatnya proses berbicara, penyumbatan oleh lendir,
pelepasan tube trakeostomi atau trakeomalasia. Neonatal yang telah melakukan
trakeostomi memerlukan monitoring 24 jam, memberikan beban yang besar kepada
pengasuh. Insiden yang berkaitan dengan komplikasi trakeostomi dilaporkan 19
sampai 49% dan mortalitas terkait 2 sampai 8.5% .31 Jika trakeostomi dilakukan
sebagai penatalaksanaan kegawat daruratan jalan nafas, distraksi mandibula dapat
dilakukan pada bayi untuk mempercepat dekanulasi. 6
Trakeostomi Starplasti adalah teknik baru berdasarkan geometri tiga dimensi
dari Z-plasti.34 Prosedur Starplasti adalah teknik secara langsung yang mudah dilihat,
terutama bagi ahli bedah yang sering melakukan trakeostomi. Dari insisi sampai
selesai, prosedur ini biasanya memerlukan waktu sekitar 30 menit. Jaringan harus
ditangani dengan lembut, dengan memperhatikan hemostasis, mengontrol bidang
jaringan dan melakukan penjahitan yang akurat. Lampu penerangan diperlukan, dan
Gambar 10A. Insisi berbentuk ”X” pada pertengahan lekukan sternal dengan kartilago krikoid34
Gambar 10C. Stoma berbentuk bintang34
3.1.1.2 Adhesi lidah-bibir/ Glosopeksi
Pada obstruksi minor dapat dikoreksi dengan pertimbangan adhesi lidah-bibir.
Pembedahan adhesi dilakukan antara lidah, bibir dan anterior mandibula.6
Adhesi lidah-bibir diawali oleh Douglas tahun 1946. Insisi transversal dibuat
pada permukaan ventral lidah dan permukaan lingual dari bibir bawah. Harus
dilakukan secara hati-hati untuk menghindari bukaan kelenjar sublingual. Dilakukan
penjahitan pada permukaan tersebut, tambahan jahitan retensi ditempatkan dari dasar
lidah tepat di atas epiglotitis ke permukaan anterior dagu, biasanya melalui kancing
untuk mencegah erosi pada permukaan lidah dan kulit. Adhesi sementara ini
dilakukan untuk memperbaiki kesulitan bernafas sampai selesai dilakukan distraksi
Tingkat keberhasilan lidah-bibir adhesi dalam menghilangkan obstruksi jalan
napas dilaporkan kira-kira 33% hingga 83%. Komplikasi yang paling sering
dilaporkan adalah terbukanya adhesi. Polisomnografi pasca operasi menunjukkan
adanya perbaikan obstructive apnea. Diperlukan pemeriksaan jangka panjang dan
tindak lanjut pasien yang menunjukkan perlunya prosedur sekunder pada kebanyakan
obstruksi berulang dan kesulitan menelan.33
Gambar 11. Adhesi lidah bibir36
3.1.2 Penatalaksanaan pemberian makan (feeding)
Bayi dengan Sindroma Treacher Collins juga mengalami kesulitan dalam
pemberian makan. Karena posisi mandibula yang abnormal, bayi dengan mandibula
puting ibu. Pada beberapa kasus, adanya celah palatum menghambat produksi
tekanan negatif untuk menghisap selama menyusui. Selain itu, adanya celah palatum
menimbulkan hubungan yang luas antara kavitas oral dan nasal yang menimbulkan
resiko tersedak dan masalah pemberian makan lainnya sehingga disarankan
melakukan konsultasi dengan spesialis. Pada beberapa kasus, seperti diinstruksikan,
seorang ibu dapat memberi makan dengan botol ketika bayi dalam posisi setengah
duduk. Pada pasien dengan keluhan yang lebih parah, diperlukan pemberian makan
secara temporer dengan gavage atau feeding tube. Jika tidak adanya peningkatan
selama berbulan-bulan, bayi memerlukan tube gastrotomi. Setelah anak
mengembangkan kemampuan untuk makan secara oral, tube tersebut dapat
disingkirkan.33
Gambar 12. Feeding tube
3.2 Perawatan definitif
Perawatan definitif terhadap deformasi kraniofasial umumnya ditunda sampai
ini akan dibahas tentang perawatan definitif terhadap hipoplasia zigoma dan
mandibula dengan rekonstruksi malar dan bedah orthognatik.
3.2.1 Penatalaksanaan hipoplasia tulang zigoma
Rekonstruksi zigoma dapat dilakukan sekitar usia 8 tahun. Rekonstruksi pada
usia awal tidak memperoleh keuntungan dan sosial, psikologi dan perkembangan
pendidikan anak merupakan hal penting dalam menentukan waktu untuk melakukan
rekonstruksi zigoma. Metode yang lebih disukai untuk rekonstruksi malar adalah
penggunaan outer table calvarial bone graft yang difiksasi pada tempatnya dengan
sekrup, plat atau keduanya.3
Gambar 13 A. Bone graft kranial ketebalan penuh untuk rekonstruksi lengkung zigoma dan lateral orbita rim37
13 B. Bone graft diosteotomi agar sesuai dengan bentuk microplate37
Perencanaan pembedahan difasilitasi oleh penggunaan CT Scan tiga dimensi
untuk merekonstruksi. Model stereolithoraphic juga mungkin berguna. CT Scan
memberikan informasi tentang deformasi celah palatum dan juga tentang ketebalan
calvarium di area donor. Jika tulang tampak terlalu tipis untuk memperoleh outer
table calvarial bone graft, dapat dilakukan kraniotomi, dan tulang tersebut dapat
dibagi di meja samping. Tempat donor dipilih untuk ketebalan dan kurvatura, CT
Scan tiga dimensi dapat memfasilitasi perencanaan ini. Jika satu lapisan tulang calvarial tidak cukup tebal sesuai yang dikehendaki untuk mengkontour dalam
rekonstruksi zigoma, maka dapat dilapisi dan difiksasi dengan menggunakan sekrup,
plat atau keduanya. 3
Insisi kulit bikoronal dilakukan pada tempat donor dan area yang akan
direkonstruksi. Jika defisiensi zigoma melibatkan komponen anterior, kelopak mata
bawah, infraorbital, diperlukan insisi transconjunctival. Pemilihan insisi tergantung
pada perluasan dan bentuk deformasi kelopak mata.3
Rekonstruksi tulang zigoma dengan kombinasi dari graft onlay dan inlay
untuk memproduksi kontur tulang normal. Sejumlah kecil overkoreksi diperlukan
untuk mengkompensasi apabila terdapat resorpsi dan jaringan lunak yang tipis. Pada
beberapa kasus jaringan lunak dapat diaugmentasi dengan penggunaan flap dua sisi
perikranial yang dilipat dan dilapisi di atas bone graft.3
Pada beberapa kasus rekonstruksi malar awal perlu dilakukan osteotomi malar
untuk mengoptimalkan posisi mereka. Untuk melakukan osteotomi malar
konvensional, perlu ditunda sampai selesai pertumbuhan. Jika diperlukan reoperasi,
perlu dinilai jumlah tulang sebelum operasi dengan CT Scan tiga dimensi karena
memerlukan bone graft lebih lanjut.3
Strategi lain untuk merekonstruksi malar meliputi rib grafts, dimana,
walaupun lebih mudah untuk dimanipulasi dan dikontour dibandingkan tulang
Pada beberapa kasus dimana rekonstruksi malar dilakukan secara bersamaan
dengan pembedahan ramus mandibula, ada resiko terjadinya ankilosis. Resiko ini
meningkat apabila digunakan fiksasi intermaksila. Untuk menghindari hal tersebut,
sebaiknya kedua prosedur dilakukan terpisah.3
Beberapa spesialis menggunakan bahan alloplastik untuk merekonstruksi
malar. Teknik ini sering menghasilkan bentuk dan kontur yang sangat baik, namun
dapat menimbulan resiko infeksi seumur hidup. Walaupun tidak sering, infeksi ini
dapat menghasilkan jaringan parut dan kerusakan yang signifikan karena sifat dasar
hipoplastik jaringan sehingga lebih disukai rekonstruksi secara autologous. 3
3.2.2 Penatalaksanaan hipoplasia mandibula
Setelah selesai pertumbuhan, dilakukan perawatan definitif bedah orthognatik.
Rencana ini harus mengikuti urutan konvensional dan harus didahului perawatan
ortodonti yang diperlukan. Penatalaksanaan hipoplasia mandibula dapat berupa
distraksi osteogenesis dan bedah orthognatik.
3.2.2.1 Distraksi Osteogenesis
Distraksi dapat dilakukan pada bayi baru lahir untuk mencegah trakeostomi
atau dapat dilakukan kemudian untuk menyingkirkan tube trakeostomi dan distraksi
osteogenesis awal harus disediakan untuk pasien yang memiliki hambatan jalan nafas
yang parah (misalnya pada pasien yang tergantung pada trakeostomi atau mengalami
sleep apnea yang sedang sampai parah). Pada kasus tersebut, distraksi mandibula secara bilateral dapat menyelamatkan hidup jika ia dapat menyebabkan dekanulasi.
menyebabkan jaringan parut pada kulit. Benih gigi yang sedang berkembang dan
bundel neurovaskular inferior gigi juga mempunyai resiko. Makin muda dan makin
kecil mandibula, makin membutuhkan teknik dalam melakukan prosedur ini, dan
makin besar resiko terjadi komplikasi. Jika distraksi dilakukan dalam periode
sebelum selesainya pertumbuhan, mungkin diperlukan pembedahan mandibula
selanjutnya. 3
Untuk memperoleh kesuksesan perawatan, diperlukan kooperasi dan
komitmen yang tinggi dari pasien dan orang tua. Alat distraksi ini diputar setiap hari,
dijaga tetap bersih untuk mencegah infeksi dan dilindungi dari trauma. Prosedur
harus direncanakan secara hati-hati, dengan memperhitungkan jumlah pemanjangan,
tempat pemanjangan (misalnya pada ramus, badan mandibula atau kombinasi), dan
oklusi akhir yang diinginkan. Desain dari alat distraksi dan tempat pemanjangan yang
diinginkan menentukan tempat ideal untuk peletakan pin dan tempat serta sudut
potongan kortikotomi. Perencanaan ini dilakukan mengikuti pemeriksaan klinis dan
radiografi, dan perawatan ortodonti juga bermanfaat dilaksanakan pada tahap ini.3
Prosedur bedah dilakukan di bawah anestesi umum. Permukaan medial dan
lateral mandibula diekspos secara transoral dan melindungi bundel neurovaskular
inferior. Potongan kortikotomi ditandai dan diletakkan pin superior untuk alat
distraksi secara transkutan. Titik entri kulit untuk pin superior harus caudal terhadap
titik entri tulang untuk meminimalkan jaringan parut. Setelah kortikotomi, border
posterior dari mandibula perlu diberikan perhatian khusus. Pin bawah dimasukkan
dan sekali lagi menarik kulit, dimana kali ini dalam arah kranial. Luka intraoral
pemberian makanan cair dan distraksi dimulai pada hari ke-5. Distraksi 1mm per
hari merupakan jumlah yang dapat diterima. Pasien harus meneruskan makanan
semisolid dan alat distraksi dilepas 3 sampai 4 minggu setelah selesai didistraksi.
Pemeriksaan radiografi dilakukan setelah 3 hari distraksi untuk memastikan distraksi
yang tepat dan tidak ada yang menggantung di kortikotomi. Jika ada yang
menggantung, kortikotomi kembali dilakukan di bawah anestesi umum untuk
membantu distraksi yang telah direncanakan.3
Mengingat distraksi osteogenesis menambah massa jaringan keras, jaringan
lunak di sekeliling termasuk otot terutama diregangkan, tetapi otot dapat beradaptasi
pada waktunya. Karena kualitas jaringan lunak di sekitarnya memainkan peranan
penting dalam perkembangan tulang wajah, pengaruh terhadap struktur jaringan
lunak oleh distraksi osteogenesis harus mengarah pada hasil estetis dan fungsional
jangka panjang. Selain bedah, ortodonti dan fisioterapeutik mengupayakan berbagai
malformasi sindroma mandibula dengan problem yang berbeda, sehingga cukup sulit
untuk memprediksikan hasil yang diperoleh pasien setelah perawatan jangka panjang.
Terjadinya relaps tampaknya tidak terelakkan, karena overkoreksi tidak mampu
mengkompensasi gangguan pertumbuhan sentral ataupun malfungsi muskular.
Namun demikian, distraksi osteogenesis mandibula tidak hanya merupakan metode
yang sangat berguna untuk mengatasi masalah pernafasan dan penelanan pada
defisiensi mandibula yang parah pada usia awal, tetapi juga meningkatkan
Gambar 14 A. Gambaran radiografi lateral kanan pasien Sindroma Treacher Collins yang berusia 18bulan menggunakan alat distraksi internal6
[image:34.612.152.482.84.299.2]Gambar 15 A.Tampilan frontal preoperatif menunjukkan pasien yang mengalami sindroma Treacher Collins, B. Tampilan frontal postoperatif dari pasien yang sama setelah osteotomi dan penempatan alat. Distraksi dimulai 5-7 hari setelah insersi alat dengan pengaktifan 1mm per hari, C. Pandangan lateral perioperatif pasien dengan trakeostomi menunjukkan mikrogenia retrognatik, D. Pandangan lateral postoperatif menunjukkan pemanjangan dalam arah vertikal dan horizontal. Setelah 5 minggu dilakukan distraksi, pasien akhirnya mampu lepas dari trakeostomi.39
3.2.2.2 Bedah orthognatik
Selain distraksi osteogenesis, osteotomi mandibular dapat dilakukan untuk
memperpanjang rahang dan menyeimbangkan oklusi gigi. Tahap rekonstruksi ini
adalah tahap yang paling invasif dan memberatkan fisik pasien. Prosedur tambahan
seperti rhinoplasti dan genioplasti dapat dilakukan setelah osteotomi mayor.5 Tidak
ada timbul kesulitan tambahan yang muncul selain perlunya melepas alat metal jika
prosedur yang sama diulangi. Pada kasus ringan dapat digunakan osteotomi sagital
split, namun tetap lebih disukai osteotomi inverted L.3
Keuntungan dari osteotomi inverted L ini adalah dapat memperpanjang
mandibula atau memperluas mandibula ketika digunakan dengan tulang atau tulang
sintetis untuk grafting, mengkoreksi prognatism mandibula atau asimetri, prosesus
koronoid dan otot temporal tetap berada di posisi yang sebenarnya. Sedangkan
kerugiannya yaitu prosedur ini membutuhkan cangkok tulang dengan tulang ataupun
[image:35.612.151.562.85.211.2]tulang sintetis untuk memperpanjang ramus mandibula, proses penyembuhan
membutuhkan waktu lebih lama. 3
Prosedur ini tidak mempengaruhi laju pertumbuhan, tetapi perubahan posisi
dan orientasi dari segmen proksimal dapat mengubah vektor pertumbuhan rahang
berikutnya. Diperkirakan prosedur ini dapat dilakukan kira-kira pada anak berusia di
atas 12 tahun.3
Gambar 16 A. Tampilan frontal preoperatif pasien berumur 16 tahun dengan sindroma Treacher Collins. Tidak pernah dilakukan koreksi sebelumnya, B. Pandangan frontal postoperatif, C.Pandangan lateral preoperative pasien yang sama, D. Pandangan lateral postoperatif 1 tahun setelah dilakukan bedah orthognatik25
[image:36.612.112.529.279.402.2]BAB 4 KESIMPULAN
Sindroma Treacher Collins adalah kelainan yang diwariskan secara autosomal
dominan yang timbul akibat penyimpangan dalam pengembangan struktur wajah
selama morfogenesis histodiferensiasi antara 20 hari dan minggu ke-12 IU.9,11
Penyebab terjadinya Sindroma Treacher Collins dapat berupa mutasi de novo (60%
dari kasus) atau diwariskan oleh orang tua kepada anaknya (40% dari kasus).2,3,4,10
Patogenesis terjadinya Sindroma Treacher Collins disebabkan karena adanya
mutasi dari gen TCOF1. Gen TCOF1 terpeta dalam kromosom band 5q31.3-33.3.
Gen ini mengkode protein treacle, yang diperlukan dalam perkembangan kraniofasial
yang normal. Haploinsufisiensi TCOF1 mengurangi biogenesis ribosom, dimana
defisiensi biogenesis ribosom berhubungan dengan kurangnya proliferasi dalam sel
neural crest dan sel neuroepithelial yang diamati pada mutan TCOF1. Sebagai stabilisasi diaktifkan banyak gen efektor proapoptotik sehingga terjadi tingkat
kematian jaringan yang tinggi yang diamati dalam patogenesis Sindroma Treacher
Collins. 2,6,10
Manifestasi klinis Sindroma Treacher Collins dapat terjadi pada tulang
tengkorak, hidung dan jaringan lunak wajah, mata, telinga, mandibula dan maksila.
Gambaran yang sangat umum dari Sindroma Treacher Collins yaitu mata yang miring
ke bawah (89% dari kasus), hipoplasia mandibula (78% dari kasus) dan tulang
zigoma (81 % dari kasus).6,9 Diagnosa Sindroma Treacher Collins dapat berdasarkan
Penatalaksanaan pasien dibagi menjadi perawatan emergensi dan perawatan
definitif. Perawatan emergensi umumnya ditujukan untuk menangani jalan nafas bayi
yang baru dilahirkan dengan Sindroma Treacher Collins, dimana karena adanya
hipoplasia mandibula dan lidah yang retroposisi, menghambat jalan nafas bayi.
Penatalaksanaan jalan nafas dapat dengan melakukan trakeostomi, pembedahan
adhesi lidah-bibir, dan distraksi osteogenesis. Sedangkan perawatan definitif
ditujukan untuk mengkoreksi cacat kraniofasial yang diderita pasien, biasanya
dilakukan bedah orthognatik pada pasien yang telah selesai
DAFTAR RUJUKAN
1. Anonymous. Treacher Collins syndrome. <http://en.wikipedia.org/wiki/ Treacher_Collins_syndrome> (22 Agustus 2009).
2. Trainor PA, Dixon J, Dixson MJ. Treacher collins syndrome : etiology,
pathogenesis and prevention. European Journal of Human Genetics 2009; 17: 275-83.
3. Koppel DA, Moos KF. Treacher Collins Syndrome . In : Booth PW, Schendel
SA, Hausamen JE. Maxillofacial Surgery. Vol II. 2nd ed St. Louis : Churchill
Livingstone Elsevier, 2007 : 947-59.
4. Junior HM, Colette RD, Miranda RT, et al. Orofacial features of treacher collins
syndrome. 2009.
5. Posnick JC, Ruiz DL. Treacher collins syndrome : current evaluation, treatment,
and future directions. The Cleft Palate-Craniofacial Journal 2000; 37(5): 434. 6. Tolarova MM, Wong GB. Mandibulofasial dysostosis (treacher collins
syndrome). 2007. Agustus 2009).
7. Avery GB. Neonatology pathophysiology and management of the newborn. 2nd
Philadelphia : J.B. Lippincott Company, 1981 : 881.
8. Marks MW. Fundamental of plastic surgery. Philadelphia : W.B. Saunders
9. Belet N, Oztruk P, Belet U, et al. Treacher Collins Syndrome associated with foot
deformity and genital anomalies. Ankara Üniversitesi Tıp Fakültesi Mecmuası 2006; 59:19-22.
10.Sakai D, Trainor PA. Unmasking the role of Tcof1/treacle. The International
Journal of Biochemistry and Cell Biology 2009; 41 : 1229-32.
11.Magalhaes MHCG, Moreira CR, Paulo S. Clinical and imaging correlations of
treacher Collins syndrome. Oral Surg Oral Med Oral Patholl Pral Radiol Endod 2007; 103 : 836-42
12.Johnson C. A guide to understanding treacher collins syndrome.
<http://www.ccakids.com/Syndrome/TreacherCollins.pdf> (23 Agustus 2009)
13.Cleft Palate Foundation. Treacher collins syndrome. <http://www.cleftline.org/publications/treacher_collins> (23 Agustus 2009)
14.Anonymous. TCS genetic. <http://www.treachercollins.co.uk/gene/genes.htm>
(20 September 2009)
15.Sapp JP, Eversole LR, Wysocki GP. Contemporary oral and maxillofacial
pathology. 2nd ed St. Louis : Mosby, 2004 : 39.
16.Arvedson JC, Brodsky L. Pediatric swallowing and feeding assessment and
management. Delhi : A.IT.B.S. Publishers & Distributors, 1993 : 25.
17.Tunnessen WW . Signs and symptoms in pediatrics. Philadelphia : J.B. Lippincott
Company, 1983 : 159, 193, 245.
18.Shafer WG, Hine MK, Leny BM. A textbook of oral pathology. 4th ed Canada :
19.David DJ. Treacher collins syndrome. In: Plastic and reconstructive surgery.
England : Bailliere Tindall, 1986 : 103-117.
20.Barton S. What is treacher collins syndrome.?. <http://
www.associatedcontent.com/image/482060/index.html?cat=25> (24 Oktober
2009)
21.International Archives of Otolaryngology. Treacher collins syndrome: review of
the literature. <http://www.arquivosdeorl.org.br/conteudo/acervo_eng.asp?id= 492> (24 Oktober 2009)
22.Marszalek B, et al. Clinical features, treatment and genetic background of
treacher collins syndrome. J Appl Genet 2002; 43(2) : 223-33.
23.Katsanis SH, Cutting GR. Treacher collins syndrome.
<http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=tcs> (20
Oktober 2009)
24.Behrents RG, McNamara JA, Avery JK. Prenatal mandibulofacial dysostosis
(treacher collins syndrome). Cleft Palate Journal 1997; 14(1) : 13-34.
25.Kliegman RM, et al. Nelson textbook of pediatrics. 18th ed Philadelphia : Saunders
Elsevier, 2007 : 1533-4.
26.Anonymous. Congenital craniofacial disorders: other craniofacial diagnoses.
<http://www.chsd.org/body.cfm?id=555753> (24 Oktober 2009)
27.Anonymous. Notching of lower eyelid. <http://www.jisppd.com/ articles/2008/26/2/images/JIndianSocPedodPrevDent_2008_26_2_88_41625_5.jp
28.Sanders B. Pediatric oral and maxillofacial surgery. St. Louis : The C.V. Mosby
Company, 1979 : 67.
29.El-Dawlatly AA, Alshimy A, Alhassan K. Difficult laryngoscopy made easy
using wuscope-univent tube technique for treacher collin's syndrome . The Internet Journal of Anesthesiology 2003; 1 (7).
30.Gale T. Treacher collins syndrome.
<http://www.novelguide.com/a/discover/gegd_0002_0002_0/gegd_0002_0002_0
_00422.html> (26 Oktober 2009)
31.Thimmappa B, Hopkins E, Schendel SA. Management of micrognathia.
American Academy of Pediatrics 2009; 10(10).
32.Stastny P. Back to sleep. <http://pennystastny.com/sids/back-to-sleep/> (4
November 2009)
33.Tolarova MM. Pierre robin malformation: treatment & medication.
<http://emedicine.medscape.com/article/995706-treatment> (4 November 2009)
34.Eliashar R, Gross M, Attal P, Hocwald E,
Sichel JY. “Starplasty” prevents tracheostomy complications in infants.
International Journal of Pediatric Otorhinolaryngology 2004; 68: 325-9.
35.Koltai JP. Starplasty: a new technique of pediatric tracheostomy. Archives of
Otolaryngology - Head & Neck Surgery 1998; 124 (10) :1105-12.
36.Parsons RW, Smith DJ. A modified tongue lip adhesion for pierre robin
anomalad. Cleft Palate Journal 1980. 17 (2). 144-7
37.Jackson TI, Malhotra G. Congenital syndromes. <
38.Klein C. Importance of distraction osteogenesis for treacher collins syndrome
and syndromic severe mandibular hypoplasia. Int. J. Oral Maxillofac. Surg. 2005; 34 : 53-4.
39.International Craniofacial Institute. Treacher collins syndrome. <http://www.craniofacial.net/treacher_collins_syndrome.htm > (20 Oktober
DAFTAR RIWAYAT HIDUP Nama Lengkap : Jupita Frantiska
Tempat/ Tanggal Lahir : Tanjung Balai / 17 Juli 1988
Jenis Kelamin : Perempuan
Agama : Buddha
Alamat : Jl. Veteran No 19 K, Medan
Orangtua
Ayah : Rudy Setiawan
Ibu : Fan Mirah
Alamat : Jl. Asahan No 106 , Tanjung Balai
Riwayat Pendidikan
1. 1993-1994 : TK Sisingamangaraja XII, Tanjung Balai
2. 1994-2000 : SD Sisingamangaraja XII, Tanjung Balai
3. 2000-2003 : SLTP Swasta Sutomo 1, Medan
4. 2003-2006 : SMA Swasta Sutomo 1, Medan