www.elsevier.com / locate / bres
Interactive report
1
Alzheimer’s disease: a dysfunction of the amyloid precursor protein
*
Rachael L. Neve , Donna L. McPhie, Yuzhi Chen
Department of Psychiatry, Harvard Medical School, MRC 223 McLean Hospital, 115 Mill St., Belmont, MA 02478, USA
Accepted 25 August 2000
Abstract
In this review, we argue that at least one insult that causes Alzheimer’s disease (AD) is disruption of the normal function of the amyloid precursor protein (APP). Familial Alzheimer’s disease (FAD) mutations in APP cause a disease phenotype that is identical (with the exception that they cause an earlier onset of the disease) to that of ‘sporadic’ AD. This suggests that there are molecular pathways common to FAD and sporadic AD. In addition, all individuals with Down syndrome, who carry an extra copy of chromosome 21 and overexpress APP several-fold in the brain, develop the pathology of AD if they live past the age of 40. These data support the primacy of APP in the disease. Although APP is the source of theb-amyloid (Ab) that is deposited in amyloid plaques in AD brain, the primacy of APP in AD may not lie in the production of Abfrom this molecule. We suggest instead that APP normally functions in the brain as a cell surface signaling molecule, and that a disruption of this normal function of APP is at least one cause of the neurodegeneration and consequent dementia in AD. We hypothesize in addition that disruption of the normal signaling function of APP causes cell cycle abnormalities in the neuron, and that these abnormalities constitute one mechanism of neuronal death in AD. Data supporting these hypotheses have come from investigations of the molecular consequences of neuronal expression of FAD mutants of APP or overexpression of wild type APP, as well as from identification of binding proteins for the carboxyl-terminus (C-terminus) of APP.
2000 Elsevier Science B.V. All rights reserved.
Theme: Disorders of the nervous system
Topic: Degenerative disease: Alzheimer’s – beta amyloid
Keywords: Alzheimer’s disease; Amyloid precursor protein; Neuronal death
1. Introduction
possession of an extra copy of chromosome 21 (Down
syndrome); or it can be caused by mutations in the amyloid
1.1. APP, A
b
, and Alzheimer disease
precursor protein (APP) gene on chromosome 21 or by
mutations in the presenilin genes on chromsome 1 and 14.
All individuals with Alzheimer disease (AD) experience
Additional genetic complexicity is conferred on it by the
a progressive loss of cognitive function, resulting from a
fact that the
e
4 allele of the APOE gene is a major risk
neurodegenerative process characterized classically by the
factor for the development of AD. Thus, it is not likely that
deposition of
b
-amyloid (A
b
) in plaques and in the
AD is caused by a single molecular event.
cerebrovasculature, and the formation of neurofibrillary
Numerous mechanisms for the neuronal cell death in
tangles in neurons. Additional pathological hallmarks of
AD have been proposed. One of these is the amyloid
AD include granulovacular degeneration, loss of synapses
hypothesis, which suggests that deposition of A
b
is a
and decreases in cell density in distinct regions of the
primary event in the pathological cascade for AD. This
brain. Alzheimer disease does not have a simple etiology.
argument is based on in vitro studies showing that A
b
is
It can occur as a ‘sporadic’ event; it can result from the
toxic to neurons and on the measurement of increased
release of A
b
by cells carrying familial AD (FAD) mutant
genes. There are two major carboxyl-terminal variants of
1Published on the World Wide Web on 11 September 2000.
A
b
. A
b
1 – 40is the major species secreted from cultured
*Corresponding author. Tel.: 11-617-855-2413; fax:11-617-855-cells and found in cerebrospinal fluid, while A
b
is the
3793. 1 – 42
E-mail address: [email protected] (R.L. Neve).
major component of amyloid deposits (reviewed in Ref.
[118]). Cells expressing FAD mutants of APP and the
transmission [28]. There has been some question of
presenilins are reported to secrete increased amounts of
whether C100 exerts its neurotoxic effects from the inside
A
b
1 – 42, suggesting a link of this variant of A
b
to AD
or the outside of the cell [23,117]. Our data of the past 6
pathogenesis. Consequently, a leading hypothesis for the
years suggest strongly that C100 kills from inside the cell;
etiology of AD is that increased A
b
1 – 42is a shared
this is supported by the observation that C100 is not
molecular correlate of FAD mutations, and that it repre-
secreted, even when it carries a signal peptide [19,14,66].
sents a gain of deleterious function that can cause FAD
Although at least one group has reported neurotoxicity due
[38] and may be an essential early event in AD [118].
to the addition of C100 to the culture medium [50], we
While this ‘amyloid hypothesis’ is attractive, molecular
believe that that type of neurotoxicity is mechanistically
mechanisms other than those mediated by extracellular A
b
different from the neurodegeneration that we observe upon
could also lead to AD neurodegeneration.
expression of C100 within primary neurons.
These mechanisms are likely to be linked in some way
The findings that APP interacts with the signaling
to the
b
-amyloid protein precursor (APP), the source of
molecule G , that FAD mutants of APP can cause G -
o oA
b
. One of the most compelling pieces of evidence that
mediated apoptosis in neuronal cells, and that these same
links AD neurodegeneration to APP and / or its A
b
-con-
FAD mutants of APP cause the intracellular accumulation
taining derivatives is the early finding that the APP gene is
of C100, suggested to us the following working
hypoth-on chromosome 21: virtually all individuals trisomic for
esis: In the brain a portion of APP is present as an integral
this chromosome show AD-like neuropathology by the age
plasma membrane protein that mediates the transduction of
of 40. Additionally, it has been discovered that specific
extracellular signals into the cell via its C-terminal tail, and
mutations in APP cause some forms of familial FAD.
abnormal accumulation of its A
b
-containing C-terminal
These data have raised the possibility that AD may result
tail in the neuron causes progressive dysfunction of APP
from an alteration in the normal function of APP [74,76],
signaling in AD, resulting in apoptosis. This hypothesis has
and have refocused attention on the delineation of the
been supported by the finding that the intracellular
C-function that APP subserves in the brain. It has been
terminal tail of APP interacts with the cell cycle protein
shown [47,83] that in the brain a percentage of APP is
APP-BP1 [10,9], and with members of the Fe65 family of
present on the cell surface, and it is proposed [76,83] that
adaptor proteins (reviewed in Ref. [89]). Additional
sup-this cell surface APP mediates the transduction of extracel-
port for this hypothesis emerged with the recent report [60]
lular signals into the cell via its C-terminal tail.
that the C31 peptide of APP, which is derived from C100
Nishimoto and his colleagues [75] showed that APP
and within which are contained the binding sites for the
binds to the brain-specific signal transducing G protein G ;
oabove proteins, is elevated in AD brain and is a potent
independent confirmation of this finding has subsequently
inducer of apoptosis.
been published [4,5]. It was then discovered [113] that
V642 (‘London’) FAD mutants of APP induce neuronal
DNA fragmentation, a feature of apoptosis, in a neuronal
2. Processing of APP
cell line. This fragmentation is independent of A
b
1 – 42production [114] and is mediated by the G
b g
2 2complex of
Most of what we know about APP processing has come
G [29]. These data support the notion that APP has an
ofrom work with cultured cells. APP matures through the
intrinsic signaling function in the neuron, which becomes
constitutive secretory pathway. Some fraction of the APP
ligand-independent when APP is mutated at V642.
is endoproteolytically cleaved at the cell surface within the
To examine the mechanism by which FAD APP might
A
b
sequence by the
a
-secretase, which generates the
cause apoptosis in neurons, we [66] expressed five differ-
neuroprotective
secreted
amyloid
precursor
protein
ent Alzheimer mutations of APP in primary neurons via
(APP ) and nonamyloidogenic 3 kDa A
sab
secreted
prod-recombinant herpes simplex virus (HSV) vectors, and
ucts [81,37,65]. APP
Sais readily detected in human plasma
quantified the levels of APP metabolites. The predominant
and cerebrospinal fluid.
can be detected intracellularly. C100-like amyloidogenic
and colleagues have shown that
b
-secretase cleavage
fragments have been detected only intracellularly.
products of APP are present in fetal and neonatal Down
It is important to note that APP processing is cell
syndrome brain at twice normal levels ([93] and personal
type-specific. LeBlanc and colleagues have reported that
communication with Dr. Russo). We have hypothesized
human neurons secrete more 4 kDa than 3 kDa A
b
, and
that abnormal accumulation of the A
b
-containing
C-termi-metabolize approximately 40% of newly-synthesized APP
nal tail (C100) of APP in neurons also occurs in Alzheimer
through the
a
-secretase pathway [53,55]. Moreover,
disease.
human neurons produce five C-terminal fragments of APP
in a pattern seen uniquely in human brain [21,53]. The two
largest C-terminal derivatives have the entire A
b
sequence
3. APP as a signaling molecule
at or near their amino terminus [21], and most likely
represent endogenous ‘C100’ fragments. Thus, C100 is a
The possibility that APP may act as a signaling receptor
physiologically relevant fragment of APP in the human
was first proposed on the basis of its predicted amino acid
brain. In contrast to human neurons, most APP-transfected
sequence, which suggested that APP was a type 1 intrinsic
human or nonhuman cell lines produce more 3 kDa than 4
membrane protein consistent with the structure of a ‘cell
kDa A
b
and show a relatively nonamyloidogenic pattern
surface receptor.’ [49]. However, subsequent studies of the
of C-terminal fragments [31,36,37,54].
function of APP concentrated largely on the secreted
Analyses of
b
-amyloid (A
b
) in genetically engineered
ectodomain, because of a lack of direct evidence that
cell lines expressing FAD mutations in both APP and the
mature APP exists on the cell surface with intact
intracel-presenilins (PS) have shown that all of the mutations cause
lular, transmembrane, and intracellular domains. Surface
either increased overall secretion of A
b
or secretion of the
APP was inferred to exist on a variety of cultured cells
‘long’ (42–43-amino acid) form of A
b
(A
b
1 – 42) relative to
[36,95,114], but some laboratories could not detect it
the shorter 40-amino acid form (reviewed in Ref. [38]).
[90,1]. Nevertheless some reports demonstrating
in-Increases in A
b
1 – 42have also been detected in transgenic
volvement of APP in neuronal development,
synap-mice expressing FAD mutations of both APP and PS
togenesis, and synaptic plasticity [62,68,69,86,70,85] did
(reviewed in Ref. [38]). A
b
1 – 42is the major component of
not restrict the observed function to secreted APP, raising
brain amyloid deposits in AD. Consequently, a leading
the possibility that some aspects of synaptic plasticity are
hypothesis for the etiology of AD is that increased A
b
1 – 42mediated by cell-associated APP. Indeed, it has now been
is a shared molecular correlate of FAD mutations which
demonstrated directly that a percentage of APP is found on
may also be operative in ‘sporadic’ AD. Increases in
the cell surface in neurons [47,99,83]. Cell-surface APP
A
b
1 – 42have not been shown directly in human AD brain
possesses a neurite-promoting activity that is distinct from
homogenates, although it is clear that amyloid plaques
that of the secreted APP [85], co-localizes with adhesion
contain a disproportionate amount of A
b
1 – 42. Furthermore,
plaque components [99,114], and participates in synaptic
analyses of levels of this peptide in the plasma and
vesicle recycling [63], suggesting that a percentage of APP
cerebrospinal fluid (CSF) of AD patients have revealed no
may function as a cell surface receptor, transducing signals
differences between AD patients and controls in the
from the extracellular matrix to the interior of the cell.
plasma [43], and a reduction of A
b
1 – 42in the CSF of AD
The growth cone G protein G
o[75], the presumptive
patients relative to controls [67,43]. However, increased
adaptor proteins Fe65 and X11 (reviewed in Ref. [89]),
release of A
b
( 1 – 42 )from fibroblasts of AD patients with
and APP-BP1 [10], a protein involved in cell cycle
presenilin mutations, as well as increased levels of
regulation [9], have been reported to interact with the
A
b
( 1 – 42 )in their plasma, have been demonstrated [94].
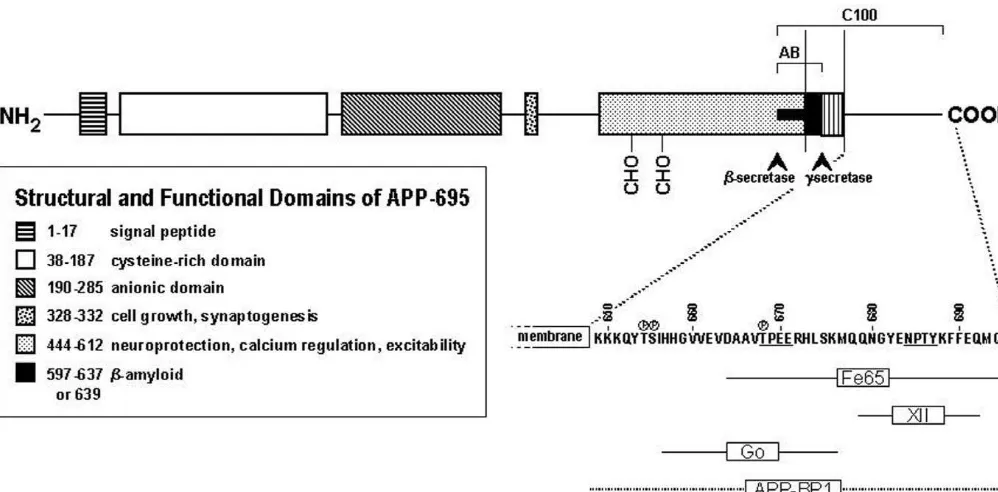
C-terminus of APP (Fig. 1), presumably to initiate
intracel-The
b
-secretase cleavage product of APP, C100, is
lular signaling. While the functions of Fe65 and X11 are
increased in cell lines expressing the Swedish FAD
not known, Fe65 has the characteristics of adaptor
pro-mutation of APP [12–14,8], but not in cell lines expressing
teins, which are thought to link signal transduction events
the London V642 mutation of APP [102]. Because neurons
emanating from plasma membrane receptors to
intracellu-process APP differently from cell lines, we expressed all
lar molecules, by forming complexes of these proteins.
known FAD mutants of APP in primary neurons in culture,
Therefore, one could envision APP being part of a G
oFig. 1. Schematic depicting the structural and functional domains of the amyloid precursor protein.
defined in greatest detail is G . Nishimoto and colleagues
orespective roles of G , APP-BP1, Fe65 and X11, and
o657 676
have demonstrated that the His
–Lys
domain of APP-
UV-DDB in the normal function of APP, to test the
695 activates the heterotrimeric GTP-binding protein G in
ohypothesis that progressive dysfunction of these roles
a GTP S-inhibitable manner [75,52]. Their demonstration
goccurs in AD.
that an antibody to the extracellular domain of APP
(22C11) that acts as a ligand mimetic [79] causes
activa-tion of G , argues that APP may be a G protein-coupled
o4. Apoptosis in Alzheimer disease
receptor. As noted above, the ‘London’ mutation of APP,
V642I, causes DNA fragmentation when expressed in a
The notion that a form of cell suicide called apoptosis
neuronal cell line [113]. Notably, expression of V642I
participates in the neuropathology of AD was raised by Su
657 676
APP deleted for residues His
–Lys
in these cells did
et al. [100], when they reported evidence for DNA
not cause DNA fragmentation. Pertussis toxin (PTX), an
fragmentation in neurons in AD brain. Although other
inhibitor of G and G , blocked the DNA fragmentation
o igroups have also detected this feature of apoptosis in AD
caused by V642I, as did co-transfection of V642I APP and
brain, many in the field have been skeptical of the idea that
a cDNA encoding a dominant negative mutant of G
a
o, but
the neurons that die in AD undergo apoptosis, partly
not with a cDNA encoding a dominant negative mutant of
because DNA fragmentation can also be caused by
oxida-G
a
i. Inhibition of A
b
1 – 42production from the V642I APP
tive damage [106] or by postmortem autolysis [98].
by mutating the
g
-secretase cleavage site did not have any
However, a report from the laboratory of Mark Mattson
effect on the DNA fragmentation caused by V642I.
[34] revived interest in the possibility that apoptosis is
These data suggest that G
omediates the DNA frag-
operative in AD. These investigators found that levels of a
mentation caused by the V642 mutants of APP; and
marker of apoptosis, Par-4 (prostate apoptosis response-4)
indeed, a subsequent paper from Nishimoto’s group re-
protein, are increased 15–20-fold over control in
vulner-vealed that the DNA fragmentation was mediated by the
able neurons in AD brain. They also showed that Par-4
bg
complex of G
o[29]. A
b
does not appear to play a
expression is increased in cultured neurons undergoing
causative role in inducing DNA fragmentation in this
apoptosis, and that inhibition of Par-4 expression in these
experimental paradigm. The data support the notion that
neurons blocks apoptosis.
differentially affected. PS-1 is reported to be expressed
primarily in CNS neurons in the brain, suggesting that this
protein may perform a neuron-specific function [20]. In
fact, in AD, neurons that express PS-1 antigen are less
vulnerable to the disease than are neurons that do not
express it [30], and inhibition of PS-1 expression results in
apoptosis [88], suggesting a protective role for this protein.
Although the precise role of PSs in regulation of apoptosis
in the neuron is still unclear, the evidence that they do play
a role in this pathway is strong. These data implicate both
APP and PSs in the control of apoptotic death in the brain,
and it is not unreasonable to suppose that FAD mutations
in these genes may cause dysfunction in this pathway.
It has been noted [82] that since the apoptotic process
proceeds to completion within 16–24 h, the extent of
apoptosis reported in AD brain would predict a complete
loss of neurons within a very brief period of time. Clearly,
this does not happen in AD. Cotman has suggested [15]
that the induction of compensatory responses to apoptosis
in the AD brain protects the neurons from terminal
apoptosis, and that a dynamic competition between cell
death processes and compensatory responses exists in AD
brain.
5. Cell cycle abnormalities in Alzheimer’s disease
The implication of apoptosis in AD etiology is
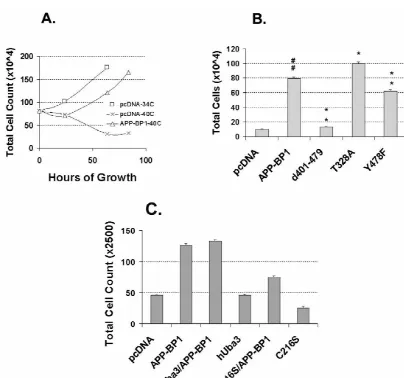
con-Fig. 2. The yeast two-hybrid reporter assay reveals that APP-BP1sistent with the numerous findings of cell cycle
abnor-interacts with hUba3. Top: schematic diagram showing the deletion andmalities in AD. Apoptosis and the cell cycle are closely
point mutants used for the assay. Below: a table indicating the strength oftied together, and the reexpression of cell cycle markers
each interaction, based on length of time for the X-gal substrate to turnhas been linked with the occurrence of certain types of
blue.neuronal cell death [40,39,22]. One interpretation of these
findings [56] is that a neuron is committed to the
perma-APP, V642I, causes DNA fragmentation when expressed in
nent cessation of cell division, so if for any reason it is
a neuronal cell line [113]. Luo et al. [61] showed that the
forced to reenter the cell cycle after this commitment, it
same mutation, as well as two additional FAD APP
dies. Notably, ectopic expression of cell cycle proteins and
mutations, induced apoptosis in differentiated PC12 cells.
their associated kinases in AD brain have been reported
Barnes et al. [2] reported that levels of APP are increased
[84,59,108,109]. More recently, Busser et al. [7] found
in motoneurons dying of apoptosis, and that APP is
abnormal appearance of cell cycle markers in regions of
cleaved by caspase-3, a caspase activated in apoptotic
AD brain where cell death is extensive, and Chow et al.
motoneurons. Interestingly, we [6] and others [77] have
[11] found increases in expression of genes encoding cell
shown that overexpression of wild-type APP causes apo-
cycle proteins in single neurons in late-stage relative to
ptotic death of neurons, although to a lesser degree than
early-stage AD brain. The phosphoepitope S214 of the
does expression of FAD mutants of APP.
microtubule associated protein tau, that appears in the
Approximately half of inherited AD cases are caused by
neurofibrillary tangles in AD, is a prominent
phosphoryla-mutations in the presenilin genes PS1 and PS2. It has been
tion site in metaphase but not in interphase of dividing
reported that overexpression of these genes in transfected
cells expressing tau [44], supporting the view that
reactiva-cell lines can cause apoptosis [45] or result in an increased
tion of the cell cycle machinery may be involved in tau
susceptibility to apoptosis [112,32–34,16]. On the other
hyperphosphorylation in AD brain. The possibility that
hand, we have found [6] that expression of PS1 in primary
phosphorylation-dependent events occurring during the cell
neurons does not cause or enhance apoptosis; rather, it
cycle affect the normal function of APP is suggested by
protects neurons against experimentally-induced apoptosis.
the finding that regulation of the phosphorylation and
Thus, the ability of PS-1 to induce apoptosis appears to be
metabolism of this protein occurs in a cell-cycle dependent
cell type specific; and this may have important implica-
manner [103,78].
by APP may be one cause of the reactivation of cell cycle
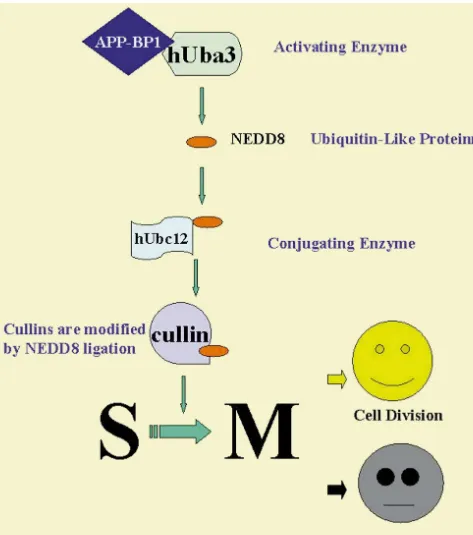
appear to have much in common with those of ubiquitin,
proteins in AD brain. In particular, we have isolated a
but the Ubls have novel regulatory functions not
necessari-binding protein for APP, termed APP-BP1 [10], and have
ly linked to proteolysis. APP-BP1 is a member of one of
shown that it is a cell cycle protein that normally regulates
these pathways. APP-BP1 is homologous to the ubiquitin
negatively the progression of cells into the S phase and
activating enzyme E1, but lacks the catalytic site. It has
regulates positively their progression into mitosis [9]. Over
been found, by our lab [9], and others, that APP-BP1 acts
the past few years, it has emerged that eukaryotes express
in concert with a second protein that possesses an E1-like
a set of ubiquitin-like proteins (Ubls) that are significantly
catalytic site, so that the two-molecule complex behaves
diverged from ubiquitin itself yet are also ligated to other
like E1, except that this complex activates the
ubiquitin-proteins [35,46]. The reactions involving these variants
like protein NEDD8 rather than ubiquitin itself:
Fig. 3. APP-BP1 rescues the ts41 cell phenotype at the nonpermissive temperature. (A) Time course of viability of ts41 cells transfected with the vector alone or with the APP-BP1 construct at the nonpermissive temperature (408C). Data for the vector-transfected cells at the permissive temperature (348C) are shown for comparison. Counts were performed at 32, 64, and 80 h except for the vector-transfected cells at the permissive temperature, which were counted at 32 and 64 h only. Note that cells transfected with a vector expressing human wild type APP-BP1 maintained a rate of growth at 408C that was similar (albeit slightly shifted to the right) to that of cells transfected with the vector alone and maintained at 348C. Vector-transfected cells that were shifted to the nonpermissive temperature showed a decreased mitotic index and eventually died. (B) Quantification of viability of wild type and mutant APP-BP1-transfected ts41 cells grown at 408C for 81 h. Note that the d401–479 deletion mutant of APP-BP1, which does not interact with hUba3, is unable to rescue the ts41 phenotype (Scheffe post hoc t tests;[[or ** indicates P,0.01, and * indicates P,0.05;[comparison with pcDNA3; *
temperature, after which they were neutralized in 0.1 M
E1-like(Ub activating) Ubiquitin-likesodium borate buffer (pH 8.2) for 10 min. After two 5-min
APP-BP11hUBA3 activates NEDD8→targets (e.g., cullin-4A)washes the neurons were incubated in 5% horse serum in
NEDD8 then forms a thiol ester linkage with hUbc12, a
PBS plus 0.1% Triton-X 100 (blocking buffer) for 1 h at
human protein that has a function parallel to that of the
room temperature, after which they were incubated
over-ubiquitin-conjugating enzyme, prior to its modification of
night in a 1:1000 dilution of the anti-BrdU monoclonal
target proteins such as cullin-4A. The functions of this
antibody BU-33 (Sigma) in blocking buffer. After three
pathway (reviewed in Ref. [41]) are still unclear; but it
5-min washes in blocking buffer, the neurons were
incu-does in some cases lead to modification of ubiquitin-like
bated for 1.5 h at room temperature in a 1:200 dilution of
proteins that are linked to cell cycle regulation [46,51].
biotinylated secondary anti-mouse antibody (Vector
Lab-We have shown that APP-BP1 plays a role in the cell
oratories) in blocking buffer, washed, and visualized using
cycle [9]. APP-BP1 co-immunoprecipitates with hUba3
DAB. Stained cells were visualized under a 40
3
objective.
from mammalian cells and binds to a region between
Ten random fields were counted from each condition, and
amino acids 443 and 479 in hUba3 (Fig. 2). Wild type
the data were expressed as the percentage ratio of stained
APP-BP1 rescues the cell cycle S–M checkpoint defect in
cells to total cells.
ts41 hamster cells [9] and this rescue is dependent on the
The results (Fig. 7) indicated that expression of FAD
binding of APP-BP1 to hUba3 (Fig. 3). Dominant negative
mutants of APP in cortical neurons caused a significant
mutants of hUba3 and Ubc12 prevent the rescue. Notably,
increase over controls in the number of cells undergoing
overexpression of APP-BP1 in primary neurons (Fig. 4)
DNA synthesis. Overexpression of wild type APP in the
causes apoptosis by a pathway that also involves hUba3
cultures also caused an increase in DNA synthesis,
al-and hUbc12 (Fig. 5). We hypothesize that overexpression
though to a lesser extent.
of APP-BP1 pushes neurons into the S phase of the cell
cycle (Fig. 6), causing DNA synthesis and, for example,
aberrant expression of mitotic cdc2 / cyclin B1 kinase, as is
6. Conclusions
observed in degenerating neurons in AD brain [109].
To test this hypothesis, we expressed FAD mutants of
We propose that mechanisms other than accumulation of
APP in neurons using HSV-1 vectors. Consistent with our
A
b
may be the cause of AD neurodegeneration and
prediction, neurons expressing FAD mutants of APP
cognitive impairment. In particular, we suggest that the
showed increased expression of APP-BP1 relative to
disease may be a consequence of disruption of function of
controls (unpublished data of Y. Chen and R. Neve). We
APP. Convincing data have accumulated that support that
next evaluated DNA synthesis in the infected cultures. Ten
idea that APP is a functional receptor linked to a G
ouM 5-bromo-2
9
-deoxyuridine BrdU (Sigma) was added to
signaling cascade. Apoptosis is induced in neuronal cells
the primary cortical cultures at the time of infection.
expressing FAD mutants of APP, and this phenotype is
Sixteen hours after infection the cells were fixed in 4%
independent of the production of A
b
1 – 42. Expression of
paraformaldehyde in 100 mM phosphate buffer pH 7.4 for
FAD mutants of APP in neurons causes not only apoptosis
20 min. The cultures were washed twice for 5 min each in
but also intracellular accumulation of carboxyl terminal
PBS, and were treated with 2 N HCl for 10 min at room
fragments of APP. These data suggest that the FAD
Fig. 5. Schematic showing the ubiquitin-like pathway in which APP-BP1 is involved. The family of genes known as the cullins are the known targets for this pathway. APP-BP1 negatively regulates the G1 to S transition and positive regulates the S to M transition via this pathway. In dividing cells, APP-BP1 promotes cell division, but in primary neurons, it causes apoptosis.
AG12954.
References
[1] B. Allinquant, K.L. Moya, C. Bouillot, A. Prochiantz, Amyloid precursor protein in cortical neurons: coexistence of two pools differentially distributed in axons and dendrites and association with cytoskeleton, J. Neurosci. 14 (1994) 6842–6854.
[2] N.Y. Barnes, L. Li, K. Yoshikawa, L.M. Schwartz, R.W. Oppenheim, C.E. Milligan, Increased production of amyloid precursor protein provides a substrate for caspase-3 in dying motoneurons, J. Neuro-sci. 18 (1998) 5869–5880.
[3] J. Berger-Sweeney, D.L. McPhie, J.A. Arters, J. Greenan, M.L. Oster-Granite, R.L. Neve, Impairment in spatial learning accom-panied by neurodegeneration in mice transgenic for the carboxyl-terminus of the amyloid precursor protein, Mol. Brain Res. 66 (1999) 150–162.
[4] S.L. Borowicz, L.A. Dokas, Association of the amyloid precursor protein, B-50 (GAP-43), and Go in neuronal membranes, Soc. Neurosci. Abst. 21 (GAP-43) (1995) 207.
Fig. 6. Diagram depicting the stages of the cell cycle at which APP-BP1
[5] E. Brouillet, A. Trembleau, D. Galanaud, M. Volovitch, C. Bouillot, act. Note that the cdc / cyclin B1 kinase acts downstream of APP-BP1; if
C. Valenza, A. Prochiantz, B. Allinquant, The amyloid precursor dysfunction of APP-BP1 occurs in AD brain, this may explain the
protein interacts with Go heterotrimeric protein within a cell aberrant expression of cdc / cyclin B1 in neurons in AD.
compartment specialized in signal transduction, J. Neurosci. 19 (1999) 1717–1727.
[6] S. Bursztajn, R. DeSouza, D.L. McPhie, S.A. Berman, J. Shioi, N.K. Robakis, R.L. Neve, Overexpression in neurons of human
in the neuron, and that these abnormalities constitute one
presenilin-1 or a presenilin-1 familial Alzheimer disease mutant
mechanism of neurodegeneration in AD.
does not enhance apoptosis, J. Neurosci. 18 (1998) 9790–9799.[7] J. Busser, D.S. Geldmacher, K. Herrup, Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer’s disease brain, J. Neurosci. 18 (1998) 2801–2807.
Acknowledgements
[8] X.D. Cai, T.E. Golde, S.G. Younkin, Release of excess amyloidb protein from a mutant amyloid b protein precursor, Science 259
We thank Dawn Morrissey for assistance in preparation
(1993) 5140516.of the manuscript. The work from our laboratory that is
[9] Y. Chen, D.L. McPhie, J. Hirschberg, R.L. Neve, The amyloiddescribed in this review was funded by NIH grant
precursor protein-binding protein APP-BP1 drives the cell cyclethrough the S–M checkpoint and causes apoptosis in neurons, J. [28] O. Ghiribi, L. Lapierre, M. Girard, M. Ohayon, J. Nalbantoglu, G. Biol. Chem. 275 (2000) 8929–8935. Massicotte, Hypoxia-induced loss of synaptic transmission is ex-acerbated in hippocampal slices of transgenic mice expressing C-[10] N. Chow, J.R. Korenberg, X.-N. Chen, R.L. Neve, APP-BP1, a
terminal fragments of Alzheimer amyloid precursor protein, Hip-novel protein that binds to the carboxyl-terminal region of the
pocampus 9 (1999) 201–205. amyloid precursor protein, J. Biol. Chem. 271 (1996) 11339–11346.
[29] U. Giambarella, T. Yamatsuji, T. Okamoto, T. Matsui, T. Ikezu, Y. [11] N. Chow, C. Cox, L.M. Callahan, J.M. Weimer, L. Guo, P.D.
Murayama, M.A. Levine, A. Katz, N. Gautam, I. Nishimoto, G Coleman, Expression profiles of multiple genes in single neurons of
protein bg complex-mediated apoptosis by familial Alzheimer’s Alzheimer’s disease, Proc. Natl. Acad. Sci. USA 95 (1998) 9620–
disease mutant of APP, EMBO J. 16 (1997) 4897–4907. 9625.
[30] P. Giannakopoulos, C. Bouras, E. Kovari, J. Shioi, N. Tezapsidis, [12] M. Citron, T. Oltersdorf, C. Haas, L. McConlogue, A.Y. Hung, P.
P.R. Hof, N.K. Robakis, Presenilin-1-immunoreactive neurons are Seubert, C. Vigo-Pelfrey, I. Lieberburg, D.J. Selkoe, Mutation of the
preserved in late-onset Alzheimer’s disease, Am. J. Pathol. 150 b-amyloid precursor protein in familial Alzheimer’s disease
in-(1997) 429–436. creasesb-protein production, Nature 360 (1992) 672–674.
[31] T. Golde, S. Estus, S.G. Younkin, Processing of the amyloid protein [13] M. Citron, D.B. Teplow, D.J. Selkoe, Generation of amyloid b
precursor to potentially amyloidogenic derivatives, Science 255 protein from its precursor is sequence specific, Neuron 14 (1995)
(1992) 728–730. 661–670.
[32] Q. Guo, K. Furukawa, B.L. Sopher, D.G. Pham, J. Xie, N. [14] M. Citron, T.S. Diehl, A. Capell, C. Haass, D.B. Teplow, D.J.
Robinson, G.M. Martin, M.P. Mattson, Alzheimer’s PS-1 mutation Selkoe, Inhibition of amyloidb-protein production in neural cells by
perturbs calcium homeostasis and sensitizes PC12 cells to death the serine protease inhibitor AEBSF, Neuron 17 (1996) 1–9.
induced by amyloid beta-peptide, Neuroreport 8 (1996) 379–383. [15] C.W. Cotman, Apoptosis decision cascades and neuronal
degenera-[33] Q. Guo, B.L. Sopher, K. Furukawa, D.G. Pham, N. Robinson, G.M. tion in Alzheimer’s disease, Neurobiol. Aging 19 (1998) S29–S32.
Martin, M. P Mattson, Alzheimer’s presenilin mutation sensitizes [16] G. Deng, C.J. Pike, C.W. Cotman, Alzheimer-associated presenilin-2
neural cells to apoptosis induced by trophic factor withdrawal and confers increased sensitivity to apoptosis in PC12 cells, FEBS Lett.
amyloid beta peptide: involvement of calcium and oxyradicals, J. 397 (1996) 50–54.
Neurosci. 17 (1997) 4212–4222. [17] T. Dyrks, A. Weidemann, G. Multhaup, J.M. Salbaum, H.G.
[34] Q. Guo, W. Fu, J. Xie, H. Luo, S.F. Sells, J.W. Geddes, V. Bondada, Lemaire, J. Kang, B. Muller-Hill, C.L. Masters, K. Beyreuther,
V.M. Rangnekar, M.P. Mattson, Par-4 is a mediator of neuronal Identification, transmembrane orientation and biogenesis of the
degeneration associated with the pathogenesis of Alzheimer disease, amyloid A4 precursor of Alzheimer’s disease, EMBO J. 7 (1988)
Nature 4 (1998) 957–962. 949–957.
[35] A.L. Haas, T.J. Siepmann, Pathways of ubiquitin conjugation, [18] T. Dyrks, E. Dyrks, T. Hartmann, C. Masters, K. Beyreuther,
FASEB J. 11 (1997) 1257–1268. Amyloidogenicity of bA4 and bA4-bearing amyloid protein
pre-[36] C. Haass, E. Koo, A. Mellon, A. Jung, D. Selkoe, Targeting of cursor fragments by metal-catalyzed oxidation, J. Biol.Chem. 267
cell-surface b-amyloid precursor protein to lysosomes: alternative (1992) 18210–18217.
processing into amyloid-bearing fragments, Nature 357 (1992) 500– [19] T. Dyrks, E. Dyrks, U. Monning, B. Urmoneit, J. Turner, K.
503. Beyreuther, Generation ofbA4 from the amyloid protein precursor
[37] C. Haass, A. Hung, M. Schlossmacher, D. Teplow, D. Selkoe, and fragments thereof, FEBS Lett. 335 (1993) 89–93.
b-amyloid peptide and a 3 kDa fragment are derived by distinct [20] G.A. Elder, N. Tezapsidis, J. Carter, J. Shioi, C. Bouras, H.C. Li,
cellular mechanisms, J. Biol. Chem. 268 (1993) 3021–3024. J.M. Johnson, S. Efthimiopoulos, V.L. Friedrich Jr., N.K. Robakis,
[38] J. Hardy, The Alzheimer family of diseases: many etiologies, one Identification and neuron specific expression of the PS-1 / presenilin
pathogenesis?, Proc. Natl. Acad. Sci. USA 18 (1997) 2095–2097. I protein in human and rodent brains, J. Neurosci. Res. 45 (1996)
308–320. [39] N. Heintz, Cell death and the cell cycle: a relationship between
transformation and neurodegeneration?, Trends Biochem Sci. 18 [21] S. Estus, T. Golde, T. Kunishita, D. Blades, D. Lowery, M. Eisen,
(1993) 157–159. M. Usiak, X. Qu, T. Tabira, B. Greenberg, S. Younkin, Potentially
amyloidogenic, carboxy-terminal derivatives of the amyloid protein [40] K. Herrup, J.C. Busser, The induction of multiple cell cycle events precursor, Science 255 (1992) 726–728. precedes target-related neuronal death, Development 121 (1995)
2385–2395. [22] R. Freeman, S. Estus, E. Johnson, Analysis of cell cycle-related
gene expression in postmitotic neurons: selection induction of cyclin [41] M. Hochstrasser, There’s the Rub: a novel ubiquitin-like modi-D1 during programmed cell death, Neuron 12 (1994) 343–355. fication linked to cell cycle regulation, Genes Dev. 12 (1998)
901–907. [23] K. Fukuchi, B. Sopher, G.M. Martin, Neurotoxicity of b-amyloid,
Nature 361 (1993) 122. [42] I. Hussain, D. Powell, D.R. Howlett, D.G. Tew, T.D. Meek, C.
Chapman, I.S. Gloger, K.E. Murphy, C.D. Southan, D.M. Ryan, T.S. [24] K. Fukuchi, B. Sopher, C.E. Furlong, A.C. Smith, N.T. Dang, G.M.
Smith, D.L. Simmons, F.S. Walsh, C. Dingwall, G. Christie, Martin, Selective neurotoxicity of COOH-terminal fragments of the
Identification of a novel aspartic protease (Asp2) as b-secretase, b-amyloid precursor protein, Neurosci. Lett. 154 (1993) 145–148.
Mol. Cell. Neurosci. 14 (1999) 419–427. [25] K. Fukuchi, D.D. Kunkel, P.A. Schwartzkroin, K. Kamino, C.E.
[43] N. Ida, T. Hartmann, J. Pantel, J. Schroder, R. Zerfass, H. Forstl, R. Ogburn, C.E. Furlong, G.M. Martin, Overexpression of a C-terminal
Sandbrink, C.L. Masters, K. Beyreuther, Analysis of heterogeneous portion of the b-amyloid precursor protein in mouse brains by
bA4 peptides in human cerebrospinal fluid and blood by a newly transplantation of transformed neuronal cells, Exptl. Neurol. 127
developed sensitive western blot assay, J. Biol. Chem. 271 (1996) (1994) 253–264.
22908–22914. [26] D. Games, D. Adams, R. Alessandrini, R. Barbour, P. Berthelette, C.
[44] S. Illenberger, Q. Zheng-Fischhofer, U. Preuss, K. Stamer, K. Blackwell, T. Carr, J. Clemens, T. Donaldson, F. Gillespie,
Al-Baumann, B. Trinczek, J. Biernat, R. Godemann, E.M. Mandelkow, zheimer-type neuropathology in transgenic mice overexpressing
E. Mandelkow, The endogenous and cell cycle-dependent phos-V717Fb-amyloid precursor protein, Nature 373 (1995) 523–527.
phorylation of tau protein in living cells: Implications for Alzheim-[27] J.E. Gardella, G.A. Gorgone, L. Candela, J. Ghiso, E.M. Castano, B.
er’s disease, Mol. Biol. Cell 9 (1998) 1495–1512. Frangione, P.D. Gorevic, High-level expression and in vitro
muta-[45] S. Janicki, M.J. Monteiro, Increased apoptosis arising from in-genesis of a fibrillogenic 109-amino-acid C-terminal fragment of
creased expression of Alzheimer’s disease associated presenilin-2 Alzheimer’s disease amyloid precursor protein, Biochem. J. 294
[46] P.R. Johnson, M. Hochstrasser, SUMO-1: ubiquitin gains weight, ing of cell-surface b-amyloid precursor protein: Evidence that a Trends Cell Biol. 7 (1997) 408–413. sorting intermediate participates in synaptic vesicle recycling, J.
Neurosci. 17 (1997) 140–151. [47] S.S. Jung, J. Nalbantoglu, N.R. Cashman, Alzheimer’sb-amyloid
precursor protein is expressed on the surface of immediately ex vivo [64] K. Maruyama, K. Terakado, M. Usami, K. Yoshikawa, Formation of brain cells: a flow cytometric study, J. Neurosci. Res. 46 (1996) amyloid-like fibrils in COS cells overexpressing part of the
Al-336–348. zheimer amyloid protein precursor, Nature 347 (1990) 566–569.
[48] A. Kammesheidt, F.M. Boyce, A.F. Spanoyannis, B.J. Cummings, [65] M. Mattson, B. Cheng, A. Culwell, F. Esch, I. Lieberburg, R. Rydel, M. Ortegon, C.W. Cotman, J. Vaught, R.L. Neve, Amyloid deposi- Evidence for excitoprotective and intraneuronal calcium-regulating tion and neuronal pathology in transgenic mice expressing the roles for secreted forms of theb-amyloid precursor protein, Neuron carboxyterminal fragment of the Alzheimer amyloid precursor in the 10 (1993) 243–254.
brain, Proc. Natl. Acad. Sci. USA 89 (1992) 10857–10861. [66] D.L. McPhie, R.K.K. Lee, C.B. Eckman, D.H. Olstein, S.P. Durham, [49] J. Kang, H.G. Lemaire, A. Unterbeck, J.M. Salbaum, C.L. Masters, D. Yager, S.G. Younkin, R.J. Wurtman, R.L. Neve, Neuronal K.H. Grzeschik, K. Beyreuther, B. Muller-Hill, The precursor of expression of b-amyloid precursor protein Alzheimer mutations Alzheimer’s disease amyloid A4 protein resembles a cell-surface causes intracellular accumulation of a C-terminal fragment con-receptor, Nature 325 (1987) 733–736. taining both the amyloidband cytoplasmic domains, J. Biol. Chem.
272 (1997) 24743–24746. [50] S.H. Kim, Y.H. Suh, Neurotoxicity of a carboxy-terminal fragment
of the Alzheimer’s amyloid precursor protein, J. Neurochem. 67 [67] R. Motter, C. Vigo-Pelfrey, D. Kholodenko, R. Barbour, K.
Johnson-(1996) 1172–1182. Wood, D. Galasko, L. Chang, B. Miller, C. Clark, R. Green,
Reduction of b-amyloid peptide in the cerebrospinal fluid of
[51] E.T. Kipreos, L.E. Lander, J.P. Wing, W.W. He, E.M. Hedgecock, 42
patients with Alzheimer’s disease, Ann. Neurol. 38 (1995) 643–648. Cul-1 is required for cell cycle exit in C. elegans and identifies a
novel gene family, Cell 85 (1996) 829–839. [68] K.L. Moya, L.I. Benowitz, G.E. Schneider, B. Allinquant, The amyloid precursor protein is developmentally regulated and corre-[52] J. Lang, I. Nishimoto, T. Okamoto, R. Regazzi, C. Kiraly, U. Weller,
lated with synaptogenesis, Dev. Biol. 171 (1994) 597–603. C.B. Wollheim, Direct control of exocytosis by receptor-mediated
activation of the heterotrimeric GTPases G and Gi o or by the [69] L. Mucke, E. Masliah, W.B. Johnson, M.D. Ruppe, M. Alford, E.M. expression of their active G alpha subunits, EMBO J. 14 (1995) Rockenstein, S. Forss-Petter, M. Pietropaolo, M. Mallory, C.A.
3635–3644. Abraham, Synaptotrophic effects of human amyloid b protein
precursors in the cortex of transgenic mice, Brain Res. 666 (1994) [53] A.C. LeBlanc, Increased production of 4 kDa amyloidbpeptide in
151–167. serum deprived human primary neuron cultures: possible
in-¨ ¨
volvement of apoptosis, J. Neurosci. 15 (1995) 7837–7846. [70] U. Muller, N. Cristina, A.W. Li, D.P. Wolfer, H.P. Lipp, T. Rulick, S. Brandner, A. Aguzzi, C. Weissmann, Behavioral and anatomical [54] A.C. LeBlanc, R. Xue, P. Gambetti, APP metabolism in primary cell
deficits in mice homozygous for a modified b-amyloid precursor cultures of neurons, astrocytes and microglia, J. Neurochem. 66
protein gene, Cell 79 (1994) 755–765. (1996) 2300–2310.
[71] J. Nalbantoglu, G. Tirado-Santiago, A. Lahsaini, J. Poirier, O. [55] A.C. LeBlanc, M. Papadopoulos, C. Belair, W. Chu, M. Crosato, J.
Goncalves, G. Verge, F. Momoli, S.A. Weiner, G. Massicotte, J.P. Powell, C. Goodyer, Amyloid precursor protein metabolism in
Julien, M.L. Shapiro, Impaired learning and LTP in mice expressing human neurons, astrocytes and microglia, J. Neurochem. 68 (1997)
the carboxy terminus of the Alzheimer amyloid precursor protein, 1183–1190.
Nature 387 (1997) 500–505. [56] E.Y.-H.P. Lee, C.Y. Chang, N. Hu, Y.C.J. Wang, C.C. Lai, K. Herrup,
[72] R.L. Neve, A. Kammesheidt, C.F. Hohmann, Brain transplants of W.H. Lee, A. Bradley, Mice deficient for Rb are nonviable and show
cells expressing the carboxyterminal fragment of the Alzheimer defects in neurogenesis and haematopoiesis, Nature 359 (1992)
amyloid protein precursor cause specific neuropathology in vivo, 288–294.
Proc. Natl. Acad. Sci. USA 89 (1992) 3448–3452. [57] Q.X. Li, C. Maynard, R. Cappai, C.A. McLean, R.A. Cherny, T.
[73] R.L. Neve, M.R. Kozlowski, The carboxyl-terminal 100 amino acids Lynch, J.G. Culvenor, J. Trevaskis, J.E. Tanner, K.A. Bailey, C.
of the b-amyloid protein precursor: Role in Alzheimer disease Czech, A.I. Bush, K. Benreuther, C.L. Masters, Intracellular
ac-neurodegeneration, Dev. Brain. Dysfunction. 8 (1995) 13–24. cumulation of detergent-soluble amyloidogenic Ab fragment of
Alzheimer’s disease precursor protein in the hippocampus of aged [74] R.L. Neve, N.K. Robakis, Alzheimer disease: A re-examination of transgenic mice, J. Neurochem. 72 (1999) 2479–2487. the amyloid hypothesis, Trends Neurosci. 21 (1998) 15–19. [58] Y.M. Li, M. Xu, M.T. Lai, Q. Huang, J.L. Castro, J. DiMuzio- [75] I. Nishimoto, T. Okamoto, Y. Matsuura, T. Okamoto, Y. Murayama,
Mower, T. Harrison, C. Lellis, A. Nadin, J.G. Neduvelil, R.B. E. Ogata, Alzheimer amyloid protein precursor complexes with Register, M.K. Sardana, M.S. Shearman, A.L. Smith, X.P. Shi, K.C. brain GTP-binding protein G , Nature 362 (1993) 75–79.o Yin, J.A. Shafer, S.J. Gardell, Photoactivatedg-secretase inhibitors [76] I. Nishimoto, A new paradigm for neurotoxicity by FAD mutants of directed to the active site covalently label presenilin 1, Science 405 bAPP: a signaling abnormality, Neurobiol. Aging 19 (1998) S33–
(2000) 689–694. S38.
[59] W.K. Liu, R. Williams, F. Hall, D. Dickson, S.H. Yen, Detection of a [77] I. Nishimura, T. Uetsuki, S.U. Dani, Y. Ohsawa, I. Saito, H. cdc2-related kinase associated with Alzheimer paired helical fila- Okamura, Y. Uchiyama, K. Yoshikawa, Degeneration in vivo of rat ments, Am. J. Pathol. 146 (1995) 228–238. hippocampal neurons by wild-type Alzheimer amyloid precursor [60] D.C. Lu, S. Rabizadeh, S. Chandra, R.F. Shayya, L.M. Ellerby, X. protein overexpressed by adenovirus-mediated gene transfer, J.
Ye, G.S. Salvesen, E.H. Koo, D.E. Bredesen, A second cytotoxic Neurosci. 18 (1998) 2387–2398.
proteolytic peptide derived from amyloid b-protein precursor, [78] M. Oishi, A.C. Nairn, A.J. Czernik, G.S. Lim, T. Isohara, S.E.
Nature Med. 6 (2000) 397–404. Gandy, P. Greengard, T. Suzuki, The cytoplasmic domain of
[61] J.J. Luo, W. Wallace, T. Riccioni, D.K. Ingram, G.S. Roth, J.W. Alzheimer’s amyloid precursor protein is phosphorylated at thr654, Kusiak, Death of PC12 cells and hippocampal neurons induced by ser655, and thr668 in adult rat brain and cultured cells, Mol. Med. 3 adenoviral-mediated FAD human amyloid precursor protein gene (1997) 111–123.
expression, J. Neurosci. Res. 55 (1999) 629–642. [79] T. Okamoto, S. Takeda, Y. Murayama, E. Ogata, I. Nishimoto, [62] L. Luo, T. Tully, K. White, Human amyloid precursor protein Ligand-dependent G protein coupling function of amyloid
trans-ameliorates behavioral deficit of flies deleted for Appl gene, Neuron membrane precursor, J. Biol. Chem. 270 (1995) 4205–4208.
9 (1992) 595–605. [80] M.L. Oster-Granite, D.L. McPhie, J. Greenan, R.L. Neve,
the carboxyl-terminus of the amyloid precursor protein, J. Neurosci. [96] S. Sinha, J.P. Anderson, R. Barbour, G.S. Basi, R. Caccavello, D. Davis, M. Doah, H.F. Dovey, N. Frigon, J. Hong, K. Jacobson-16 (1996) 6732–6741.
Croak, N. Jewett, P. Keim, J. Knops, I. Lieberburg, M. Power, H. [81] M. Palmert, S. Siedlak, M. Podlisny, B. Greenberg, E. Shelton, H.
Tan, G. Tatsuno, J. Tung, D. Schenk, P. Seuberg, S.M. Suomensaari, Chan, M. Usiak, D. Selkoe, G. Perry, S. Younkin, Soluble
deriva-S. Wang, D. Walker, J. Zhao, L. McConlogue, V. John, Purification tives of theb-amyloid protein precursor of Alzheimer’s disease are
and cloning of amyloid precursor protein b-secretase from human labeled by antisera to the b-amyloid protein, Biochem. Biophys.
brain, Nature 402 (1999) 537–540. Res. Commun. 165 (1989) 7533–7539.
[97] B.L. Sopher, K. Fukuchi, A.C. Smith, K.A. Leppig, C.E. Furlong, [82] G. Perry, A. Nunomura, P. Lucessen, H. Lassmann, M.A. Smith,
G.M. Martin, Cytotoxicity mediated by conditional expression of a Apoptosis and Alzheimer’s disease, Science 282 (1998) 1268–1269.
carboxyl-terminal derivative of the b-amyloid precursor protein, [83] R.G. Perez, H. Zheng, L.H.T. Van der Ploeg, E.H. Koo, The
Mol. Brain Res. 26 (1994) 207–217. b-amyloid precursor protein of Alzheimer’s disease enhances
neu-[98] C. Stadelmann, W. Bruck, C. Bancher, K. Jellinger, H. Lassmann, ron viability and modulates neuronal polarity, J. Neurosci. 17 (1997)
Alzheimer disease: DNA fragmentation indicates increased neuronal 9407–9414.
vulnerability, but not apoptosis, J. Neuropathol. Exp. Neurol. 57 [84] W. Pope, M. Lambert, B. Leypole, R. Seupaul, L. Sletten, G. Krafft,
(1998) 456–464. W. Klein, Microtubule-associated protein tau is hyperphosphorylated
[99] E. Storey, T. Spurck, J. Pickett-Heaps, K. Beyreuther, C.L. Masters, during mitosis in the human neuroblastoma cell line SH-SY5Y, Exp.
The amyloid precursor protein of Alzheimer’s disease is found on Neurol. 126 (1994) 185–194.
the surface of static but not actively motile portions of neurites, [85] W. Q Qiu, A. Ferreira, C. Miller, E.H. Koo, D.J. Selkoe,
Cell-Brain Res. 735 (1996) 59–66. surfaceb-amyloid precursor protein stimulates neurite outgrowth of
[100] J.H. Su, A.J. Anderson, B.J. Cummings, C.W. Cotman, Immuno-hippocampal neurons in an isoform-dependent manner, J. Neurosci. histochemical evidence for DNA fragmentation in neurons in the
15 (1995) 2157–2167. AD brain, NeuroReport 5 (1994) 2529–2533.
[86] J.M. Roch, E. Masliah, A.C. Roch-Levecq, M.P. Sundsmo, D.A. [101] Y.H. Suh, An etiological role of amyloidogenic carboxyl-terminal Otero, I. Veinbergs, T. Saitoh, Increase of synaptic density and fragments of the b-amyloid precursor protein in Alzheimer’s memory retention by a peptide representing the trophic domain of disease, J. Neurochem. 68 (1997) 1781–1791.
the amyloidb/A4 protein precursor, Proc. Natl. Acad. Sci. USA 91 [102] N. Suzuki, T.T. Cheung, X.D. Cai, A. Odaka, L. Otvos Jr., C.
(1994) 7450–7454. Eckman, T.E. Golde, S.G. Younkin, An increased percentage of
[87] T.T. Rohn, K.J. Ivins, B.A. Bahr, C.W. Cotman, D.H. Cribbs, A long amyloid beta protein secreted by familial amyloidbprotein monoclonal antibody to amyloid precursor protein induces neuronal precursor (bAPP717) mutants, Science 264 (1994) 1336–1340. apoptosis, J. Neurochem. 74 (2000) 2331–2342. [103] T. Suzuki, M. Oishi, D.R. Marshak, J. Czernik, A.C. Nairn, P. [88] J.P. Roperch, V. Alvaro, S. Prieur, M. Tuynder, M. Nemani, F. Greengard P, Cell cycle-dependent regulation of the phosphoryla-Lethrosne, L. Piouffre, M.C. Gendron, D. Israeli, J. Dausset, M. tion and metabolism of the Alzheimer amyloid precursor protein, Oren, R. Amson, A. Telerman, Inhibition of presenilin 1 expression EMBO J. 13 (1994) 1114–1222.
is promoted by p53 and p21WAF-1 and results in apoptosis and [104] B. Tate, K.S. Aboody-Guterman, A.M. Morris, E.C. Walcott, R.E. tumor suppression, Nature Med. 4 (1998) 835–838. Majocha, C. A Marotta, Disruption of circadian regulation by brain [89] T. Russo, R. Faraonio, G. Minopoli, P. De Candia, S. De Renzis, N. grafts that overexpress Alzheimerb/A4 amyloid, Proc. Natl. Acad.
Zambrano, Fe65 and the protein network centered around the Sci. USA 89 (1992) 7090–7094.
cytosolic domain of the Alzheimer’s b-amyloid precursor protein, [105] L.O. Tjernberg, J. Naslund, J. Thyberg, S.E. Gandy, L. Terenius,
FEBS Lett. 434 (1998) 1–7. C. Nordstedt, Generation of Alzheimer amyloidbpeptide through
[90] K. Sambamurti, J. Shioi, A.P. Anderson, M. A Pappolla, N.K. nonspecific proteolysis, J. Biol. Chem. 272 (1997) 1870–1875. Robakis, Evidence for intracellular cleavage of the Alzheimer’s [106] S.Y. Tsang, S.C. Tam, I. Bremner, M.J. Burkitt, Copper-1,10-amyloid precursor in PC12 cells, J. Neurosci. Res. 33 (1992) phenanthroline induces internucleosomal DNA fragmentation in
319–329. HepG2 cells, resulting from direct oxidation by the hydroxyl
[91] M. Sato, T. Kawarabashi, M. Shoji, T. Kobayashi, N. Tada, E. radical, Biochem. J. 317 (1996) 13–16.
Matsubara, S. Hirai, Neurodegeneration and gliosis in transgenic [107] R. Vassar, B.D. Bennett, S. Babu-Khan, S. Kahn, E.A. Mendiaz, P. mice overexpressing a carboxy-terminal fragment of Alzheimer Denis, D.B. Teplow, S. Rose, P. Amarante, R. Loeloff, Y. Lui, S. amyloid-beta protein precursor, Dement. Geriatr. Cogn. Disord. 8 Fisher, J. Fuller, S. Edenson, J. Lile, M.A. Jarosinski, A.L. Biere,
(1997) 296–307. E. Curran, T. Burgess, J.C. Louis, F. Collins, J. Treanor, G.
[92] G. Sberna, J. Saez-Valero, Q.X. Li, C. Czech, K. Beyreuther, C.L. Rogers, M. Citron, b-secretase cleavage of Alzheimer’s amyloid Masters, C.A. McLean, D.H. Small, Acetylcholinesterase is in- precursor protein by the transmembrane aspartic protease BACE, creased in the brains of transgenic mice expressing the C-terminal Science 286 (1999) 735–741.
fragment (CT100) of theb-amyloid protein precursor of Alzheim- [108] I. Vincent, M. Rosado, P. Davies, Mitotic mechanisms in Alzheim-er’s disease, J. Neurochem. 71 (1998) 723–731. er’s disease?, J. Cell Biol. 132 (1996) 413–425.
[93] G. Schettini, C. Russo, W. Sannita, P. Gambetti, Characterization of [109] I. Vincent, G. Jicha, M. Rosado, D.W. Dickson, Aberrant expression carboxy-terminal APP derivatives in Alzheimer’s disease and Down of mitotic cdc2 / cyclin B1 kinase in degenerating neurons of syndrome, Soc. Neurosci. Abstr. 25 (1999) 837. Alzheimer’s disease brain, J. Neurosci. 17 (1997) 3588–3598.
[110] T. Watanabe, J. Sukegawa, I. Sukegawa, S. Tomita, K. Iijima, S. [94] D. Scheuner, C. Eckman, M. Jensen, X. Song, M. Citron, N. Suzuki,
Oguchi, T. Suzuki, A.C. Nairn, P. Greengard, A 127-kDa protein T.D. Bird, J. Hardy, M. Hutton, W. Kukull, E. Larson, E.
Levy-(UV-DDB) binds to the cytoplasmic domain of the Alzheimer’s Lahad, M. Viitanen, E. Peskind, P. Poorkaj, G. Schellenberg, R.
amyloid precursor protein, J. Neurochem. 72 (1999) 549–556. Tanzi, W. Wasco, L. Lannfelt, D. Selkoe, S. Younkin, Secreted
[111] D. Wolf, D. Quon, Y. Wang, B. Cordell, Identification and charac-amyloidb-protein similar to that in the senile plaques of
Alzheim-terization of C-terminal fragments of the b-amyloid precursor er’s disease is increased in vivo by the presenilin 1 and 2 and APP
produced in cell culture, EMBO J. 9 (1990) 2079–2084. mutations linked to familial Alzheimer’s disease, Nat. Med. 2
[112] B. Wolozin, K. Iwasaki, P. Vito, J.K. Ganjei, E. Lacana, T. (1996) 864–870.
Sunderland, B. Zhao, J.W. Kusiak, W. Wasco, L. D’Adamio, [95] M. Simons, E. Ikonen, P.J. Tienari, A. Cid-Arregui, U. Monning, K.
Participation of presenilin 2 in apoptosis enhanced basal activity Beyreuther, C.G. Dotti, Intracellular routing of human amyloid
conferred by an Alzheimer mutation, Science 274 (1996) 1710– protein precursor: Axonal delivery followed by transport to the
[113] T. Yamatsuji, T. Okamoto, S. Takeda, H. Fukumoto, T. Iwatsubo, Gurney, Membrane-anchored aspartyl protease with Alzheimer’s N. Suzuki, A. Asami-Odaka, S. Ireland, T.B. Kinane, I. Nishimoto, diseaseb-secretase activity, Nature 402 (1999) 533–537. G protein-mediated neuronal DNA fragmentation induced by [116] B.A. Yankner, L.R. Dawes, S. Fisher, L. Villa-Komaroff, M.L. familial Alzheimer’s disease-associated mutants of APP, Science Oster-Granite, R.L. Neve, Neurotoxocity of a fragment of the
272 (1996) 1349–1352. amyloid precursor associated with Alzheimer’s disease, Science
[114] T. Yamazaki, D.J. Selkoe, E.H. Koo, Trafficking of cell surface 245 (1989) 417–420.
b-amyloid precursor protein: retrograde and transcytotic transport [117] K. Yoshikawa, Neurotoxicity of b-amyloid (reply), Nature 361 in cultured neurons, J. Cell Biol. 129 (1995) 432–442. (1993) 122–123.
[115] R. Yan, M.J. Bienkowski, M.E. Shuck, H. Miao, M.C. Tory, A.M. [118] S.G. Younkin, Evidence that Ab42 is the real culprit in Alzheim-Pauley, J.R. Brashier, N.C. Stratman, W.R. Mathews, A.E. Buhl, er’s disease, Ann. Neurol. 37 (1995) 287–288.