www.elsevier.com / locate / bres
Research report
Human neurons generate C-reactive protein and amyloid P:
upregulation in Alzheimer’s disease
*
Koji Yasojima, Claudia Schwab, Edith G. McGeer, Patrick L. McGeer
Kinsmen Laboratory of Neurological Research, Department of Psychiatry, University of British Columbia, Vancouver, BC, Canada V6T 1Z3
Accepted 12 September 2000
Abstract
C-reactive protein (CRP) and amyloid P (AP) are pentraxins which are associated with many pathological lesions, including the amyloid deposits and neurofibrillary tangles (NFTs) of Alzheimer disease (AD). It has always been assumed that they are generated by liver and delivered to their sites of action by serum. Here we report by in situ hydridization, reverse transcriptase–polymerase chain reaction analysis, Western blotting and immunohistochemistry that the mRNAs and proteins of both CRP and AP are concentrated in pyramidal neurons and are upregulated in affected areas of AD brain. Controlling pentraxin production at the tissue level may be important in reducing inflammatory damage in AD. 2000 Elsevier Science B.V. All rights reserved.
Theme: Disorders of the nervous system
Topic: Degenerative disease: Alzheimer’s — other
Keywords: Pentraxin; Pyramidal neuron; Hippocampus; Entorhinal cortex; Liver; Cerebellum; Western blot; In situ hybridization; mRNA; Immuno-histochemistry
1. Introduction these reactants, with serum levels rising as much as 1000-fold following injury or infection. It is therefore intimately C-reactive protein (CRP) was named after it was involved in all forms of damage to host tissue [35]. identified as a factor which binds to the C-polysaccharide One of the important functions of these pentraxins is to of pneumococcus. Amyloid P (AP) was named for its activate the classical complement pathway in an antibody-amyloid binding capacity. AP is an invariant component of independent fashion [12,40]. Complement is also a phylo-all amyloid deposits, including those of Alzheimer’s genetically ancient host defense mechanism which, like the disease (AD) brain. CRP and AP are referred to as pentraxins, can be traced back to the horseshoe crab [21]. pentraxins because of their capacity to aggregate, in a When activated, complement opsonizes targets for phago-non-covalent fashion, into flat pentameric discs. Pentraxins cytosis and directly kills pathogens by insertion of the are highly conserved, being traced back as far as the membrane attack complex (MAC) into their cell mem-horseshoe crab. They are hypothesized to be duplicated branes. But the MAC can also insert itself into host cells, from a common ancestral gene as early as 200 million causing autodestruction in a process called bystander lysis. years ago [35]. Their genes are located on band q2.1 of The association of CRP [6,15] and AP [1,3,7] with chromosome 1 in a region rich in host protective genes. activated complement fragments attached to host tissue in They are presumed to have great survival value and to be such degenerative conditions as AD, myocardial infarction intimately associated with innate immune defense (for [20] and atherosclerosis [38] is suggestive of their partici-reviews see [8,9,35]). The proteins are also major acute pation, with complement, in a negative, autodestructive phase reactants. In humans, CRP is the most sensitive of role. It may therefore be important to learn methods of
controlling pentraxin production at the cellular level. It has always been assumed that CRP and AP are *Corresponding author. Tel.: 11-604-822-7377; fax: 1
1-604-822-primarily, if not exclusively, produced by liver hepatocytes 7086.
E-mail address: [email protected] (P.L. McGeer). [8,9,35]. The presence of AP in AD brain has been taken
as evidence of impaired blood–brain barrier function in guanidium thiocyanate–phenol–chloroform method, and AD [3,7,17], but direct measurement using tomographic single-strand cDNA synthesis performed on 5mg of total techniques has failed to demonstrate evidence of any RNA extract as previously described [42]. The cDNA (1 deficit [2,31]. ml) was amplified in a 50ml reaction buffer containing 20 Since innate immunity must function at a local level mM Tris–HCl (pH 8.4), 50 mM KCl, 200mM dNTPs and [23], and complement proteins in brain have previously 2 mM MgCl . In addition to 0.52 mM of each specific been shown to be generated mainly by neurons [33,37,42], oligonucleotide primer, 1% Tween 20, and 2% DMSO it is logical to hypothesize that brain pentraxins might be were added as enhancers. The first amplification step derived from a similar source. Identification of neurons as consisted of denaturation at 948C for 5 min, followed by a a source of pentraxins would have implications for control hot start addition of 2.5 U Taq DNA polymerase at 808C of inflammation not just in brain, but in chronic in- (1–3 min). This was followed by an annealing step at 558C flammatory conditions in the periphery where production for 1 min and an extension step at 728C for 3 min, and then by cells other than hepatocytes may be central to the by 35 cycles of a denaturation step at 948C for 1 min, an inflammatory process. annealing step at 558C for 1 min, and an extension step at
728C for 1 min.
The primers for AP (GenBank Acc[X04608) were:
2. Methods forward CCTTTGCTCACACAGACCTCAGTGG, and re-verse AGGACTCCCAGCTCACACACAGATGTG. These 2.1. Cases studied primers span the intron and are designed to yield a product of 309 bp. Endonuclease cut sites are present for Aci I Fifteen autopsied cases were chosen for CRP and AP (228, 81 bp fragments) and Spe I (191, 118 bp fragments). mRNA analysis by RT–PCR. There were eight non-AD The primers for CRP (GenBank Acc[M11725) were: cases (age 73.664.9; four males, four females) and seven forward TCGTATGCCACCAAGAGACAAGACA, and AD cases (age 69.662.5; two males and five females). reverse AACACTTCGCCTTGCACTTCATACT. These Post-mortem delay (PMD) in the AD cases varied from 6 primers were designed to yield a product of 440 bp with to 16 h. PMD in the control cases varied from 8 to 96 h. endonuclease cut sites for Hae III (377, 63 bp fragments) For purposes of analysis, the control cases were grouped and Mse I (356, 84 bp fragments).
into short PMD (four cases, average 14.2 h) and long PMD The primers for cyclophilin (GenBank Acc[Y00052) (four cases, average 48 h). Due to the post-mortem were, as previously reported [42]: forward stability of the mRNAs being analyzed, no significant ATGGTCAACCCCACCGTGTTCTTCG, and reverse differences were found between the two control groups CGTGTGAAGTCACCACCCTGACACA. These primers (see Results). Standard neuropathological analysis showed were designed to yield a product of 206 bp with a Hae III that all the non-AD cases were free of typical AD lesions. endonuclease cut site (153, 53 bp fragments).
All of the AD cases had extensive pathology, easily A linear relationship was found between the log of PCR meeting CERAD criteria for definite AD [25], and with product intensity and cycle number for cyclophilin be-typical regional distribution of the lesions. The hippocam- tween 20 and 29 cycles, for CRP between 26 and 37 pus and midtemporal gyrus were heavily affected with cycles, and for AP between 24 and 32 cycles, after which classical senile plaques and NFTs. The cerebellum was plateaus were reached. A linear relationship was also found free of such lesions. Intermediate pathology was found in between the amount of cDNA product obtained and the the frontal cortex, while only moderate lesions were found original cDNA added within the range corresponding to in the motor cortex. Blocks of tissue of 0.5–1 g weight 0.01 to 0.5 mg total RNA. Accordingly, standard con-were dissected from the fresh brains, and homogenized. ditions were followed in which cDNA (1 ml) corre-Aliquots were taken for extraction of RNA and protein. sponding to 0.1 mg total RNA was added and the Adjacent areas were taken for standard neuropathology, cyclophilin product amplified for 27 cycles and, in parallel immunohistochemistry, and in situ hybridization. In four of experiments, the CRP product amplified for 35 cycles and the normal and four of the AD cases, other organs were the AP amplified for 30 cycles. Each PCR product was available, permitting comparable analyses to be done on electrophoresed through a 6% polyacrylamide gel and the
liver specimens. product visualized by incubation for 10 min in a solution
In all experiments, the presence of possible contami- Hybridization was performed for 16 h at 508C in a nants was checked by control reactions in which amplifica- humidified chamber. Test sections were hybridized with tion was carried out for up to 35 cycles on samples in the antisense cRNA probes; and controls were probed with which we omitted from the RT–PCR reaction mixture the sense cRNA probes. After hybridization, the sections either (i) the reverse transcriptase or (ii) a template cDNA. were washed twice with 50% deionized formamide in No product was obtained under these conditions. 23standard saline citrate (SSC) at 558C for 30 min, followed by incubation with 20mg / ml RNase A (Sigma) 2.3. cRNA probe preparation and in situ hybridization for 30 min at 378C. After being washed in 23SSC for 20 min and then in 0.23SSC for 20 min twice at 558C, the AP and CRP cRNA probes were prepared starting with sections were incubated in a blocking buffer containing the RT–PCR products. The 309 bp AP cDNA fragment 1.5% blocking reagent (DIG Nucleic Acid Detection Kit, was subcloned into the pGEM-T Easy plasmid vector Roche, Canada), 100 mM Tris (pH 7.5), and 150 mM (Promega). The product was linearized with Nco I and Sal NaCl overnight at 48C. Alkaline phosphatase-conjugated I (NEB) to produce sense and antisense DNA templates, anti-DIG antibody (Roche, Canada) diluted 1:1000 in respectively. The AP cRNA probes were synthesized at blocking buffer was incubated with sections overnight at 378C for 3 h in a mixture composed of 1 mg linearized 48C. Before detection of alkaline phosphatase / DIG-labeled template DNA, 2 ml of ATP, CTP, GTP and digoxigenin RNAs, sections were prewashed in 100 mM Tris (pH 9.5), (DIG)-labeled UTP, 2 ml transcription buffer; and 20 U 100 mM NaCl and 50 mM MgCl , and then incubated for2
RNase inhibitor with 20 U SP6 RNA polymerase for the 5 to 11 h in the dark at 48C in color substrates (nitroblue sense strand or 20 U T7 RNA polymerase for the antisense tetrazolium salt and 5-bromo-4-chloro-3-indolyl-phosphate strand (DIG RNA labeling Kit SP6 / T7, Roche, Canada). toluidine salt in dimethylformamide) diluted in the prewash The 440 bp CRP cDNA fragment was similarly treated to buffer as described by the manufacturer. Once the desired produce sense and antisense CRP cRNA probes. color intensity was attained, the color reaction was stopped After labeling, the AP and CRP cRNA probes were by washing the sections in 10 mM Tris / 1 mM EDTA (pH treated with 10 U RNase-free DNase I for 45 min at 378C, 8.0). The slides were photographed.
ethanol-precipitated, and resuspended in diethyl
pyrocarbo-nate (DEPC)-treated distilled water containing 20 U RNase 2.4. Western blots inhibitor. The transcripts were analyzed on agarose gels
after ethidium bromide staining, and the yields were Western blots were performed on extracts of the soluble estimated densitometrically by comparison with a control fraction of homogenates of control and AD hippocampus RNA of known concentration. Immunohistochemical de- and liver, as well as on normal serum, following previous-tection of DIG-labeled RNAs on nylon membranes ly reported methodology [42]. Briefly, tissue samples were (Hybond-N1, Amersham, UK) revealed equivalent label- homogenized in 5 times v / w extraction buffer (0.02 M ing efficiency between sense and antisense cRNAs. Tris–HCl, pH 7.5, and 0.1% Triton X) containing 1 mM In situ hybridization was carried out on paraformal- EDTA and the protease inhibitors PMSF (10mg / ml) and dehyde-fixed, paraffin-embedded blocks of tissue. Fifteen aprotinin (10 mg / ml). Homogenates were centrifuged at micrometer sections were deparaffinized with xylene and 18,000 g at 48C for 30 min. The protein content of the rehydrated in a graded series of ethanol solutions (100%, supernatants was determined, the samples diluted in SDS 95%, 90%, 85%, 80%,and 70% in DEPC-treated distilled sample buffer (60 mM Tris, pH 6.8, 2.5% SDS, 5% water) for 5 min at each step. Tissue sections were then 2-mercaptoethanol) to a final protein concentration of 10 mounted on Silane-coated slides and treated with 2mg / ml mg /ml and were boiled for 3 min. Samples containing 100 proteinase K (Sigma) at 378C for 30 min. They were mg of protein were loaded onto 10% acrylamide minigels. further fixed for 30 min in 4% paraformaldehyde at 48C, For serum, samples containing 1–10 mg of protein were followed by treatment with 0.25% acetic anhydride in 0.1 diluted in 5 ml of Tris buffer. An equal volume of SDS M triethanolamine (pH 8.0). The sections were washed sample buffer was added and the mixture boiled for 3 min with phosphate-buffered saline (PBS) before dehydration before loading on 10% acrylamide minigels. Life Tech-in a graded series of ethanol as described above for 30 s at nologies high range prestained standards were used as
each step. molecular weight markers. After 45 min of electrophoresis
The sections were prehybridized for 2 h at 508C in a (200 V), the proteins were transferred onto PVDF mem-hybridization mixture [50% deionized formamide, 10 mM branes (Immobilon P, Millipore Corp., MA) at 7 V for 45 Tris (pH 7.4), 200 mg / ml yeast tRNA, 13 Denhardt’s min using a semi-dry blotter. Membranes were blocked in solution, 10% dextran sulfate, 600 mM NaCl, 0.25% SDS 5% skim milk overnight at 48C. The immunoblots were and 1 mM EDTA]. The probes (30 ng / ml) were added treated for 6 h at 48C with a primary antibody, followed by into the hybridization mixture, which was heated at 808C treatment for 1 h with a secondary antibody labeled with for 10 min and cooled before addition to the tissue horseradish peroxidase. Immunoreactivity was visualized
chemilumines-cent substrate (Pierce Chemical Co., Rockford, IL). After containing 0.6% nickel ammonium sulfate and 0.001% draining, the membranes were covered in clear plastic H O in 0.05 Tris–HCl buffer, pH 7.6. When a dark blue2 2
wrapping and exposed to X-ray film (Hyperfilm ECL, color developed, sections were washed, mounted on glass Amersham Life Science, UK) for 0.3 to 2 min, depending slides and coverslipped with Entellan.
on the strength of the signal. The primary antibodies against human CRP were rabbit anti-CRP (DAKO, 1:500),
sheep anti-CRP (WAKO, 1:1000), mouse monoclonal anti- 3. Results
CRP 6804 (Medix Biochimica, 1:100), and mouse
mono-clonals anti-CRP-1 and anti-CRP-4 (kind gifts of Dr 3.1. Relative mRNA levels Gregory Lee, each at 1:100). Each antibody gave a single
band on tissue extracts comigrating with authentic CRP The results of reverse transcriptase–polymerase chain standard. The primary antibody against human AP was reaction (RT–PCR) analysis on the liver and five brain rabbit anti-AP (Calbiochem, 1:3000). The secondary anti- regions from AD and neurologically normal cases are bodies were from Sigma (1:8000). shown in Fig. 1 and Table 1 (see Materials and methods for details). Fig. 1 shows polaroid photographs of typical 2.5. Immunohistochemistry ethidium bromide-stained gels. Fig. 1a is for CRP mRNA in a normal case, and Fig. 1b for an AD case. Signals were Control cases without neurological symptoms or neuro- obtained from all brain areas and from liver in both normal pathological findings and confirmed AD cases were
select-ed for study from our brain bank at the University of British Columbia. Brain tissues for routine immunocytoch-emistry had been fixed in 4% paraformaldehyde and, after 3 to 4 days, transferred to a 15% buffered sucrose maintenance solution. Some blocks were further embedded in paraffin after fixation. Other tissue blocks were fixed in 70% ethanol in saline for 1 day before transfer to the sucrose maintenance solution. The monoclonal antibodies to CRP required such fixation to give positive immuno-staining. Sections from paraformaldehyde and ethanol fixed tissues were cut at 30mm on a freezing microtome. The paraffin blocks were cut at 10–15 mm on a standard microtome. The paraffin sections were deparaffinized in a series of xylene and ethanol concentrations. All sections were incubated free floating for immunocytochemistry as previously reported [1,32,37].
Sections for processing with the rabbit polyclonal anti-human CRP antibody (DAKO) were treated for 1–5 min with concentrated formic acid. Such pretreatment greatly enhanced the immunohistochemical staining [6,15]. These and other sections not exposed to formic acid were treated for 30 min with 0.5% H O solution in 0.01 M phosphate-2 2
buffered saline, pH 7.4, containing 0.3% Triton X-100 (PBS–T). They were transferred into 5% skim milk in
PBS–T for 30 min and incubated for 72 h at 48C or Fig. 1. Representative polaroid photomicrographs of ethidium bromide overnight at room temperature with one of the anti-human stained electrophoretic gels of RT–PCR products of CRP (a, b) and AP (c, primary antibodies. These were anti-CRP (DAKO, d) RT–PCR products. Gels from an AD case (b, d) are compared with a normal case (a, c). Lane 1, size markers; lane 2, hippocampus; lane 3, 1:5000); anti-CRP 5G4 (kind gift of Dr W.K. Lagrand,
midtemporal gyrus; lane 4, midfrontal gyrus; lane 5, motor cortex; lane 6, Amsterdam, MAB, 1:1000); anti-CRP 6804 (Medix
Bio-cerebellum; lane 7, liver. A single band of 440 bp was obtained for the chimica, MAB, 1:100); anti-CRP 6405 (Medix Bio- CRP product and 309 bp for the AP product (see Materials and methods chimica, MAB, 1:500); and rabbit anti-AP (Calbiochem, for details). Notice in the normal case for CRP that only weak bands were 1:3000). Sections were next treated with appropriate obtained from brain areas, with a much stronger band from liver. By contrast, in the AD case, stronger bands were obtained from brain areas, biotinylated secondary antibodies for 2 h at room
tempera-especially the heavily affected hippocampus and midtemporal gyrus ture, followed by incubation in avidin–biotinylated
Table 1
Relative levels (mean6S.E.M.) of the mRNAs for amyloid P, C-reactive protein (CRP) and the housekeeping gene cyclophilin (CP) in five brain
a
regions and liver in AD and controls
Tissue Amyloid P CRP CP
Midtemporal-AD 41.0462.37*** 51.9162.21*** 84.3560.70 Control 17.1661.19 2.1360.28 85.5060.68 Hippocampus-AD 43.2861.96*** 58.6662.43*** 85.7561.07 Control 21.0461.15 3.1360.66 85.0660.60 Midfrontal-AD 39.4462.37*** 30.3462.18*** 85.9660.92 Control 17.9662.24 3.7260.72 84.9960.63 Motor-AD 40.8463.04*** 16.1163.42** 84.9160.80 Control 18.4461.27 3.1560.40 85.5560.91 Cerebellum-AD 32.4863.10* 9.3762.92 85.2761.02 Control 19.2061.40 3.8260.42 86.4360.76 Liver-AD 146.163.2 5.8861.31 97.0660.54 Control 148.067.6 7.4360.73 96.9160.58
a
***P,0.001, **P,0.01, *P,0.05 for difference between AD and control cases; all corrected by Holm’s stepdown procedure [13] for multiple analyses. The calculations for CRP and AP mRNA levels used data normalized to the cyclophilin values.
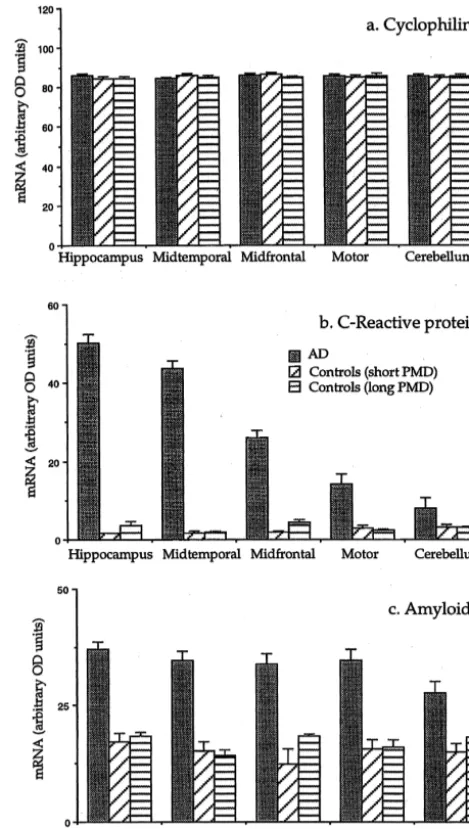
and AD cases. In normal brain, however, these signals were very faint, but in AD cases, as illustrated in Fig. 1b, intense signals were observed in the hippocampus and midtemporal gyrus. Intermediate signals were seen in the midfrontal and motor cortices, with only a weak signal in the cerebellum. As illustrated in the figure, the AD brain areas showed a stronger signal than the liver.
Figs. 1c and 1d show comparable data for AP. Fig. 1c shows a polaroid photograph of a typical ethidium bromide gel for a normal case and Fig. 1d for an AD case. Signals for AP mRNA were clearly visible from all areas in the normal case and in AD they were not increased as dramatically as those for CRP mRNA. Moreover, the signals from liver were much stronger than from any of the brain areas.
Fig. 2 illustrates the lack of effect of PMD and agonist
Fig. 2. Bar graphs showing the mean6S.E.M. of the mRNAs in arbitrary causes of death on the mRNA levels of the constituents
OD units for cyclophilin (A), CRP (B) and AP (C) for the seven AD and under study. The housekeeping gene cyclophilin gave
eight control cases. The eight controls were divided into four cases with a almost identical values in every region of brain in AD post-mortem delay of 20 h or less (average 14.2 h) and four with a cases, controls with short PMD and controls with long post-mortem delay of 24 h or more (average 48 h).
PMD. For CRP and AP, the levels varied widely according to disease and area but again values for short-term PMD
were almost identical with those for long-term PMD. cerebellum. These differences were highly significant Accordingly, the eight control cases were combined for except for the cerebellum (P50.065) which had a rela-overall statistical analysis. tively large S.E.M. In the liver, there was no significant Table 1 summarizes the semiquantitative mRNA values difference between AD and control values. Surprisingly, in the AD and the combined control cases for CRP, AP and the liver mRNA levels were lower than in all areas the housekeeping gene cyclophilin. Data were analyzed measured of AD brain.
liver levels were approximately 3.6-fold higher than in AD Western blots of serum up to a concentration of 10mg brain. The stronger signal for AP compared with CRP in of total protein failed to show a detectable band for either liver is consistent with their differing base values in CRP or AP (data not shown). Since this level of serum normal serum. These are typically of the order of 20–30 protein is well beyond the residual level that would remain mg / ml for AP [28] but usually less than 2 for CRP [8], in the volume of brain tested [4], it can be concluded that indicating a higher level of liver AP production compared the Western blots of Fig. 3 reflect proteins derived from
with CRP. brain substance itself. The relative intensity of the bands in
Fig. 3 compared with authentic standards indicate a level 3.2. Western blotting of AP in normal hippocampus of approximately 0.6 mg / mg protein and in AD a level of about 1mg / mg protein. Examples of Western blotting results are shown in Fig. CRP was below detection levels in normal hippocampus, 3a for CRP and Fig. 3b for AP. Protein extracts of the but in AD it was approximately 1.4 mg / mg of protein. hippocampus in typical AD and control cases were com- These Western blots are consistent with the mRNA data of pared, as well as liver extracts from the same cases. Table 1, indicating that, for the areas examined, the Extracts were run in parallel with authentic protein stan- mRNAs were being translated into their protein products in dards. Identical results were obtained with three commer- approximate proportion to their relative mRNA levels, cial anti-CRP antibodies; a rabbit polyclonal (DAKO), a with normal levels for CRP in brain being below the limits sheep polyclonal (WAKO) and a murine monoclonal of Western blot detection.
(6804, Medix Biochmica) and two other murine
mono-clonals (CRP-1 and CRP-4, gifts of Dr G. Lee). There was 3.3. In situ hybridization no detectable band for CRP from normal hippocampus but
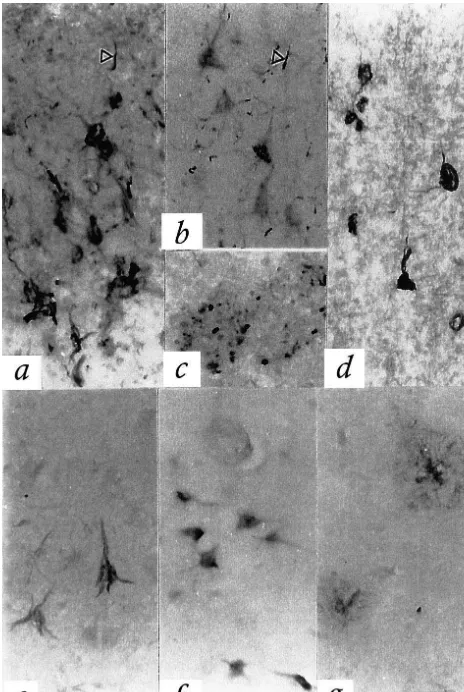
a single strong band was observed in AD, with a weaker Strong in situ hybridization signals for both CRP and band being seen in liver. The tissue extracts migrated AP mRNAs were detected over neurons in the AD equally with authentic CRP standard (ca. 25 kDa). Fig. 3b hippocampus / entorhinal cortex and adjacent temporal shows a comparable analysis for AP with a polyclonal neocortex. The distribution of positive signals was the rabbit antibody (Calbiochem). A single AP band was same for both pentraxins. Signals were mostly present in picked up from normal hippocampus, with a stronger band pyramidal cells although granule cells of the dentate gyrus from AD hippocampus, and a considerably more intense were strongly positive for both pentraxins. Typical results band from liver. All bands migrated equally with the are shown in Fig. 4. Fig. 4a is a photomicrograph of the authentic AP standard (ca. 25 kDa). CA4 region of the hippocampus in an AD case stained with the anti-sense CRP probe. A strong signal is observed over many pyramidal neurons. Fig. 4b is an adjacent section stained with the sense probe. No signal is observ-able. Fig. 4c is a photomicrograph of the CA4 hippocam-pal region in an AD case stained with the anti-sense AP probe. Again, a strong signal is observable over many pyramidal neurons. Fig. 4d is an adjacent section stained with the AP sense probe, with no signal being observable. Figs. 4e and g illustrate hybridization of granule cells in the dentate gyrus with the CRP and AP antisense probes respectively. Figs. 4f and h illustrate hybridization in the hippocampal CA3 and CA1 pyramidal cells with CRP and AP antisense probes, respectively, and Figs. 4i and j hybridization in the entorhinal cortex and adjacent tempo-ral neocortex using CRP and AP antisense probes. No signal was detected in any of these regions using the CRP and AP sense probes. Very weak signals in layer 1 of the entorhinal and neocortex may indicate some mRNA pro-duction by glial cells.
Fig. 3. Western blots of the hippocampus of a normal (lane 1), AD (lane 3.4. Immunohistochemistry 2) and AD liver (lane 3) compare with authentic standards (lane 4, 0.5
mg) for CRP (a) and AP (b). Single bands were detected co-migrating Largely comparable immunohistochemical staining was with the standards in each case (ca. 25 kDa). Notice the much stronger
observed with antibodies to CRP and AP, but there were bands for both CRP and AP in the AD case compared with the control.
some differences. All antibodies tested showed intense Detection was with the anti-CRP-4 MAB but similar data were obtained
Fig. 4. In situ hybridization for CRP anti-sense (a, e, f, i), CRP sense (b), AP antisense (c, g, h, j), and AP sense (d) (see Materials and methods for details). Strong signals are seen over pyramidal neurons in hippocampus CA4 for both the CRP (a) and AP (c) anti-sense probes, but no signals are seen for the CRP (b) and AP (d) sense probes. Dentate granule cells also give strong signals for CRP (e) and AP (g). Hippocampal CA1 pyramidal neurons can be seen to be positive for the CRP (f) and AP (h) antisense probes. Pyramidal neurons of the entorhinal cortex are also positive with the CRP antisense probe (i), and those of the adjacent temporal cortex with the AP antisense probe (j). Sense probes in all of these areas gave no signal.
immuno-employed coupled with higher sensitivity primers. The signal from liver is considerably stronger than from brain, and insensitive techniques could easily miss tissues other than the liver.
The data show that, at least for the mRNAs being studied here, there was no evidence of post-mortem deterioration for periods up to 96 h after death. Such stability in tissues that have not been frozen has previously been reported by us [41,42], as well as others [16,26]. However, the methods reported here would only detect cleavage within the regions amplified by RT–PCR and would be insensitive to cleavage at other sites.
There have been many previous reports on the immuno-histochemical detection of AP in fixed brain tissue. But CRP has been much more difficult to identify even though it is easily detected by Western blot. Accordingly, we tested two polyclonal and three monoclonal antibodies for their ability to detect CRP at the cellular level. The three monoclonal antibodies recognized CRP only on ethanol-fixed tissue, and one polyclonal only on ethanol-fixed tissue following formic acid pretreatment. The reasons are un-known.
CRP and AP have relatively high turnover rates. The half-life of CRP in serum is about 19 h [39], with comparable values being obtained for serum AP [10].
123
Scintigraphy with I-labeled AP showed uptake and retention of AP in peripheral amyloid deposits [10]. However, there was no measurable uptake and labeling of AD brain amyloid deposits [22]. Nevertheless, AP has been detected in normal CSF, with upregulation in AD [11,18]. This is consistent with serum AP failing to cross Fig. 5. Immunohistochemistry for CRP (a–d) and AP (e–g). Sections
were formic acid treated and stained with polyclonal anti-CRP antibody the blood–brain barrier and with production of AP in brain (a–c), ethanol-fixed and treated with 5G4 MAB (d), or paraffin embedded accounting for much of the CSF content. There was also and treated with the polyclonal anti-AP antibody (e–g) (see Materials and 123
no detectable uptake of I-labeled CRP into brain [39], methods for details). Photomicrographs show strong CRP immunostaining
although CRP has been detected in normal CSF, with of eNFTs (a), of neurons (b), plaques (c) and iNFTs (d). The senile plaque
dramatic upregulation in bacterial meningitis [34]. Again, in c shows granular staining for CRP, probably in dystrophic nerve
endings. Dystrophic neurites are also visible in a and b (arrowheads). The these results are consistent with brain production of CRP AP antibody stained tangles (e), normal appearing neurons (f) and senile accounting for much of its presence in normal CSF. plaques (g). Note, however, the different appearance of senile plaque
CRP has previously been reported to be produced by staining of AP compared with CRP. The AP immunostaining (g) is
alveolar macrophages in addition to hepatocytes [5], but stronger and appears to be localized to the extracellular amyloid material.
AP has been reported only to be produced by hepatocytes Scale bar in g applies to a–g550mm.
[17]. In results to be reported elsewhere, we have found mRNAs for both CRP and AP in lung, heart, arteries, kidney and spleen, so many types of cells in addition to histochemical results are consistent with the proteins being hepatocytes, macrophages and neurons may produce these generated primarily within neurons and then becoming pentraxins.
deposited on extracellular lesions. Numerous functions have been associated with CRP, all of them related to inflammatory processes and host defense [36]. But CRP may not always play a protective role. CRP
[3] F. Coria, E. Castano, F. Prelli, M. Larrondo-Lillo, S. van Duinen, [30]. Thus CRP may, through interaction with
comple-M.L. Shelanski, B. Frangione, Isolation and characterization of ment, promote autodestruction of tissue.
amyloid P component from Alzheimer’s disease and other types of AP is reported to be a normal constituent of basement cerebral amyloidosis, Lab. Invest. 58 (1988) 454–458.
membranes and may have a physiological role apart from [4] B.L. Dean, C. Lee, J.E. Kirsch, V.M. Runge, R.M. Dempsey, L.C. being a response protein in pathological situations [43]. Pettigrew, Cerebral hemodynamics and cerebral blood volume: MR assessment using gadolinium contrast agents and T1-weighted That would explain its higher base level of production
Turbo-FLASH imaging, Am. J. Neurol. 13 (1992) 39–48. compared to CRP. Its strong binding to
glycosamino-[5] Q. Dong, J.R. Wright, Expression of C-reactive protein by alveolar glycans would explain its appearance in many forms of macrophages, J. Immunol. 156 (1996) 4815–4820.
amyloid, but it would not explain its association with [6] T. Duong, M. Nikolaeva, P.J. Acton, C-reactive protein-like im-pathological structures such as the eNFTs of AD which are munoreactivity in the neurofibrillary tangles of Alzheimer’s disease,
Brain Res. 749 (1997) 152–156. devoid of amyloidogenic constituents. AP is an activator of
[7] T. Duong, E.C. Pommier, A.B. Scheibel, Immunodetection of the the classical complement pathway in vitro [12] and may
amyloid P component in Alzheimer’s disease, Acta Neuropathol. 78 act in this capacity when bound to pathological structures. (1989) 429–437.
The staining for AP generally parallels that for the [8] C. Gabay, I. Kushner, Acute-phase proteins and other systemic complement proteins C4d and C3d in neurologically responses to inflammation, New Engl. J. Med. 340 (1999) 448–454. [9] H. Gewurz, X.-H. Zhang, T.F. Lint, Structure and function of the diseased brain tissue [1] and the deposition of AP on
pentraxins, Curr. Opin. Immunol. 7 (1995) 54–64. eNFTs appears to precede the appearance of C4d [32].
[10] P.N. Hawkins, S. Richardson, D.M. Vigushin, J. David, C.R. Kelsey, These findings would be consistent with AP being an R.E. Gray, M.A. Hall, P. Woo, J.P. Lavender, M.B. Pepys, Serum activator of complement in association with eNFTs. CRP amyloid P component scintigraphy and turnover studies for diag-may also be an activator. The temporal association of CRP nosis and quantitative monitoring of AA amyloidosis in juvenile
rheumatoid arthritis, Arthritis Rheumat. 36 (1993) 842–851. with C4d on eNFTs has not yet been investigated.
[11] P.N. Hawkins, M.N. Rossor, J.R. Gallimore, B. Miller, E.G. Moore, Taken together, these data indicate a central role of the
M.B. Pepys, Concentration of serum amyloid P component in the pentraxins CRP and AP in the innate immune response of CSF as a possible marker of cerebral amyloid deposits in Alzheim-brain. This innate immune response, while designed to er’s disease, Biochem. Biophys. Res. Commun. 201 (1994) 722– protect the host, can be destructive to host tissue when 726.
[12] P.S. Hicks, L. Saunero-Nazia, T.W. Duclos, C. Mold, Serum amyloid misdirected. One in vivo evidence of such autodestructive
P component binds to histones and activates the classical comple-processes occurring in AD would be a protective effect of
ment pathway, J. Immunol. 149 (1992) 306–310. anti-inflammatory agents. There is now a wealth of
evi-[13] S. Holm, A simple sequentially repetitive multiple test procedure, dence that patients taking anti-inflammatory agents, or Scand. J. Statist. 6 (1979) 65–70.
suffering from conditions in which such agents are routine- [14] E. Iseki, N. Amano, T. Matsuishi, S. Yokoi, N. Arai, S. Yagishita, A case of familial, atypical Alzheimer’s disease: immunohistochemical ly used, have a significantly reduced prevalence of AD
study of amyloid P-component, Neuropath. Appl. Neurobiol. 14 [24]. CRP and AP, as activators of complement, may be
(1988) 169–174. important initiators of this autodestructive inflammatory
[15] N. Iwamoto, E. Nishiyama, J. Ohwada, H. Arai, Demonstration of response. As such, they may be prime targets for therapeu- CRP immunoreactivity in brains of Alzheimer’s disease: immuno-tic intervention, not just in AD, but in other inflammatory histochemical study using formic acid pretreatment of tissue
sec-tions, Neurosci. Lett. 177 (1994) 23–26. conditions.
[16] S.A. Johnson, D.G. Morgan, C.E. Finch, Extensive postmortem stability of RNA from rat and human brain, J. Neurosci. Res. 16 (1986) 267–280.
Acknowledgements [17] R.N. Kalaria, T.E. Golde, M.L. Cohen, S.G. Younkin, Serum
amyloid P in Alzheimer’s disease: implications for dysfunction of the blood–brain barrier, Ann. N.Y. Acad. Sci. 640 (1991) 145–148. This research was supported by grants from the Jack
[18] Y. Kanoh, H. Ohtani, Levels of interleukin-6, CRP anda 2-macro-Brown and Family A.D. Research Fund and the Alzheimer
globulin in cerebrospinal fluid (CSF) and serum as indicator of Societies of British Columbia and Canada, as well as
blood–CSF barrier damage, Biochem. Mol. Biol. Intl. 43 (1997) donations from The Friends of UBC and individual British 269–278.
Columbians. We thank Dr W.K. Lagrand and Dr Gregory [19] W.K. Lagrand, C.A. Visser, W.T. Hermans, H.W. Niessen, F.W. Verheugt, G.J. Wolbink, C.E. Hack, C-reactive protein as a car-Lee for the gift of anti-CRP antibodies.
diovascular risk factor, Circulation 100 (1999) 96–102.
[20] W.K. Lagrand, H.W. Niessen, G.J. Wolbink, L.H. Jaspars, C.A. Visser, F.W. Verheugt, C.J. Meijer, C.E. Hack, C-reactive protein
References colocalizes with complement in human hearts during acute
myocar-dial infarction, Circulation 95 (1997) 97–103.
[1] H. Akiyama, T. Yamada, T. Kawamata, P.L. McGeer, Association of [21] J.D. Lambris, The chemistry, biology, and phylogeny of C3, in: J.M. amyloid P component with complement proteins in neurologically Cruise, R.E. LewisJr. (Eds.), Complement Today, Karger, Basel, diseased tissue, Brain Res. 548 (1991) 349–352. New York, London, 1993, pp. 16–45.
[2] M.T. Caserta, D. Caccioppo, G.D. Lapin, A. Ragin, D.R. Groothuis, [22] L.B. Lovat, A.A. O’Brien, S.F. Armstrong, S. Madhoo, C.J. Bulpitt,
123
Blood–brain barrier integrity in Alzheimer’s disease patients and M.N. Rossor, M.B. Pepys, P.N. Hawkins, Scintigraphy with I-elderly control subjects, J. Neuropsych. Clin. Neurosci. 10 (1998) serum amyloid P component in Alzheimer disease, Alz. Dis. Assoc.
[23] P.L. McGeer, E.G. McGeer, Autotoxicity and Alzheimer disease, [33] Y. Shen, R. Li, E.G. McGeer, P.L. McGeer, Neuronal expression of Arch. Neurol. 57 (2000) 789–790. mRNAs for complement proteins of the classical pathway in [24] P.L. McGeer, M. Schulzer, E.G. McGeer, Arthritis and anti-in- Alzheimer brain, Brain Res. 769 (1997) 391–395.
flammatory agents as possible protective factors for Alzheimer’s [34] M. Stearman, H.J. Southgate, The use of cytokine and C-reactive disease: a review of 17 epidemiological studies, Neurology 47 protein measurements in cerebrospinal fluid during acute infective (1996) 425–432. meningitis, Ann. Clin. Biochem. 31 (1994) 255–261.
[25] S.S. Mirra, A. Heyman, D. McKeel, S.M. Sumi, B.J. Crain, L.M. [35] D.M. Steel, A.S. Whitehead, The major acute phase reactants: Brownlee, F.S. Vogel, J.P. Hughes, G. van Belle, L. Berg, The C-reactive protein, serum amyloid P component and serum amyloid Consortium to Establish a Registry for Alzheimer’s Disease A protein, Immunol. Today 15 (1994) 81–88.
(CERAD). II: Standardization of pathothologic assessment of Al- [36] A.J. Szalai, A. Agrawal, T.J. Greenhough, J.E. Volanakis, C-Reac-zheimer’s disease, Neurology 41 (1991) 479–486. tive protein: structural biology, gene expression and host defense [26] M.R. Morrison, W.S.T. Griffin, The isolation and in vitro translation function, Immunol. Res. 16 (1997) 127–136.
of undegraded messenger RNAs from human postmortem brain, [37] K. Terai, D.G. Walker, E.G. McGeer, P.L. McGeer, Neurons express Analyt. Biochem. 113 (1981) 318–324. proteins of the classical complement pathway in Alzheimer disease, [27] K.W. Muir, C.J. Weir, W. Alwan, I.B. Squire, K.R. Lees, C-Reactive Brain Res. 769 (1997) 385–390.
protein and outcome after ischemic stroke, Stroke 30 (1999) 981–
[38] J. Torzewski, M. Torsewski, D.E. Bowyer, M. Frolich, W. Koenig, J. 985.
Waltenberger, C. Fitzsimmons, V. Hombach, C-Reactive protein [28] M.B. Pepys, Amyloid P component and the diagnosis of
frequently colocalizes with the terminal complement complex in the amyloidosis, J. Int. Med. 232 (1992) 519–521.
intima of early atherosclerotic lesions of human coronary arteries, [29] K.O. Pietila, A.P. Harmoinen, J. Jokiniitty, A.I. Pasternack, Serum
Arterioscler. Thromb. Vasc. Biol. 18 (1998) 1386–1392. C-reactive protein concentration in acute myocardial infarction and
[39] D.M. Vigushin, M.B. Pepys, P.N. Hawkins, Metabolic and sci-its relationship to mortality during 24 months of follow-up in
ntigraphic studies of radioiodinated human C-reactive protein in patients under thrombolytic treatment, Eur. Heart J. 17 (1996)
health and disease, J. Clin. Invest. 90 (1993) 1351–1357. 1345–1349.
[40] G.J. Wolbink, M.C. Brouwer, S. Buysmann, I.J.M. ten Berge, C.E. [30] P.M. Ridker, C.H. Hennekens, J.E. Buring, N. Rifai, C-reactive
Hack, CRP-mediated activation of complement in vivo, J. Immunol. protein and other markers of inflammation in the prediction of
157 (1996) 473–479. cardiovascular disease in women, New Engl. J. Med. 342 (2000)
[41] K. Yasojima, C. Schwab, E.G. McGeer, P.L. McGeer, Human heart 836–843.
generates complement proteins that are upregulated and activated [31] N.L. Schlageter, R.E. Carson, S.I. Rapoport, Examination of blood–
after myocardial infarction, Circulation Res. 83 (1998) 860–869. brain barrier permeability in dementia of the Alzheimer type with
[42] K. Yasojima, C. Schwab, E.G. McGeer, P.L. McGeer, Upregulated [68Ga]EDTA and positron emission tomography, J. Cerebr. Blood
production and activation of the complement system in Alzheimer Flow Metab. 7 (1987) 1–8.
disease brain, Am. J. Pathol. 154 (1999) 927–936. [32] C. Schwab, J.C. Steele, E.G. McGeer, P.L. McGeer, Amyloid P