Modulation of Amphetamine-Induced Striatal Dopamine
Release by Ketamine in Humans: Implications for
Schizophrenia
Lawrence S. Kegeles, Anissa Abi-Dargham, Yolanda Zea-Ponce,
Janine Rodenhiser-Hill, J. John Mann, Ronald L. Van Heertum, Thomas B. Cooper,
Arvid Carlsson, and Marc Laruelle
Background: Recent brain imaging studies have
indi-cated that schizophrenia is associated with increased
amphetamine-induced dopamine release in the striatum. It
has long been hypothesized that dysregulation of
subcor-tical dopamine systems in schizophrenia might result from
a failure of the prefrontal cortex (PFC) to adequately
control subcortical dopaminergic function. The activity of
midbrain dopaminergic neurons is regulated, in part, by
glutamatergic projections from the PFC acting via
gluta-matergic N-methyl-
D-aspartate (NMDA) receptors. The
goal of this study was to test the hypothesis that a
pharmacologically induced disruption of NMDA
transmis-sion leads to an increase in amphetamine-induced
dopa-mine release in humans.
Methods: In eight healthy volunteers, we compared
stri-atal amphetamine-induced (0.25 mg/kg) dopamine release
under control conditions and under sustained disruption
of NMDA transmission induced by infusion of the
noncom-petitive NMDA antagonist ketamine (0.2 mg/kg
intrave-nous bolus followed by 0.4 mg/kg/hour intraveintrave-nous
infu-sion for 4 hours). Amphetamine-induced dopamine release
was determined with single photon emission computed
tomography, as the reduction in the binding potential (BP)
of the radiolabeled D
2receptor antagonist [
123
I]IBZM.
Results: Ketamine significantly enhanced the
amphet-amine-induced decrease in [
123I]IBZM BP, from
2
5.5%
6
3.5% under control conditions to
2
12.8%
6
8.8%
under ketamine pretreatment (repeated-measures analysis
of variance, p
5
.023).
Conclusions: The increase in amphetamine-induced
do-pamine release induced by ketamine (greater than
two-fold) was comparable in magnitude to the exaggerated
response seen in patients with schizophrenia. These data
are consistent with the hypothesis that the alteration of
dopamine release revealed by amphetamine challenge in
schizophrenia results from a disruption of glutamatergic
neuronal systems regulating dopaminergic cell activity.
Biol Psychiatry 2000;48:627– 640 © 2000 Society of
Bi-ological Psychiatry
Key Words: Schizophrenia, SPECT, dopamine, [
123I]IBZM,
glutamate, ketamine, amphetamine
Introduction
T
he classic dopamine hypothesis of schizophrenia
pro-poses that hyperactivity of dopamine transmission is
responsible for positive symptoms of the disorder
(Carls-son and Lindqvist 1963). This hypothesis was traditionally
supported by the correlation between clinical doses of
antipsychotic drugs and their potency to block dopamine
D
2receptors (Creese et al 1976; Seeman and Lee 1975),
and by the psychotogenic effects of dopamine-enhancing
drugs (for reviews, see Angrist and van Kammen 1984;
Lieberman et al 1987). More recently, the hypothesis of a
dysregulation of dopamine function in schizophrenia
re-ceived additional support from the observation of
in-creased striatal dopamine transmission following acute
amphetamine challenge in untreated patients with
schizo-phrenia. Amphetamine-induced increase in D
2receptor
occupancy by dopamine was measured as the reduction in
the binding potential (BP) of the single photon emission
computed tomography (SPECT) radiotracer [
123I]IBZM or
the positron emission tomography radiotracer [
11C]raclo-pride. In three independent cohorts, the
amphetamine-induced reduction in radiotracer BP was found to be
significantly increased in patients with schizophrenia, as
compared with matched healthy control subjects
(Abi-Dargham et al 1998; Breier et al 1997b; Laruelle et al
1996). This exaggerated response was also documented in
first-episode patients never previously exposed to
antipsy-chotic drugs (Laruelle et al 1999). Together, these data
support the hypothesis of a dysregulation of dopamine
release in schizophrenia (Laruelle and Abi-Dargham
1999).
From the Departments of Psychiatry (LSK, AA-D, YZ-P, JR-H, JJM, TBC, ML) and Radiology (LSK, AA-D, JJM, RLVH, ML), Columbia University College of Physicians and Surgeons and New York State Psychiatric Institute, New York, and the Department of Pharmacology, University of Go¨teborg, Go¨teborg, Sweden (AC).
Address reprint requests to Lawrence S. Kegeles, M.D., Columbia University, NYSPI, 1051 Riverside Drive, Unit 42, New York NY 10032.
Received February 17, 2000; revised June 22, 2000; accepted June 29, 2000.
The pathophysiologic mechanism underlying this
dys-regulation remains to be elucidated. The amphetamine
challenge might reveal a primary abnormality of
dopami-nergic neurons in schizophrenia or, more likely, a
defi-ciency in the feedback mechanisms that modulate
dopa-minergic cell activity following amphetamine exposure.
Abnormalities of the glutamatergic afferents from the
prefrontal cortex (PFC) to the midbrain dopaminergic cell
areas are likely to be implicated in this deficient
regula-tion, given the evidence for deficits in PFC function in
schizophrenia (for reviews, see Goldman-Rakic and
Selemon 1997; Weinberger and Berman 1996).
Since positive symptoms are more sensitive than
nega-tive symptoms to direct manipulation of the dopamine
system, hyperactivity of dopamine transmission is more
relevant to positive symptoms than to cognitive and
negative ones (Crow 1980). The hypothesis that
schizo-phrenia might be associated with a deficiency of
N-methyl-
D-aspartate (NMDA) receptor function addresses
some of the limitations of the dopamine hyperactivity
model (Javitt and Zukin 1991; Olney and Farber 1995;
Tamminga et al 1995). In contrast to dopamine agonists,
noncompetitive NMDA antagonists such as phencyclidine
(PCP) and ketamine induce positive and negative
symp-toms in healthy and schizophrenic subjects (Allen and
Young 1978; Collier 1972; Krystal et al 1994; Lahti et al
1995). Thus, NMDA receptor hypofunction might offer a
more compelling pharmacologic model of the illness than
the dopamine hyperactivity model. In addition, it has been
postulated that the dysregulation of dopamine
transmis-sion in schizophrenia might itself be secondary to an
alteration in NMDA transmission (Grace 1991; Olney and
Farber 1995).
Recent data in rodents offer a putative direct link
between the NMDA hypofunction hypothesis and the
alteration of dopamine transmission actually observed in
patients with schizophrenia (i.e., increased response to
amphetamine). Miller and Abercrombie (1996) reported
that pretreatment with the noncompetitive antagonist
MK-801 resulted in a large potentiation of
amphetamine-induced dopamine release as measured with microdialysis
in rats. This study suggested that the increased
amphet-amine-induced dopamine release observed in
schizophre-nia might result from a deficiency in NMDA-mediated
glutamate transmission.
The purpose of our study was to test the plausibility of
this model by investigating the existence of the same
modulatory interaction in humans. Given the large
be-tween-subject variability of amphetamine-induced
dopa-mine release in healthy humans (Laruelle et al 1999), the
existence of an NMDA modulation of this response was
tested using a within-subject design. We first established
that, under control conditions, the amphetamine-induced
dopamine release in healthy volunteers measured by the
SPECT method was highly reproducible and stable over
time (Kegeles et al 1999). In our study
amphetamine-induced dopamine release was measured twice, at a
1-week interval, in eight healthy volunteers. The first
measurement was performed under control conditions, and
the second measurement was obtained under conditions of
sustained NMDA transmission deficiency, as induced by
prolonged infusion of the noncompetitive NMDA
antag-onist ketamine. From the microdialysis data of Miller and
Abercrombie (1996), we postulated that
amphetamine-induced dopamine release measured under conditions of
impaired NMDA transmission would be significantly
elevated compared with amphetamine-induced dopamine
release measured under control conditions.
Methods and Materials
Subjects
Eight healthy subjects (three female, five male; ages 27 6 2 years), never previously exposed to psychostimulants, were studied twice (control and ketamine conditions), at intervals of 8 6 4 days (range, 5–16 days). Two subjects were African American, four white, and two Hispanic. Inclusion criteria were 1) absence of past or present neurologic or psychiatric illness, 2) no current or past cardiovascular conditions, 3) no pregnancy, and 4) no prior exposure to psychostimulants.
Intravenous (IV) administration of both [123
I]IBZM and am-phetamine were approved by the Food and Drug Administration under an Investigational New Drug protocol. The protocol was approved by the institutional review boards of Columbia Pres-byterian Medical Center and New York State Psychiatric Insti-tute. All subjects gave written informed consent for the study, after detailed explanation of the nature and risks of the study. The information relative to the risks of the study included discussion of risks associated with radioactivity exposure, cardiovascular side effects (transient increase in blood pressure), expected side effects of the challenges (transient induction of dissociative or psychotic state), and exposure to drugs of abuse potential (ketamine and amphetamine).
Radioligand Preparation
[123
I]IBZM was prepared by direct electrophilic radioiodination of the phenolic precursor BZM [(S)(2 )-N-[(1-ethyl-2-pyrrolidi-nyl)methyl]-2-hydroxy-6-methoxybenzamide] as described (Kung and Kung 1990; Zea-Ponce and Laruelle, in press). Labeling yield was 69%67%, the radiochemical yield 63%6
8%, and the radiochemical purity 98%60.6% (n516).
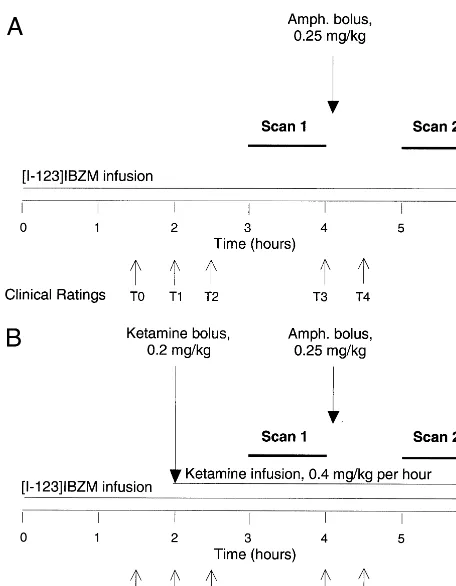
Study Design
A schematic representation of the study protocol is presented in Figure 1. The previously described [123
scanning sessions were obtained on each experimental day. On the test day, [123
I]IBZM BP was measured under control condi-tions and 60 min following administration of amphetamine (0.25 mg/kg, IV) according to our standard protocol (Kegeles et al 1999). The comparison between the baseline scan and the postamphetamine scan provided a measure of the decrease in [123
I]IBZM BP resulting from amphetamine-induced dopamine release under control conditions. On the ketamine day, both scanning sessions were obtained under conditions of NMDA deficiency, as achieved by a bolus plus constant infusion of ketamine. The comparison between the baseline and postamphet-amine scan allowed measurement of amphetpostamphet-amine-induced do-pamine release under conditions of NMDA deficiency.
Both amphetamine and ketamine increase the blood pressure, raising concern about the cardiovascular response to a challenge with both drugs. Two precautions were therefore implemented to minimize this risk. First, we used a dose of amphetamine slightly lower (0.25 mg/kg, IV) than our standard dose (0.3 mg/kg, IV). Second, the order of the pharmacologic intervention
(amphet-amine alone vs. ket(amphet-amine plus amphet(amphet-amine) was not counter-balanced. The observation of the cardiovascular response of these subjects to amphetamine alone was felt to be important before administering the ketamine plus amphetamine challenges. Importantly, we previously performed a test/retest study in which healthy volunteers received two amphetamine injections (0.3 mg/kg) at a 2-week interval. In this study, the amphetamine-induced dopamine release measured as the displacement of [123
I]IBZM was similar on the test day (8%68%) and retest day (9% 6 7%), indicating absence of tolerance or sensitization. Moreover, the individual [123
I]IBZM displacement values mea-sured on the test day were highly predictive of these values measured on the retest day (r2
5.76, p5.023). The availability of this information avoided the necessity of a counterbalanced design for this study.
Imaging Protocol
CONTROL DAY. To decrease radiation exposure to the thyroid gland, subjects received 0.6 g potassium iodide 60 min before [123
I]IBZM injection. Four fiducial markers each contain-ing 3mCi of [99m
Tc]NaTcO4were attached to each side of the
subject’s head at the level of the cantho-meatal line. An IV catheter was inserted in each arm, one for blood sampling and the other for amphetamine injection and ligand administration. [123
I]IBZM was administered as a priming bolus, immediately followed by a continuous infusion at a constant rate (bolus/ infusion ratio, 3.92 6 0.01 hours) for the duration of the experiment (360 min). The total injected dose was 10.16 0.7 mCi, decay corrected to the beginning of the experiment. We previously established that, using this administration schedule, stable activity levels in plasma and brain are observed from 150 min until the end of the infusion. During the first 180 min of the infusion subjects were allowed to relax in a comfortable setting, in a room adjacent to the camera room. The first scanning session (preamphetamine) was initiated at 180 min and lasted 60 min (Figure 1A). Single photon emission computed tomography data were obtained with the triple-head PRISM 3000 (Picker, Cleve-land), equipped with low-energy ultra– high-resolution fan beam collimators (full width at half maximum 8 –10 mm). Scanning was performed with the following acquisition parameters: con-tinuous acquisition mode; matrix, 64364332; angular range, 120; angular steps, three; seconds per step, 18; frame duration, 12 min; number of frames, five; and radius of rotation, 13.5 cm. Amphetamine (0.25 mg/kg, IV) was injected shortly after com-pletion of the first scanning session (240 min). Amphetamine injection was followed by 60 min of behavioral ratings, as detailed below, and vital signs monitoring. The second scan session (postamphetamine) was obtained using similar parame-ters from 300 to 360 min.
KETAMINE DAY. The only change to the above protocol consisted of the use of one additional IV catheter for ketamine administration. Ketamine hydrochloride (S-R-racemic) was ad-ministered as a bolus (0.2 mg/kg) at 60 min before the first SPECT scan (i.e., 120 min after beginning the radioligand infusion), followed by a constant infusion of 0.4 mg/kg/hour for the remaining 4 hours of the study (Figure 1B). All subjects were
Figure 1. Protocol timeline of the first study day (A, control day) and of the second study day (B, ketamine day). The control day included two single photon emission computed tomography scans (60-min acquisition time) separated by 1 hour with intervening amphetamine bolus injection (0.25 mg/kg), all under sustained equilibrium administration of [123
admitted to the Irving Center for Clinical Research at Columbia Presbyterian Medical Center on the ketamine condition day and were discharged after an overnight stay and physician clearance the next morning.
Image Analysis
Image analysis was performed as previously described (Laruelle et al 1995). Briefly, frames were transferred into the MEDx software system (Sensor Systems, Sterling, VA) and corrected for attenuation assuming uniform attenuation (attenuation coef-ficientm 50.10 cm2
/g) within an ellipse drawn around the skull as identified by fiducial markers. Frames were realigned to each other, using a least-squares algorithm for within-modalities coregistration (automated image registration; Woods et al 1992). Standard regions of interest of constant size were used to analyze all studies. Nonspecific activity was estimated as the average of the frontal and occipital regional activities, since these regions can be identified with greater reliability than the cerebellum. For each scanning session, the specific to nonspecific equilibrium ratio (V399) was calculated as the ratio of striatal minus
nonspe-cific to nonspenonspe-cific activity (Laruelle et al 1994a). Under steady-state conditions, and assuming that amphetamine does not affect [123
I]IBZM nonspecific binding, the percent decrease in V399is equal to the percent decrease in BP (Laruelle et al 1995).
Amphetamine-induced decrease in [123
I]IBZM BP was ex-pressed as a percentage of the preamphetamine value.
Amphetamine and Ketamine Plasma Measurement
Amphetamine plasma levels were measured in three venous samples obtained at 10, 20, and 40 min after amphetamine injection. Amphetamine was quantified as its N-heptafluorobu-tyryl derivative via gas chromatography/mass spectroscopy uti-lizing a capillary column with mass spectrometer with simulta-neous ion monitoring in the negative chemical ionization mode and reactant gas methane/ammonia (95:5). The method is essen-tially the same as described by Reimer et al (1993) with the following modifications: a 30-m DB-17 capillary column was substituted to improve separation and peak symmetry. Trideuter-ated amphetamine was used as the internal standard. Standard curves for both compounds are uniformly linear (r..99) over the range tested (0.1–500 ng/mL) with negligible intercepts. Sensitivity is,0.1 ng/mL for each when 1 mL of plasma is extracted. Interassay RSD% is 5.2% at 5 ng/mL.
Ketamine plasma levels were measured in three venous samples obtained at 1, 2, and 3 hours after beginning the ketamine infusion. Plasma ketamine and norketamine were assayed using a validated liquid chromatographic (LC) procedure with UV detection. Following the addition of 500 ng of internal standard (2-phenylmorpholinol, BW306U), ketamine and the metabolite norketamine were extracted from 1 mL of plasma, made alkaline with 0.5 mol/L NaOH, with 5.0 mL of 1.5% isoamyl alcohol in n-heptane. The organic extract was back-extracted with 0.25 mL of 0.01 mol/L HCl and transferred to inserts for injection on the LC. Chromatography was performed using a trimethylsilyl-bonded silica column (LC-1, Supelco, Bellefonte, PA) with a mobile phase consisting of 85%
phos-phate buffer and 15% acetonitrile, adjusted to pH of 2.4 with phosphoric acid, triethylamine, and heptane sulfonate. At a flow rate of 2.0 mL/min, ketamine, norketamine, and the internal standard were separated and detected at a UV wavelength of 210 nm in less than 12 min. Within-day coefficient of variation of ketamine and norketamine did not exceed 10.6% (range, 2000 –25 ng/mL; n 5 12 for each of seven concentrations). Day-to-day variation of ketamine and norketamine quality con-trols at 1250, 250, and 50 ng/mL did not exceed 5.6% and 5.8%, respectively (n5 20 days). The minimum quantifiable limits were set at 10 ng/mL for both ketamine and norketamine. Plasma levels were not obtained in one subject on the ketamine day because of difficulties with IV access for blood sampling.
Behavioral Responses
Behavioral responses to the challenges were measured with the Clinician Administered Dissociative Symptom Scale (CADSS; Krystal et al 1994), the Brief Psychiatric Rating Scale (BPRS; Overall and Gorham 1962), and a modified version of the Amphetamine Interview Rating Scale (Laruelle et al 1995). The CADSS consists of 27 items rated from 0, “not at all,” to 4, “extremely,” and includes 19 subjective items and eight observer items (range, from 0 to 108). The BPRS consists of 18 items rated from 1, “not present,” to 7, “extremely severe.” A positive symptom subscale (Bowers et al 1980; Kane et al 1988; Krystal et al 1993, 1994) was evaluated consisting of the four symptoms conceptual disorganization, suspiciousness, hallucinatory behav-ior, and unusual thought content. This positive subscale ranges from 4 to 28. A negative symptom subscale (Krystal et al 1994; Thiemann et al 1987) was also separately evaluated containing the three items blunted affect, emotional withdrawal, and motor retardation, ranging from 3 to 21.
The CADSS and BPRS were administered at six time points during the study (times are relative to the beginning of the [123
I]IBZM infusion; Figure 1): Time 0, 90 min; Time 1, 120 min; Time 2, 160 min; Time 3, 240 min; Time 4, 270 min; and Time 5, 360 min. On the control days, Times 0, 1, 2, and 3 corresponded to baseline ratings and Times 4 and 5 corresponded to postamphetamine ratings (amphetamine was administered at 240 min). On the ketamine day, Time 0 corresponded to the baseline rating; Times 1, 2, and 3 corresponded to ratings under ketamine alone; and Times 4 and 5 corresponded to ratings under both amphetamine and ketamine.
Statistical Analyses
Results
Imaging Data
Table 1 lists [
123I]IBZM V
399
values for both pre- and
postamphetamine scans on the control day and the
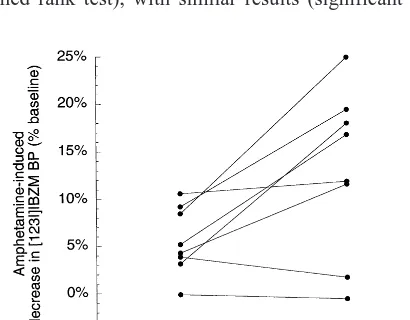
ket-amine day. Figure 2 shows the individual amphetket-amine-
amphetamine-induced percent decrease in BP under control conditions
and under ketamine pretreatment, with each subject’s two
data points connected.
On the test day, amphetamine induced a significant
5.5%
6
3.5% reduction in [
123I]IBZM BP
[repeated-measures ANOVA, F(7,1)
5
18.4, p
5
.003]. On the
ketamine day, amphetamine reduced [
123I]IBZM BP by
12.8
6
8.8% [repeated-measures ANOVA, F(7,1)
5
17.4,
p
5
.004]. The amphetamine-induced decrease in
[
123I]IBZM BP was significantly larger on the ketamine
day, as compared with the control day [repeated-measures
ANOVA, F(7,1)
5
8.3, p
5
.023]. The variance of the
amphetamine effect was significantly higher on the
ket-amine day [F test of variance ratio, F(7,7)
5
0.16, p
5
.03]. Because of this difference in variance, we also
analyzed the significance of the between-day difference in
amphetamine effect using a nonparametric test (Wilcoxon
signed rank test), with similar results (significant
differ-ence between amphetamine effect on test and retest day,
p
5
.049).
No differences were found between preamphetamine
[
123I]IBZM V
399
on the control day (0.74
6
0.06) and
ketamine day [0.74
6
0.07; repeated measures ANOVA,
F(7,1)
5
0.026, p
5
.87]. Thus no effect of ketamine alone
was detected on [
123I]IBZM V
3
99
.
The ketamine modulation of the amphetamine effect on
[
123I]IBZM V3
99
was assessed by calculating the relative
increase in the amphetamine effect due to ketamine. A
larger amphetamine-induced reduction in [
123I]IBZM BP
on the ketamine day, as compared with the control day,
was observed in six out of eight subjects. Ketamine
increased the amphetamine effect by 183%
6
171%
(range, from
2
11% to 474%). Thus, a large
between-subject variability was observed in the ketamine
modula-tion of the amphetamine effect.
We observed a nonsignificant association between the
amphetamine effect on [
123I]IBZM V
399
on control and
ketamine days (r
25
.38, p
5
.09). The amphetamine
effect under control conditions was not predictive of the
magnitude of the ketamine modulation of this effect (r
25
.28, p
5
.17).
The increased effect of amphetamine on [
123I]IBZM
V
399
on the ketamine day was not due to differences in
amphetamine plasma levels. On the control day, plasma
amphetamine levels averaged over the three measurements
for each subject were 35
6
10 ng/mL (n
5
8); on the
ketamine day they were 33
6
5 ng/mL [n
5
7;
repeated-measures ANOVA, F(7,1)
5
0.026, p
5
.87]. No
rela-tionships were observed between amphetamine plasma
levels and amphetamine-induced decrease in [
123I]IBZM
V
399
, either on the control day (r
2
5
.04, p
5
.61) or on the
ketamine day (r
25
.04, p
5
.66).
Ketamine plasma levels, measured 2 hours from
begin-ning of ketamine infusion (before amphetamine injection)
and at 3 hours (after amphetamine injection), were 191
6
38 ng/mL (n
5
6) and 197
6
52 ng/mL (n
5
7),
respectively. These values were not significantly different
by repeated-measures ANOVA [F(5,1)
5
2.1, p
5
.20].
Thus, the injection of amphetamine did not affect
ket-amine plasma levels. Ketket-amine plasma levels were not
significantly correlated with the ketamine modulation of
Figure 2. Ketamine modulation of striatal amphetamine-induced dopamine release, showing a significantly larger release in eight healthy volunteers pretreated with intravenously administered ketamine (repeated-measures analysis of variance, p 5 .023). BP, binding potential.
Table 1. Amphetamine Effect on D2Receptor Availability on Control and Ketamine Days
Experimental day
Preamphetamine [123I]IBZM V
399 (unitless)
Postamphetamine [123I]IBZM V
399 (unitless)
Amphetamine-induced decrease in [123I]IBZM BP
(% baseline)
Amphetamine plasma level (ng/mL)
Control day 0.7460.06 0.7060.06 25.5%63.6% 35610
Ketamine day 0.7460.07 0.6560.07 212.8%68.8%a 3365
BP, binding potential.
the amphetamine effect (r
25
.28, p
5
.22). The levels of
the ketamine metabolite norketamine increased with time,
with preamphetamine levels of 134
6
39 ng/mL (n
5
6)
and postamphetamine levels of 169
6
52 ng/mL [n
5
7;
repeated-measures ANOVA, F(5,1)
5
12.7, p
5
.02].
Behavioral Responses
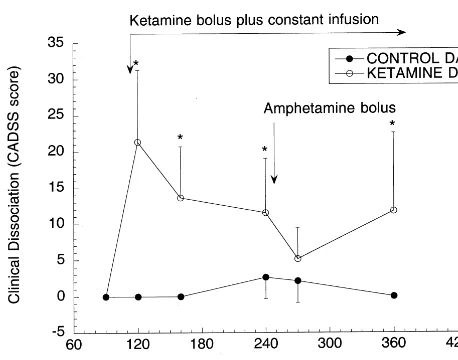
DISSOCIATION.
Amphetamine alone did not induce
dissociation. On control day, CADSS mean total scores
showed no significant changes with amphetamine
admin-istration by repeated-measures ANOVA [Figure 3;
repeat-ed-measures ANOVA, F(7,5)
5
1.2,p
5
.28]. In contrast,
ketamine produced significant levels of dissociation. On
ketamine day, CADSS mean total scores showed
signifi-cant changes [repeated-measures ANOVA, F(7,5)
5
3.9,
p
5
.006]. Post hoc analysis revealed that CADSS scores
were significantly elevated relative to baseline score (0
6
0) at every time point during ketamine administration,
with the exception of the first score obtained following
amphetamine bolus injection (Time 4): Time 0, 0
6
0;
Time 1, 21
6
20, p
5
.002; Time 2, 14
6
14, p
5
.013;
Time 3, 12
6
15, p
5
.034; Time 4, 5
6
9, p
5
.33; Time
5, 12
6
21, p
5
.031. Figure 3 illustrates the average
changes in dissociation over time in the control and
ketamine days. Thus, amphetamine injection resulted in a
transient improvement of ketamine-induced dissociation,
which, as noted, was not due to an impact of amphetamine
on ketamine plasma levels.
POSITIVE SYMPTOMS.
Amphetamine alone did not
elicit positive symptoms: no significant changes in the
total of four positive items of the BPRS were observed on
the control day [repeated-measures ANOVA, F(7,5)
5
1.0, p
5
.43]. On the ketamine day, ketamine
administra-tion was followed by a mild increase in positive symptoms
that
did
not
reach
significance
[repeated-measures
ANOVA, F(7,5)
5
1.3, p
5
.28]: Time 0, 4.0
6
0; Time
1, 5.2
6
1.5; Time 2, 5.2
6
1.6; Time 3, 5.6
6
3.1; Time
4, 5.1
6
2.2; Time 5, 4.6
6
1.7.
NEGATIVE SYMPTOMS.
Amphetamine alone did not
elicit negative symptoms: on the control day, the sum of
the three negative symptoms of the BPRS remained at its
minimum of 3
6
0 at every time point. On the ketamine
day, significant changes were observed
[repeated-mea-sures ANOVA, F(7,5)
5
3.74, p
5
.008]. Post hoc
analysis revealed that negative scores were significantly
elevated relative to baseline scores (3
6
0) during
ket-amine administration before the amphetket-amine injection
(Times 1, 2, and 3), but not after amphetamine (Times 4
and 5): Time 1, 4.1
6
1.6, p
5
.03; Time 2, 5.0
6
1.8, p
5
.0004; Time 3, 4.1
6
1.6, p
5
.03; Time 4, 3.8
6
1.0, p
5
.15; Time 5, 3.4
6
0.7, p
5
.46.
In summary, infusion of low-dose ketamine induced a
significant increase in dissociative and negative
symp-toms, and these symptoms were transiently improved
following the bolus injection of amphetamine.
SAFETY INFORMATION.
No synergy between
am-phetamine and ketamine was observed on the
cardiovas-cular response (data not shown). The subjective effects of
ketamine vanished within minutes of discontinuation of
the infusion. Subjects were observed overnight in a
re-search-dedicated inpatient unit and were discharged the
next day after medical evaluation, which was
unremark-able in all cases. Subjects were contacted by phone 3
months after completion of the study. None of the subjects
reported any side effects of the study during this time
interval.
Discussion
This study indicates that, in humans, acute administration
of the noncompetitive NMDA antagonist ketamine
in-duces a significant increase in the effect of amphetamine
on [
123I]IBZM BP. On the test day, amphetamine (0.25
mg/kg) induced a small but significant 5.5%
6
3.5%
Figure 3. Dissociative symptoms: dissociative state measured by the Clinician Administered Dissociative Symptom Scale (CADSS) mean total score on the control day and on the ketamine day as a function of time (n 5 8). No significant dissociation was observed on the control day, either before or after amphetamine challenge (arrow). On the ketamine day, dissociation was rapidly induced by the bolus of ketamine. This dissociation stabilized at lower levels during the ketamine constant infusion. Amphetamine bolus administration was fol-lowed by a brief improvement in dissociation. As the effect of the amphetamine bolus waned, the ketamine-induced dissociative state reemerged. *Rating significantly different from baseline; repeated-measures analysis of variance followed by post hoc test,
reduction in [
123I]IBZM BP. This response was smaller
than reported previously for healthy volunteers (
2
7.6%
6
8.0%, n
5
15 [Laruelle et al 1996];
2
7.11%
6
6.3%, n
5
15 [Abi-Dargham et al 1998]; and
2
8.2%
6
7.7%, n
5
6
[Kegeles et al 1999]), presumably because of the lower
dose of amphetamine injected in our study (0.25 mg/kg),
as compared with the previous studies (0.3 mg/kg). On the
ketamine
day,
amphetamine
(0.25
mg/kg)
reduced
[
123I]IBZM BP by 12.8%
6
8.8%, an effect significantly
larger than on the test day. This result suggests that
disruption of NMDA transmission significantly increases
amphetamine-induced dopamine release.
This conclusion is supported by several considerations.
First, the larger amphetamine effect observed on the
second study day (ketamine day) was not due to
sensiti-zation to amphetamine induced by the first amphetamine
injection (control day), since we previously observed an
absence of sensitization or tolerance to
amphetamine-induced dopamine release in healthy volunteers studied
twice at a 2-week interval under similar conditions
(Keg-eles et al 1999). Second, this effect was mediated by
disruption of NMDA transmission. The selectivity of
racemic ketamine for the NMDA receptor (K
d5
0.6
m
mol/L; Tam and Zhang 1988) is only relative, since
racemic ketamine also displays affinities for other
recep-tors known to affect dopamine release such as mu opioid
receptors (K
d5
27
m
mol/L), sigma receptors (K
d5
66
m
mol/L), or dopamine transporters (K
d5
63
m
mol/L;
Nishimura et al 1998; Smith et al 1987). Yet, at the
steady-plasma level achieved in this study (195
6
45
ng/mL) the intracerebral free ketamine concentration is
expected to be in the range of 0.38
6
0.09
m
mol/L (given
a molecular weight of 274.19 and a plasma free fraction of
53%; Dayton et al 1983). At this concentration, ketamine
should affect mainly, if not exclusively, NMDA receptors.
Third, the larger amphetamine-induced reduction in
[
123I]IBZM BP stimulated by ketamine could result from a
larger amphetamine-induced dopamine release, an
in-creased affinity of D
2receptors for dopamine induced by
ketamine, or some combination of both factors (for a
review, see Laruelle 2000a). Yet, we did not observe any
effect of ketamine alone on [
123I]IBZM binding,
suggest-ing that ketamine does not significantly alter the affinity of
D
2receptors for dopamine, the endogenous competitor of
[
123I]IBZM. Therefore, the data reported in this study
suggest that, in humans, alteration in NMDA transmission
results in a significant increase in amphetamine-induced
dopamine release.
Ketamine as a Pharmacologic Model of Cortical–
Subcortical Dysconnectivity
This observation confirms in humans a regulation of
dopaminergic responses by glutamatergic inputs
previ-ously observed in rodents (Miller and Abercrombie 1996)
and indicates that the acute response of dopaminergic
neurons to amphetamine exposure in humans is not solely
determined by the availability of dopamine stores in the
terminals, but also by the regulation of dopaminergic
neurons through glutamatergic transmission. The
enhance-ment of striatal amphetamine-induced dopamine release
induced by ketamine was comparable to the greater
response to amphetamine challenge (more than twofold
higher release; Table 2) observed in patients with
schizo-phrenia (Abi-Dargham et al 1998; Breier et al 1997b;
Laruelle et al 1996). The SD of the amphetamine effect
was larger under ketamine conditions, as compared with
the control conditions, a finding also observed in patients
with schizophrenia compared with control subjects. These
results suggest the increased amphetamine-induced
dopa-mine release measured in patients with schizophrenia
might result from an abnormal glutamatergic regulation of
dopaminergic neurons, rather than from a primary
pathol-ogy of these neurons (Weinberger 1999).
It has long been hypothesized that dysregulation of
subcortical dopamine function in schizophrenia may be
secondary to a failure of the PFC to adequately control
subcortical dopaminergic function (Grace 1991;
Wein-berger et al 1986). The PFC regulates subcortical
dopa-mine via, in part, glutamatergic projections to the midbrain
dopaminergic neurons. This regulation is mediated, among
others, by NMDA receptors. Therefore, pharmacologically
Table 2. Amphetamine Effect on [123I]IBZM Binding Potential
Study (reference)
Amphetamine
dose Percent change in binding potential (condition)
Relative increase
Test/retest in healthy control subjects (Kegeles et al 1999)
0.30 mg/kg 28.2%67.7% (test) 29.2%67.2% (retest) 1.12
(n56) (n56)
Control/NMDA deficiency in healthy control subjects (this study)
0.25 mg/kg 25.5%63.6% (test) 212.8%68.8%a(NMDA deficiency) 2.32
(n58) (n58)
Healthy control/patients with schizophrenia (Laruelle et al 1999)
0.30 mg/kg 27.5%67.1% (test-control subjects)217.1%613.2%b(test-patients) 2.28
(n536) (n534)
NMDA, N-methyl-D-aspartate.
induced disruption of NMDA receptor function provides a
model for cortical–subcortical dysconnectivity potentially
relevant to the pathophysiology of the illness. The
dem-onstration that disruption of cortical–subcortical
connec-tivity by ketamine in healthy individuals induces a
dys-regulation of dopaminergic function (i.e., increase in
amphetamine-induced dopamine release) similar to the
one observed in patients with schizophrenia supports the
relevance of these models (Grace 1991; Weinberger et al
1986).
Glutamatergic Control of Dopaminergic Cell
Response to Amphetamine
Modulation of dopamine cell activity and dopamine
re-lease by glutamatergic projections is complex. Stimulation
of NMDA,
a
-amino-3-hydroxy-5-methyl-4-isoxazole
pro-pionic acid (AMPA) and kainate receptors localized on A9
and A10 midbrain dopaminergic cells by direct application
of glutamate or glutamate agonists enhances dopaminergic
cell-firing rate and burst firing (Chergui et al 1993;
Christoffersen and Meltzer 1995; Overton and Clark 1992;
Scarnati and Pacitti 1982; Suaud-Chagny et al 1992),
leading to increased dopamine release in the terminal
regions as measured by voltametry (Suaud-Chagny et al
1992) and microdialysis (Enrico et al 1998; Kalivas et al
1989; Schilstrom et al 1998; Westerink et al 1998).
Stimulation of glutamatergic afferents from the PFC to the
ventral tegmental area (VTA) and substantia nigra (SN;
Christie et al 1985; Sesack and Pickel 1992) results in the
same effect. For example, an increase in burst-firing
activity of midbrain dopaminergic neurons and an increase
in striatal dopamine release are elicited by electrical
stimulation of the PFC (Gariano and Groves 1988),
prefrontal application of glutamate (Murase et al 1993a),
or blockade of the inhibitory effects of
g
-aminobutyric
acid (GABA) on prefrontal neurons by local application of
the GABA
Aantagonist bicuculline (Karreman and
Moghaddam 1996). Conversely, inhibition of prefrontal
cell activity by prefrontal application of the sodium
channel blocker tetrodotoxin or the monoamine releaser
amphetamine decreases dopamine release in the striatum,
an effect demonstrated both in rats (Karreman and
Moghaddam 1996) and in rhesus monkeys (Kolachana et
al 1995). This activation of dopamine transmission by the
PFC is mediated by activation of glutamatergic projections
to midbrain dopaminergic neurons rather than to the
dopaminergic terminals in the striatum, since the increased
striatal dopamine release elicited by prefrontal stimulation
is inhibited by local injection of glutamate antagonists in
the VTA but not in the striatum (Karreman and
Moghaddam 1996).
One of the apparent paradoxes of glutamatergic
modu-lation of dopamine release is that systemic administration
of noncompetitive NMDA antagonists also stimulates
dopamine cell activity. A large number of studies reported
that systemic administration of noncompetitive NMDA
antagonists such as PCP or MK-801 increases the firing
rate of midbrain dopaminergic neurons (Freeman and
Bunney 1984; French 1986; French and Ceci 1990; French
et al 1993; Murase et al 1993b; Pawlowski et al 1990;
Rouillard et al 1990; Steinfels et al 1989; Svensson et al
1995; Zhang et al 1992) and increases dopamine release
and utilization in the terminal fields (Carboni et al 1989;
Deutch et al 1987; Doherty et al 1980; Hertel et al 1995;
Hondo et al 1994; Jentsch et al 1997; Mathe et al 1996;
Miller and Abercrombie 1996; Steinpreis and Salamone
1993; Verma and Moghaddam 1996; Wedzony et al 1993).
These stimulatory effects of systemic injections of
non-competitive NMDA antagonists are more prominent on
the VTA cells than on the SN cells (Zhang et al 1992), and
the increase in dopamine release elicited by these drugs is
more pronounced in the mesolimbic system (PFC,
hip-pocampus, and nucleus accumbens) than in the
nigrostri-atal system (dorsal striatum; Carboni et al 1989; Deutch et
al 1987; Kashihara et al 1990; Verma and Moghaddam
1996; Wedzony et al 1993; Weihmuller et al 1991;
Whitton et al 1992). Three recent brain imaging studies
(Breier et al 1998; Smith et al 1998; Vollenweider et al
2000) demonstrated that, in healthy humans, ketamine
administration alone induced a slight reduction of [
11C]ra-clopride striatal BP (an effect not detected in this study,
possibly due to the lower vulnerability to endogenous
competition of [
123I]IBZM binding relative to [
11C]raclo-pride binding). Importantly, the stimulatory effect of
systemic noncompetitive NMDA antagonists on
dopami-nergic activity is not observed when these drugs are
applied directly into the VTA (Freeman and Bunney 1984;
French 1986; Rouillard et al 1990; Westerink et al 1998;
Zhang et al 1992, 1994), suggesting that the activating
effects of systemic noncompetitive antagonists on
dopa-mine systems are mediated by indirect mechanisms.
nondopa-minergic, presumably GABA-ergic interneurons in the SN
(Ceci and French 1989; Zhang et al 1993, 1994). Thus, the
activating effect of NMDA noncompetitive antagonists
might result from inhibition of glutamatergic cortical
projections to GABA-ergic midbrain interneurons or to
GABA-ergic striatomesencephalic projections (Carlsson
and Carlsson 1990; Carlsson 1993).
According to this model (Carlsson et al 1999b), activity
of midbrain dopaminergic neurons is under dual influence
of the PFC via an activating pathway (the “accelerator”)
and an inhibitory pathway (the “brake”), allowing fine
tuning of dopaminergic activity by the PFC (Figure 4).
The activating pathway is provided by direct
glutamater-gic projections onto the dopaminerglutamater-gic cells, and this
stimulatory influence is mediated by NMDA, AMPA, and
kainate receptors. The inhibitory pathway is provided by
glutamatergic projections to GABA-ergic interneurons or
striatomesencephalic GABA neurons. Since systemic
ad-ministration of AMPA– kainate antagonists do not
stimu-late dopamine release (Bubser et al 1995), this inhibitory
pathway is most likely mainly mediated by NMDA
transmission.
The inhibition of dopaminergic cell firing following
amphetamine is an important feedback mechanism by
which the brain reduces the effect of amphetamine on
dopamine release. The inhibition of dopaminergic cell
firing induced by amphetamine is mediated both by
stimulation of presynaptic D
2autoreceptors and by
stim-ulation of this inhibitory pathway (Bunney and
Aghaja-nian 1978). The model depicted in Figure 4 predicts that,
following administration of amphetamine (i.e., under
con-ditions in which the inhibitory pathway should be
activat-ed), NMDA receptor blockade would result in a failure of
activation of the inhibitory pathway, resulting in
exagger-ated amphetamine-induced dopamine release (Carlsson et
al 1999b). This prediction is consistent with observations
in rodents (Miller and Abercrombie 1996) and in humans
(this study).
More recently, a second mechanism has been proposed
to explain the paradoxical increase in dopaminergic cell
activity following systemic administration of
noncompet-itive NMDA antagonists. The basic observation leading to
this hypothesis is that systemic administration and local
VTA application of AMPA and kainate antagonists,
though having no effect on striatal dopaminergic output
per se, effectively block the increase in extracellular
dopamine induced by systemic injection of MK-801 and
ketamine (Mathe et al 1998; Moghaddam et al 1997;
Svensson et al 1998). This observation suggested that
MK-801–induced dopamine release is mediated by
acti-vation of AMPA and kainate receptors in the VTA. This
hypothesis is consistent with the recent demonstration that
systemic administration of noncompetitive NMDA
antag-onists increases extracellular concentration of glutamate in
the VTA (Svensson et al 1997), as well as in other regions
such as the PFC, the nucleus accumbens, the striatum, and
the
hippocampus
(Liu
and
Moghaddam
1995;
Moghaddam and Adams 1998). Thus, stimulation of
AMPA and kainate receptors by ketamine-induced
gluta-mate release in the VTA might antagonize the
GABA-mediated reduction in dopamine cell firing elicited by
amphetamine, leading to an exaggerated dopamine release
following amphetamine administration. This alternate,
possibly complementary mechanism is also represented in
Figure 4. The model depicted in Figure 4 emphasizes the
afferents from the PFC to the VTA, yet does not rule out
the role of other glutamatergic projections, such as
affer-ents from the amygdala or hippocampus, whose disruption
by ketamine might also contribute to the observed effect.
Implication for the NMDA Deficiency Model of
Schizophrenia
Although results from this study are consistent with an
NMDA receptor deficiency model of schizophrenia, the
direct evidence for such a deficiency is still lacking. Two
important limitations of this study should be kept in mind
regarding the relevance of these imaging data to the
NMDA deficiency hypothesis of schizophrenia.
First, the NMDA receptor hypofunction hypothesis of
schizophrenia assumes a chronic and possibly
neurodevel-opmental deficit (Olney and Farber 1995) rather than an
acute deficit such as induced in this study. Acute and
chronic NMDA receptor hypofunctions have different
consequences for brain function (for a review, see Jentsch
and Roth 1999); however, two recent studies in rodents
indicate that striatal amphetamine-induced dopamine
re-lease is also increased following chronic PCP exposure
(D.C. Javitt, personal communication, October 1999;
Jentsch et al 1998). These data suggest that both acute and
chronic NMDA hypofunction might be associated with
increased
responsivity
of
subcortical
dopaminergic
systems.
Second, the failure of glutamatergic control of
dopa-mine release might stem from mechanisms other than
NMDA hypofunction. For example, glutamatergic
projec-tions from the PFC to the VTA are under tonic inhibition
by prefrontal GABA and dopamine activity (see Karreman
and Moghaddam 1996 and references therein). It follows
that deficits in GABA-ergic or dopaminergic function in
the PFC are expected to have consequences similar to an
NMDA deficiency on the subcortical dopamine response
to amphetamine. Interestingly, both deficits have also been
implicated in schizophrenia (Benes 1991; Deutch et al
1990; Weinberger 1987). Thus, in patients with
schizo-phrenia various or multiple mechanisms (NMDA receptor
hypofunction, GABA-ergic or dopaminergic deficits in the
PFC) may lead to the dysregulation of subcortical
dopa-mine revealed by the amphetadopa-mine challenge.
Dopamine and Psychotomimetic Effects of
Low-Dose Ketamine
Besides investigating in humans neuromodulatory effects
previously demonstrated in rodents, brain imaging enables
direct assessment of the subjective effects of
pharmaco-logic manipulations in relation to neurotransmission
events. Whether the psychotomimetic effects of acute
administration of noncompetitive NMDA antagonists are
mediated entirely, partly, or not at all by an increase in
dopamine activity is a long-standing debate (Carlsson et al
1999a). Our study clearly demonstrates that the acute
dissociative state induced by this low dose of ketamine is
not caused by an increase in dopamine activity, since this
dissociative state was, if anything, reduced following
amphetamine administration. The decrease in
ketamine-induced dissociative state following amphetamine was an
interesting observation but should be viewed with caution,
given the absence of a comparison group with
ketamine-only administration. Nevertheless, functional imaging
studies suggested that the dissociative state induced by
subanesthetic ketamine might be due to perturbation of
intracortical connectivity at the level of the PFC (Breier et
al 1997a; Vollenweider et al 1997). Amphetamine-induced
dopamine release in the PFC might actually partially
compensate for this disorganized activity, since prefrontal
dopamine has been shown to augment the coherence of
cognitive processing in the PFC (Daniel et al 1991).
According to this view, the increase in PFC dopamine
following acute (but not chronic) exposure to
noncom-petitive NMDA antagonists might be viewed as a
compensatory reaction (i.e., part of the solution rather
than part of the problem). In this regard, it is interesting
to note that clozapine, a drug shown to antagonize some
of the psychotomimetic effects of ketamine (Malhotra et
al 1997), also augments dopamine release in the PFC
(Youngren et al 1999).
At this low dose, ketamine alone failed to elicit a
significant increase in the BPRS positive subscale, an
observation consistent with a previous report of lack of
significant induction of positive symptoms following this
low dose of ketamine in healthy subjects (Krystal et al
1994). In healthy volunteers, positive symptoms are
typi-cally observed following higher doses of ketamine
(Krys-tal et al 1994; Newcomer et al 1999). Neither
amphet-amine alone nor the combination of amphetamphet-amine and
ketamine elicited significant positive symptoms, despite
the marked elevation of dopamine release measured by
SPECT. This result is consistent with the well-established
observation that acute stimulation of dopamine release in
otherwise healthy subjects is not sufficient to provoke
positive psychotic symptoms (for a review, see Lieberman
et al 1987 and references therein). Rather, sustained
stimulation of dopamine release is needed for the
emer-gence of positive symptoms in healthy subjects and,
presumably, in patients with schizophrenia (Angrist and
Gershon 1970; Laruelle 2000b; Lieberman et al 1990).
Conclusion
amphetamine-induced dopamine release in humans. This
observation supports the hypothesis that, in schizophrenia,
a deficiency of glutamatergic control of dopamine
neuro-nal activity might underlie the increase in
amphetamine-induced dopamine release previously reported
(Abi-Dargham et al 1998; Breier et al 1997b; Laruelle et al
1996). The mechanism(s) responsible for this deficient
glutamatergic regulation of subcortical dopamine activity
in schizophrenia remain to be elucidated. If an alteration of
NMDA receptor function is implicated, pharmacologic
augmentation of NMDA transmission might restore the
ability of the PFC to control dopamine release and offer an
antipsychotic strategy potentially devoid of the morbidity
associated with chronic D
2receptor blockade.
Supported by the Scottish-Rite Foundation, the National Institute of Mental Health (Grant No. K02 MH01603-01), and the National Institute of Health (Grant No. M01RR00645, to the Irving Center for Clinical Research).
The authors thank Bart N.M. van Berckel, M.D., Ph.D., for helpful discussions and Analia Arevalo, Suehee Chung, Manuel DeLaNuez, Ted Pozniakoff, Mali Pratap, Yolanda Rubino, Dan Schneider, and Richard Weiss for excellent technical assistance.
References
Abi-Dargham A, Gil R, Krystal J, Baldwin R, Seibyl J, Bowers M, et al (1998): Increased striatal dopamine transmission in schizophrenia: Confirmation in a second cohort. Am J Psy-chiatry 155:761–767.
Allen RM, Young SJ (1978): Phencyclidine-induced psychosis. Am J Psychiatry 135:1081–1084.
Angrist B, van Kammen DP (1984): CNS stimulants as a tool in the study of schizophrenia. Trends Neurosci 7:388 –390. Angrist BM, Gershon S (1970): The phenomenology of
experi-mentally induced amphetamine psychosis—preliminary ob-servation. Biol Psychiatry 2:95–107.
Benes FM (1991): Toward a neurodevelopmental understanding of schizophrenia and other psychiatric disorders. In: Cichetti D, editors. Developmental Psychopathology. Hillsdale, NJ: Erlbaum, 161–184.
Bowers MB Jr, Heninger GR, Sternberg D, Meltzer HY (1980): Clinical processes and central dopaminergic activity in psy-chotic disorders. Commun Psychopharmacol 4:177–183. Breier A, Adler CM, Weisenfeld N, Su TP, Elman I, Picken L, et
al (1998): Effects of NMDA antagonism on striatal dopamine release in healthy subjects: Application of a novel PET approach. Synapse 29:142–147.
Breier A, Malhotra AK, Pinals DA, Weisenfeld NI, Pickar D (1997a): Association of ketamine-induced psychosis with focal activation of the prefrontal cortex in healthy volunteers. Am J Psychiatry 154:805– 811.
Breier A, Su TP, Saunders R, Carson RE, Kolachana BS, deBartolomeis A, et al (1997b): Schizophrenia is associated with elevated amphetamine-induced synaptic dopamine con-centrations: Evidence from a novel positron emission tomog-raphy method. Proc Natl Acad Sci U S A 94:2569 –2574.
Bubser M, Tzschentke T, Hauber W (1995): Behavioural and neurochemical interactions of the AMPA antagonist GYKI 52466 and the non-competitive NMDA antagonist dizocilpine in rats. J Neural Transm 101:115–126.
Bunney BS, Aghajanian GK (1978): d-Amphetamine-induced depression of central dopamine neurons: Evidence for medi-ation by both autoreceptors and a striato-nigral feedback pathway. Naunyn Schmiedebergs Arch Pharmacol 304:255– 261.
Carboni E, Imperato A, Perezzani L, Di Chiara G (1989): Amphetamine, cocaine, phencyclidine and nomifensine in-crease extracellular dopamine concentrations preferentially in the nucleus accumbens of freely moving rats. Neuroscience 28:653– 661.
Carlsson A, Hansson LO, Waters N, Carlsson ML (1999a): A glutamatergic deficiency model of schizophrenia. Br J Psy-chiatry Suppl 37:2– 6.
Carlsson A, Lindqvist M (1963): Effect of chlorpromazine or haloperidol on formation of 3-methoxytyramine and normeta-nephrine in mouse brain. Acta Pharmacol Toxicol 20:140 – 144.
Carlsson A, Waters N, Carlsson ML (1999b): Neurotransmitter interactions in schizophrenia—therapeutic implications. Biol Psychiatry 46:1388 –1395.
Carlsson M, Carlsson A (1990): Interactions between glutama-tergic and monoaminergic systems within the basal ganglia— implications for schizophrenia and Parkinson’s disease. Trends Neurosci 13:272–276.
Carlsson ML (1993): Are the disparate pharmacological profiles of competitive and uncompetitive NMDA antagonists due to different baseline activities of distinct glutamatergic path-ways? (Hypothesis). J Neural Transm 94:1–10.
Ceci A, French ED (1989): Phencyclidine-induced activation of ventral tegmental A10 dopamine neurons is differentially affected by lesions of the nucleus accumbens and medial prefrontal cortex. Life Sci 45:637– 646.
Chergui K, Charlety PJ, Akaoka H, Saunier CF, Brunet JL, Buda M, et al (1993):Tonic activation of NMDA receptors causes spontaneous burst discharge of rat midbrain dopamine neu-rons in vivo. Eur J Neurosci 5:137–144.
Christie MJ, Bridge S, James LB, Beart PM (1985): Excitotoxin lesions suggest an aspartatergic projection from rat medial prefrontal cortex to ventral tegmental area. Brain Res 333: 169 –172.
Christoffersen CL, Meltzer LT (1995): Evidence for N-methyl-D-aspartate and AMPA subtypes of the glutamate receptor on substantia nigra dopamine neurons: Possible preferential role for N-methyl-D-aspartate receptors. Neuroscience 67:373– 381.
Collier BB (1972): Ketamine and the conscious mind. Anaesthe-sia 27:120 –134.
Creese I, Burt DR, Snyder SH (1976): Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science 19:481– 483.
Crow TJ (1980): Molecular pathology of schizophrenia: More than one disease process? Br Med J 280:66 – 68.
Dayton PG, Stiller RL, Cook DR, Perel JM (1983): The binding of ketamine to plasma proteins: Emphasis on human plasma. Eur J Clin Pharmacol 24:825– 831.
Deutch AY, Clark WA, Roth RH (1990): Prefrontal cortical dopamine depletion enhances the responsiveness of mesolim-bic dopamine neurons to stress. Brain Res 521:311–315. Deutch AY, Tam SY, Freeman AS, Bowers MB Jr, Roth RH
(1987): Mesolimbic and mesocortical dopamine activation induced by phencyclidine: Contrasting pattern to striatal response. Eur J Pharmacol 134:257–264.
Doherty JD, Simonovic M, So R, Meltzer HY (1980): The effect of phencyclidine on dopamine synthesis and metabolic in rat striatum. Eur J Pharmacol 65:139 –149.
Enrico P, Bouma M, de Vries JB, Westerink BH (1998): The role of afferents to the ventral tegmental area in the handling stress-induced increase in the release of dopamine in the medial prefrontal cortex: A dual-probe microdialysis study in the rat brain. Brain Res 779:205–213.
Freeman AS, Bunney BS (1984): The effects of phencyclidine and N-allylnormetazocine on midbrain dopamine neuronal activity. Eur J Pharmacol 104:287–293.
French ED (1986): Effects of phencyclidine on ventral tegmental A10 dopamine neurons in the rat. Neuropharmacology 25: 241–248.
French ED, Ceci A (1990): Non-competitive N-methyl-D-aspar-tate antagonists are potent activators of ventral tegmental A10 dopamine neurons. Neurosci Lett 119:159 –162.
French ED, Mura A, Wang T (1993): MK-801, phencyclidine (PCP), and PCP-like drugs increase burst firing in rat A10 dopamine neurons: Comparison to competitive NMDA an-tagonists. Synapse 13:108 –116.
Gariano RF, Groves PM (1988): Burst firing induced in midbrain dopamine neurons by stimulation of the medial prefrontal and anterior cingulate cortices. Brain Res 462:194 –198. Goldman-Rakic PS, Selemon LD (1997): Functional and
ana-tomical aspects of prefrontal pathology in schizophrenia. Schizophr Bull 23:437– 458.
Grace AA (1991): Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: A hypothesis for the etiology of schizophrenia. Neuroscience 41:1–24. Hertel P, Mathe JM, Nomikos GG, Iurlo M, Mathe AA,
Svensson TH (1995): Effects of D-amphetamine and phen-cyclidine on behavior and extracellular concentrations of neurotensin and dopamine in the ventral striatum and the medial prefrontal cortex of the rat. Behav Brain Res 72:103– 114.
Hondo H, Yonezawa Y, Nakahara T, Nakamura K, Hirano M, Uchimura H, Tashiro N (1994): Effect of phencyclidine on dopamine release in the rat prefrontal cortex; an in vivo microdialysis study. Brain Res 633:337–342.
Javitt DC, Zukin SR (1991): Recent advances in the phencycli-dine model of schizophrenia. Am J Psychiatry 148:1301– 1308.
Jentsch JD, Elsworth JD, Redmond DE Jr, Roth RH (1997): Phencyclidine increases forebrain monoamine metabolism in rats and monkeys: Modulation by the isomers of HA966. J Neurosci 17:1769 –1775.
Jentsch JD, Roth RH (1999): The neuropsychopharmacology of phencyclidine: From NMDA receptor hypofunction to the
dopamine hypothesis of schizophrenia. Neuropsychopharma-cology 20:201–225.
Jentsch JD, Taylor JR, Roth RH (1998): Subchronic phencycli-dine administration increases mesolimbic dopaminergic sys-tem responsivity and augments stress- and psychostimulant-induced hyperlocomotion. Neuropsychopharmacology 19:105– 113.
Kalivas PW (1993): Neurotransmitter regulation of dopamine neurons in the ventral tegumental area. Brain Res Rev 18:75–113.
Kalivas PW, Duffy P, Barrow J (1989): Regulation of the mesocorticolimbic dopamine system by glutamic acid recep-tor subtypes. J Pharmacol Exp Ther 251:378 –387.
Kane J, Honigfeld G, Singer J, Meltzer HY, Group tCCS (1988): Clozapine for the treatment-resistant schizophrenic: A dou-ble-blind comparison with chlorpromazine. Arch Gen Psychi-atry 45:789 –796.
Karreman M, Moghaddam B (1996): The prefrontal cortex regulates the basal release of dopamine in the limbic striatum: An effect mediated by ventral tegmental area. J Neurochem 66:589 –598.
Kashihara K, Hamamura T, Okumura K, Otsuki S (1990): Effect of MK-801 on endogenous dopamine release in vivo. Brain Res 528:80 – 82.
Kegeles LS, Zea-Ponce Y, Abi-Dargham A, Rodenhiser J, Wang T, Weiss R, et al (1999): Stability of [123
I]IBZM SPECT measurement of amphetamine-induced striatal dopamine re-lease in humans. Synapse 31:302–308.
Kolachana BS, Saunders RC, Weinberger DR (1995): Augmen-tation of prefrontal cortical monoaminergic activity inhibits dopamine release in the caudate nucleus: An in vivo neuro-chemical assessment in the rhesus monkey. Neuroscience 69:859 – 868.
Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, et al (1994): Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry 51:199 –214.
Krystal JH, Seibyl JP, Price LH, Woods SW, Heninger GR, Aghajanian GK, Charney DS (1993): m-Chlorophenylpipera-zine (MCPP) effects in neuroleptic-free schizophrenic pa-tients: Evidence implicating serotonergic systems in the positive symptoms of schizophrenia. Arch Gen Psychiatry 50:624 – 635.
Kung M-P, Kung H (1990): Peracetic acid as a superior oxidant for preparation of [123
I]IBZM, a potential dopamine D-2 receptor imaging agent. J Lab Comp Radiopharm 27:691– 700.
Lahti AC, Koffel B, LaPorte D, Tamminga CA (1995): Subanes-thetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology 13:9 –19.
Laruelle M (2000a): Imaging synaptic neurotransmission with in vivo binding competition techniques: A critical review. J Cereb Blood Flow Metab 20:423– 451.
Laruelle M, Abi-Dargham A, Al-Tikriti MS, Baldwin RM, Zea-Ponce Y, Zoghbi SS, et al (1994a): SPECT quantification of [123
I]iomazenil binding to benzodiazepine receptors in nonhuman primates. II. Equilibrium analysis of constant infusion experiments and correlation with in vitro parameters. J Cereb Blood Flow Metab 14:453– 465.
Laruelle M, Abi-Dargham A, Gil R, Kegeles L, Innis R (1999): Increased dopamine transmission in schizophrenia: Relation-ship to illness phase. Biol Psychiatry 46:56 –72.
Laruelle M, Abi-Dargham A, van Dyck CH, Gil R, De Souza CD, Erdos J, et al (1996): Single photon emission computer-ized tomography imaging of amphetamine-induced dopamine release in drug free schizophrenic subjects. Proc Natl Acad Sci U S A 93:9235–9240.
Laruelle M, Abi-Dargham A, van Dyck CH, Rosenblatt W, Zea-Ponce Y, Zoghbi SS, et al (1995): SPECT imaging of striatal dopamine release after amphetamine challenge. J Nucl Med 36:1182–1190.
Laruelle M, van Dyck C, Abi-Dargham A, Zea-Ponce Y, Zoghbi SS, Charney DS, et al (1994b): Compartmental modeling of iodine-123-iodobenzofuran binding to dopamine D2receptors
in healthy subjects. J Nucl Med 35:743–754.
Lieberman JA, Kane JM, Alvir J (1987): Provocative tests with psychostimulant drugs in schizophrenia. Psychopharmacol-ogy 91:415– 433.
Lieberman JA, Kinon BL, Loebel AD (1990): Dopaminergic mechanisms in idiopathic and drug-induced psychoses. Schizophr Bull 16:97–110.
Liu J, Moghaddam B (1995): Regulation of glutamate efflux by excitatory amino acid receptors: Evidence for tonic inhibitory and phasic excitatory regulation. J Pharmacol Exp Ther 274:1209 –1215.
Malhotra AK, Adler CM, Kennison SD, Elman I, Pickar D, Breier A (1997): Clozapine blunts N-methyl-D-aspartate antagonist-induced psychosis: A study with ketamine. Biol Psychiatry 42:664 – 668.
Mathe JM, Nomikos GG, Hildebrand BE, Hertel P, Svensson TH (1996): Prazosin inhibits MK-801-induced hyperlocomotion and dopamine release in the nucleus accumbens. Eur J Phar-macol 309:1–11.
Mathe JM, Nomikos GG, Schilstrom B, Svensson TH (1998): Non-NMDA excitatory amino acid receptors in the ventral tegmental area mediate systemic dizocilpine (MK-801) in-duced hyperlocomotion and dopamine release in the nucleus accumbens. J Neurosci Res 51:583–592.
Miller DW, Abercrombie ED (1996): Effects of MK-801 on spontaneous and amphetamine-stimulated dopamine release in striatum measured with in vivo microdialysis in awake rats. Brain Res Bull 40:57– 62.
Moghaddam B, Adams B, Verma A, Daly D (1997): Activation of glutamatergic neurotransmission by ketamine: A novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci 17:2921–2927.
Moghaddam B, Adams BW (1998): Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science 281:1349 –1352.
Murase S, Grenhoff J, Chouvet G, Gonon FG, Svensson TH (1993a): Prefrontal cortex regulates burst firing and transmit-ter release in rat mesolimbic dopamine neurons studied in vivo. Neurosci Lett 157:53–56.
Murase S, Mathe JM, Grenhoff J, Svensson TH (1993b): Effects of dizocilpine (MK-801) on rat midbrain dopamine cell activity: Differential actions on firing pattern related to anatomical localization. J Neural Transm 91:13–25. Newcomer JW, Farber NB, Jevtovic-Todorovic V, Selke G,
Melson AK, Hershey T, et al (1999): Ketamine-induced NMDA receptor hypofunction as a model of memory impair-ment and psychosis. Neuropsychopharmacology 20:106 –118. Nishimura M, Sato K, Okada T, Yoshiya I, Schloss P, Shimada S, Tohyama M (1998): Ketamine inhibits monoamine trans-porters expressed in human embryonic kidney 293 cells. Anesthesiology 88:768 –774.
Olney JW, Farber NB (1995): Glutamate receptor dysfunction and schizophrenia. Arch Gen Psychiatry 52:998 –1007. Overall JE, Gorham DR (1962): The brief psychiatric rating
scale. Psychol Rep 10:799 – 812.
Overton P, Clark D (1992): Iontophoretically administered drugs acting at the N-methyl-D-aspartate receptor modulate burst firing in A9 dopamine neurons in the rat. Synapse 10:131– 140.
Pawlowski L, Mathe JM, Svensson TH (1990): Phencyclidine activates rat A10 dopamine neurons but reduces burst activity and causes regularization of firing. Acta Physiol Scand 139:529 –530.
Reimer ML, Mamer OA, Zavitsanos AP, Siddiqui AW, Dadgar D (1993): Determination of amphetamine, methamphetamine and desmethyldeprenyl in human plasma by gas chromatog-raphy/negative ion chemical ionization mass spectrometry. Biol Mass Spectrom 22:235–242.
Rouillard C, Chiodo LA, Freeman AS (1990): The effects of the phencyclidine analogs BTCP and TCP on nigrostriatal dopa-mine neuronal activity. Eur J Pharmacol 182:227–235. Scarnati E, Pacitti C (1982): Neuronal responses to
iontophoreti-cally applied dopamine, glutamate, and GABA of identified dopaminergic cells in the rat substantia nigra after kainic acid-induced destruction of the striatum. Exp Brain Res 46:377–382.
Schilstrom B, Nomikos GG, Nisell M, Hertel P, Svensson TH (1998): N-Methyl-D-aspartate receptor antagonism in the ventral tegmental area diminishes the systemic nicotine-induced dopamine release in the nucleus accumbens. Neuro-science 82:781–789.
Seeman P, Lee T (1975): Antipsychotic drugs: Direct correlation between clinical potency and presynaptic action on dopamine neurons. Science 188:1217–1219.
Sesack SR, Pickel VM (1992): Prefrontal cortical efferents in the rat synapse on unlabeled neuronal targets of catecholamine terminals in the nucleus accumbens septi and on dopamine neurons in the ventral tegmental area. J Comp Neurol 320:145–160.
Smith DJ, Bouchal RL, deSanctis CA, Monroe PJ, Amedro JB, Perrotti JM, Crisp T (1987): Properties of the interaction between ketamine and opiate binding sites in vivo and in vitro. Neuropharmacology 26:1253–1260.
Smith GS, Schloesser R, Brodie JD, Dewey SL, Logan J, Vitkun SA, et al (1998): Glutamate modulation of dopamine mea-sured in vivo with positron emission tomography (PET) and 11C-raclopride in normal human subjects. Neuropsychophar-macology 18:18 –25.
![Table 2. Amphetamine Effect on [123I]IBZM Binding Potential](https://thumb-ap.123doks.com/thumbv2/123dok/3143627.1383571/7.612.60.549.92.186/table-amphetamine-effect-on-i-ibzm-binding-potential.webp)
![Figure 4. Simplified circuitry diagram showing control of dopa-mine (DA) cell activity in the midbrain (ventral tegmental area[VTA] and substantia nigra [SN]) by glutamatergic (GLU)projections from the prefrontal cortex (PFC)](https://thumb-ap.123doks.com/thumbv2/123dok/3143627.1383571/9.612.315.546.82.255/simplified-circuitry-midbrain-tegmental-substantia-glutamatergic-projections-prefrontal.webp)