Inability of plasma high-density lipoproteins to inhibit cell
adhesion molecule expression in human coronary artery
endothelial cells
Anita K. Stannard
a, Shabeena Khan
c, Annette Graham
b, James S. Owen
a,*,
Sean P. Allen
caDepartment of Medicine,Royal Free and Uni6ersity College Medical School,Uni6ersity College London,Royal Free Campus,
London NW3 2PF,UK
bDepartment of Biochemistry and Molecular Biology,Royal Free and Uni6ersity College Medical School,Uni6ersity College London,
Royal Free Campus,London NW3 2PF,UK
cDepartment of Cardiothoracic Surgery,Imperial College of Science,Technology and Medicine,Harefield Hospital,Harefield,
Middlesex UB9 6JH,UK
Received 2 November 1999; received in revised form 21 February 2000; accepted 1 March 2000
Abstract
High-density lipoproteins (HDL) have several antiatherogenic actions, including the ability to sequester cellular cholesterol, to protect low-density lipoproteins from oxidation and to inhibit platelet aggregation. An early event in atherogenesis is the adhesion and recruitment of blood monocytes, a process mediated by cell adhesion molecules (CAMs), including vascular cell adhesion molecule-1 (VCAM-1) which is rapidly synthesized by endothelial cells in response to cytokines. It has been reported that HDL limits CAM expression in cultured human umbilical vein endothelial cells (HUVECs), implying that HDL also protects at an early stage in lesion development. Here, we have studied HDL suppression of CAM induction in human coronary artery endothelial cells (HCAECs), a model directly relevant to blood vessels susceptible to atherosclerosis. Arterial endothelial cells were preincubated with increasing amounts of total HDL, or different subfractions, and then activated with the inflammatory cytokine, tumor necrosis factor-alpha (TNF-a). Flow cytometric analysis failed to detect any downregulation of VCAM-1 or E-selectin expression by HDL in this model of vascular endothelium. Moreover, we were unable to confirm that HDL could suppress CAM induction in well-characterized, low-passage HUVECs, even though positive controls, 17b-estradiol or a nitric oxide donor, did cause downregulation and factors such as variability in donors and HDL preparation, or culture conditions, were excluded. We tentatively conclude that, as isolated HDL did not downregulate CAM expression in cultured HCAECs or HUVECs, attenuation of CAM induction in arterial endothelium is unlikely to contribute to HDL antiatherogenic actions in vivo. © 2001 Elsevier Science Ireland Ltd. All rights reserved.
Keywords:Atherosclerosis; E-Selectin; Human umbilical vein endothelial cells; Inflammatory cytokines; Vascular cell adhesion molecule-1 www.elsevier.com/locate/atherosclerosis
1. Introduction
Coronary heart disease (CHD) is a leading cause of death in industrialized societies and epidemiological studies have established that plasma high-density lipo-protein (HDL)-cholesterol concentrations are inversely related to the development of the disease [1]. This
protective effect of HDL may be associated with several functions, including a central role in reverse cholesterol transport [2,3] and anti-oxidant [4,5] and anti-throm-botic properties [6,7]. Studies in transgenic mice overex-pressing human apolipoprotein A-I (apo A-I), the major apolipoprotein in HDL, have confirmed the anti-atherogenic role of HDL; the animals have increased HDL-cholesterol and resist atherosclerosis induced by diet [8], by apo E-deficiency [9] or by the human lipoprotein(a) transgene [10]. A direct protective role of HDL was also shown in cholesterol-fed rabbits when infusions of HDL regressed atherosclerotic lesions [11]. * Corresponding author. Tel.: +44-20-74332853; fax: +
44-20-74332852.
E-mail address:[email protected] (J.S. Owen).
Atherosclerotic plaques develop from complex multi-cellular processes in which recruitment of circulating monocytes to focal areas of the arterial sub-endothe-lium is an early event. Localized upregulation of adhe-sive endothelial cell adhesion molecules (CAMs), a prerequisite for monocyte adherence and migration [12], is a dynamic process which is sensitive to inflam-matory cytokines, shear stress and oxidative insults. Levels of certain CAMs are elevated in human atherosclerotic tissue with vascular cell adhesion 1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-molecule-1), E-selectin and P-selectin, in particular, being indicators of inflammation and early atheroscle-rosis [13 – 15]. VCAM-1, a member of the immunoglob-ulin superfamily of CAMs, aids the selective accumulation of monocytes and T-lymphocytes and is a hallmark of early atherogenesis [16 – 18].
It has been reported that levels of cytokine-induced VCAM-1, ICAM-1 and E-selectin were reduced by HDL in human umbilical vein endothelial cells (HU-VECs) [19], a popular and relatively accessible in vitro model for CAM research [20]. Here, we have investi-gated whether HDL can downregulate CAM expression in primary human coronary artery endothelial cells (HCAECs), a model directly relevant to blood vessels susceptible to atherosclerosis. Arterial endothelial cells were preincubated with increasing amounts of total HDL, or different subfractions, and then activated with the inflammatory cytokine, tumor necrosis factor-alpha (TNF-a). However, flow cytometric analysis failed to detect any downregulation of VCAM-1 or E-selectin expression by HDL, implying that suppression of CAM induction in arterial endothelium is unlikely to con-tribute to HDL anti-atherogenic effects in vivo.
2. Materials and methods
2.1. Materials
O-Phenylenediamine (OPD) substrate tablets, IgG1 negative control antibodies, von Willebrand Factor (vWF) antibodies (clone F8/86), StreptABComplex/
HRP Duet kit, fluorescein isothiocyanate (FITC)-con-jugated antibodies and R-phycoerythrin (RPE)-conjug-ated antibodies were from DAKO (High Wycombe, UK). Monoclonal antibodies against VCAM-1 (CD106; clone BBIG-V1), ICAM-1 (CD54; clone BBIG-I1), E-selectin (CD62E; clone BBIG-E4) and platelet endothelial CAM-1 or PECAM-1 (CD31; clone 9G11) were from R&D Systems (Abingdon, UK). Other antibodies used for endothelial cell characteriza-tion were thrombomodulin (clone QB/End/40) and en-dothelial cell CD34 (clone QB/End/10) [21], both from Quantum Biosystems (Waterbeach, Cambridge, UK). M199 media, trypsin-EDTA, glutamine and penicillin/
streptomycin were purchased from Life Technologies (Paisley, UK). S-Nitroso-L-glutathione (GSNO) was supplied by Alexis (Nottingham, UK). Recombinant human TNF-a and other chemicals or tissue culture reagents were from Sigma (Poole, UK).
2.2. Cell culture
HCAECs were isolated from the vessels of four pa-tients with dilated cardiomyopathy at the time of ortho-topic heart transplantation. Informed consent was obtained from each patient and the study was approved by the Ethical Committee of Harefield Hospital. The methods used and culture conditions were as described previously [22,23]. In brief, the endothelium was scraped off the vessel lumen, collected in M199 media and cultured in plates pre-coated with 1% gelatin. Growth media was M199, supplemented with 2 mmol/l glutamine, 100 IU/ml penicillin, 100 mg/ml strepto-mycin, and 15% (v/v) foetal calf serum (FCS) and 15% (v/v) human AB serum. Cells were used between pas-sages 3 and 10 and the purity of the population was routinely confirmed by positive staining for PECAM-1, negative staining for smooth muscle a-actin and by uptake of DiI-acetylated LDL (Biogenesis, Bournemouth, UK). HUVECs were isolated and cul-tured as described previously [24]. Cultures were exten-sively characterized on the basis of ‘cobblestone’ morphology and by expression of endothelial antigens: vWF, CD34, thrombomodulin, PECAM-1 and ICAM-1 [24]. Experiments were carried out in MICAM-199 media containing 3.6 mmol/l glutamine, 100 IU/ml penicillin, 100 mg/ml streptomycin and 20% (v/v) FCS. Cells were used between passages 2 and 4.
2.3. Preparation of HDL
reagent; Bio-Rad Laboratories, Hemel Hempstead, UK).
2.4. Flow cytometry analysis of cell-surface CAMs
FACS analysis for cell-surface VCAM-1 and E-se-lectin was carried out on confluent HCAEC and HU-VEC monolayers as described previously [22,24]. Cells were preincubated for either 1 or 16 h with HDL (0.25 – 2 mg HDL protein/ml) in serum-containing me-dia, washed with warm HDL-free media and then stimulated for 4 h with TNF-a (10 ng/ml for HCAECs and 5 ng/ml or 100 U/ml for HUVECs). For positive controls showing VCAM-1 downregulation, cells were preincubated for 40 h with 10 mmol/l 17b-estradiol prior to TNF-aaddition [24]. Primary antibody binding and negative control antibody binding (at equivalent IgG1 protein concentrations) was detected using either goat anti-mouse FITC- or RPE-conjugated antibodies, and 5×103 cells were analysed per well by FACS (Coulter Epics Elite or Epics XL-MCL; Coulter, Hialeah, FL).
2.5. Cell-associated VCAM-1 ELISA in HUVECs
The ELISA for measuring total VCAM-1, that is both cell-surface and intracellular VCAM-1, by fixation prior to antibody incubations, was carried out as de-scribed previously [24]. HUVECs were grown in 96-well plates pre-coated with 1% gelatin (w/v) until confluent, preincubated for 16 h with HDL (1.5 mg/ml) and then, after washing with warm media to remove HDL, treated with 100 U/ml TNF-a (or suboptimal concen-trations as indicated) for a further 6 h. Variations to this protocol included the use of serum-free media supplemented with bovine serum albumin (BSA) (1% w/v) and the replacement of TNF-awith interleukin-1b
(IL-1b) (1 ng/ml or 100 U/ml) or lipopolysaccharide (LPS) (5 mg/ml or 50 U/ml). For positive controls of VCAM-1 downregulation, parallel wells were preincu-bated with 17b-estradiol (10 mmol/l) prior to TNF-a
addition [24] or were coincubated withS-nitroso-L -glu-tathione (GSNO) (200 mmol/l). Any binding of mouse anti-human monoclonal VCAM-1 antibodies or iso-type-matched irrelevant antibodies (at equivalent IgG1 protein concentrations) was detected by the sensitive StreptABComplex/HRP Duet kit and OPD chro-mogenic substrate. Absorbances were read at 492 nm in a Titertek Multiskan MCC/340 plate reader against blank substrate. The cell protein per well was measured using the Bradford reagent.
2.6. Statistical analysis
The statistical analysis of independent experiments was performed using Student’st-test; results are shown as mean9S.E. andPB0.05 was considered significant.
3. Results
3.1. Flow cytometric analysis of VCAM-1 and E-selectin expression in HCAECs
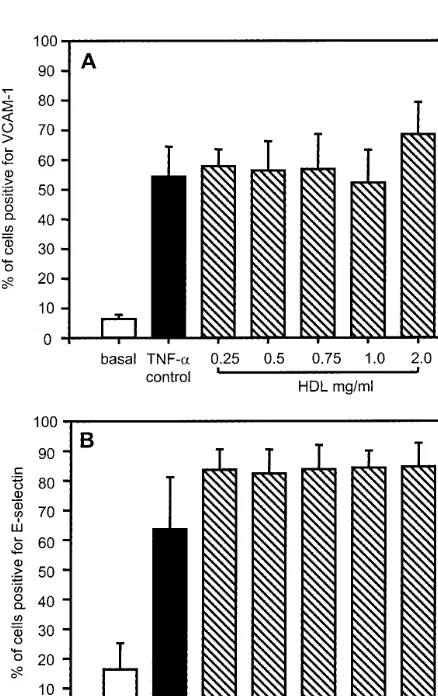
Confluent HCAEC monolayers were preincubated for 1 h with total HDL concentrations ranging from low to high physiological levels (0.25 – 2 mg HDL protein/ml) before cytokine stimulation with TNF-a(10 ng/ml) for 4 h and analysis by flow cytometry. In control wells, without HDL pretreatment, 54910% of cells treated with TNF-a were positive for VCAM-1 expression after 4 h as compared to 691% before stimulation (Fig. 1A). Increasing amounts of HDL had no effect (P\0.05) on either basal VCAM-1 (data not shown) or on the subsequent upregulation of VCAM-1 by TNF-a (Fig. 1A). Similar findings were noted for E-selectin expression: cells positive for cell-surface E-se-lectin increased from 1699 to 64918% after TNF-a
stimulation, but HDL was ineffective at suppressing this upregulation (P\0.05), even at high concentra-tions (Fig. 1B).
Although insufficient for maximum downregulation, a 1-h preincubation time with HDL is reported to effectively inhibit CAM expression in HUVECs (
50% maximal inhibition) [19]. Nevertheless, to exclude the possibility that HDL effects on HCAECs were dependent on exposure time, we extended the preincu-bation period to 16 h before addition of cytokine; no significant inhibition of either VCAM-1 or E-selectin was observed (data not shown). In other experiments we tested the major HDL subfraction, HDL3, as this is considered more inhibitory than HDL2 [27], but this also failed to downregulate TNF-a-induced VCAM-1 or E-selectin. In all experiments, the findings were not influenced by cytotoxic effects of HDL, as judged by microscopic evaluation and trypan blue exclusion tests for cell viability.
3.2. Flow cytometric analysis of VCAM-1 in HUVECs
fluorescence intensity (Fig. 2C). In parallel wells, how-ever, a 40-h preincubation with one high dose of 17b -estradiol (10 mmol/l), which is reported to maximally suppress VCAM-1 induction by TNF-a[28], did show a pronounced fall in mean fluorescence (Fig. 2D). This response was similar to other reports [28,29] and confi-rmed that the VCAM-1 expressed by our stimulated HUVECs was susceptible to downregulation.
Fig. 2. Inability of HDL to downregulate TNF-a-induced VCAM-1 in HUVECs. Anti-VCAM-1 binding to HUVEC suspensions was detected using a secondary FITC-conjugated antibody before fixation in 1% paraformaldehyde (w/v). Intact cells were gated on forward scatter versus side scatter amplifications and 5000 gated cells were analysed for FITC fluorescence. Histograms show a three-decade log10fluorescence scale against the number of gated cells or ‘events’
with corresponding fluorescence values. Basal VCAM-1 expression was negligible (A), but after 4 h exposure to TNF-a there was upregulation of cell-surface VCAM-1 (B). A 16-h preincubation with 1.5 mg HDL protein/ml failed to reduce VCAM-1 expression (C), although preincubation with 10mmol/l 17b-estradiol was an effective downregulator (D). Samples were analysed in duplicate but only single measurements are shown for a representative experiment. The results were confirmed in three independent assays using different batches of HUVEC and HDL preparations.
3.3. VCAM-1 ELISA on HUVECs
An ELISA was used to quantify total VCAM-1, that is both cell-surface and intracellular VCAM-1, in
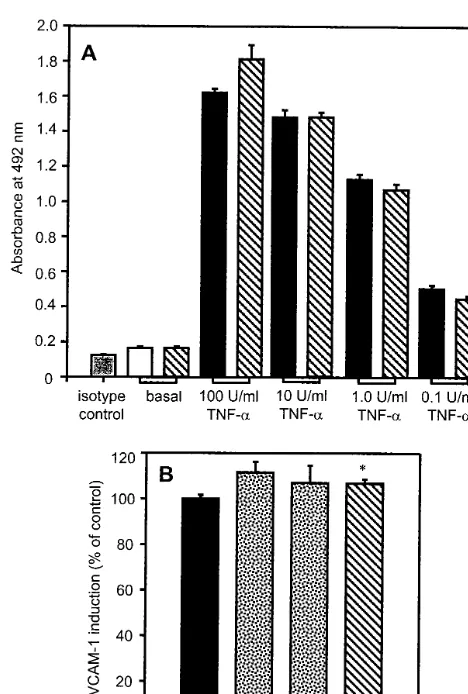
confluent HUVECs [24]. This assay verified the FACS data shown above. Thus, basal VCAM-1 was low, with the anti-VCAM-1 signal equivalent to that of the isotype control, but the HUVECs readily re-sponded to TNF-a in a dose-dependent manner when stimulated with a range of concentrations (0.1 – 100 U/ml), giving a 10-fold increased signal after 6 h with the optimum dose (Fig. 3A). Preincubating cells for 16 h with total HDL (1.5 mg protein/ml) before cy-tokine stimulation failed to restrict VCAM-1 induc-tion, even at suboptimal TNF-a concentrations (Fig. 3A). Indeed, when nine individual donors of HDL and three batches of HUVECs were tested, total HDL proved to be mildly, but significantly, stimula-tory (792% greater induction than with TNF-a
alone, PB0.01), while the HDL2 and HDL3 subfrac-tions were also ineffectual at downregulating VCAM-1 expression (Fig. 3B). By contrast, as we found previously [24], each batch of HUVECs was respon-sive to two different suppressors of CAM induction, the nitric oxide donor, S-nitroso-L-glutathione (GSNO) (200 mmol/l) [30,31], and 17b-estradiol (10
mmol/l) [28,29].
Several additional studies were carried out in which experimental conditions were varied. In each case, and using 10 different preparations of low-passage HUVECs, HDL consistently failed to suppress
TNF-a-induced induction of VCAM-1. Thus, use of a range of HDL concentrations (0.1 – 1.5 mg protein/ml) and pre-incubation times (1, 6 and 16 h), or adding TNF-a without prior removal of the HDL [19], all failed to reduce VCAM-1 induction (data not shown). Neither was a potential HDL suppressive activity on VCAM-1 upregulation counteracted by lipoproteins or other constituents of serum-containing media; par-allel experiments on HUVECs cultured in serum-free media containing BSA (1%, w/v) showed no in-hibitory effect of HDL (11590.4 vs. 10097% for control cells; (P\0.05)). Similarly, the inability of HDL to downregulate VCAM-1 was not due to the initial stimulus or to the source and mode of prepara-tion of HDL. Two other agents, the inflammatory cytokine, IL-1b (1 ng/ml or 100 U/ml) or the endo-toxin, LPS (5 mg/ml or 50 U/ml) readily achieved upregulation of VCAM-1 but these inductions were not suppressed by HDL pre-treatment (10495 vs. 10093% for IL-1b control (P\0.05); 11399 vs. 10098% for LPS control (P\0.05). Inter-donor variation of the HDL was also eliminated by using blood from a total of 14 healthy individuals, while reducing the time to isolate HDL, by precipitating apo B-containing lipoproteins before ultracentrifuga-tion and by use of desalting columns, made no differ-ence to our ELISA or FACS measurements.
Fig. 3. Effect of TNF-adose and HDL subclasses on VCAM-1 as assessed by ELISA. Confluent HUVEC monolayers in 96-well plates were preincubated for 16 h with HDL samples (1.5 mg protein/ml) and then stimulated with TNF-afor 6 h to induce VCAM-1 expres-sion. (A) Decreasing concentrations of TNF-a(100 – 0.1 U/ml) were used to examine whether an inhibitory effect of total HDL could be revealed. VCAM-1 was measured by sensitive ELISA. This had a low background signal, as shown by isotype-matched irrelevant antibod-ies (grey bar), while unstimulated cells (basal level) had insignificant VCAM-1 expression (white bar). A 10-fold induction above basal was seen after treatment with 100 U/ml TNF-aand this was reduced in a stepwise manner with decreasing additions of cytokine (black bars). Preincubation with total HDL had no effect on either basal VCAM-1 or on subsequent VCAM-1 induction (hatched bars) as compared to the corresponding TNF-acontrols. Cellular protein was unaltered by any of the treatments described. Results are from a representative experiment performed in quadruplicate (mean9S.E.) but were repro-duced in two other independent assays. (B) Preincubation with either HDL2or HDL3(stippled bars) failed to suppress VCAM-1
4. Discussion
Endothelium dysfunction is central to the develop-ment of atherosclerotic lesions [32,33]. Endothelial cells interact and communicate with macrophages, platelets, smooth muscle cells and T-lymphocytes; they are also a potential site of oxidation of LDL [34]. The emerging concept of atherosclerosis as a chronic inflammatory disease, in which an early cellular event is the expres-sion of specific CAMs on the surface of endothelial cells, endorses this view [18,35]. As such, the report [19] that HDL, whose plasma level has a strong inverse relationship to risk of coronary heart disease [1] and which has diverse antiatherogenic effects on various cell types [36], can attenuate upregulation of CAMs in cultured HUVECs has important implications for atherogenesis, and for its prevention or reversal.
However, although primary cultures of HUVECs are a well-established system to study human vascular en-dothelial cell biology in vitro and have been used extensively in CAM research [20], HUVECs are an imperfect model in which to test anti-atherogenic ac-tions of HDL; the cells are close to senescence, are isolated from hypoxic and possibly activated blood vessels [37], and are derived from an endothelium which is not susceptible to coronary atherosclerosis. Here, we have examined HDL effects on CAM expression using primary cultures of human coronary artery endothelial cells which are a model directly relevant to blood vessels affected by atherosclerosis [22,23]. Nevertheless, in four independent experiments, which used different plasma donors and batches of HCAECs, we failed to demonstrate a reduced induction of VCAM-1 or E-se-lectin by TNF-a when cells were preincubated with HDL, or with its two major subfractions, HDL2 and HDL3.
We then tried to reproduce the original observation of VCAM-1 suppression in HUVECs [19,27]. However, although our cells were carefully characterized and of low-passage number, neither ELISA or FACS analysis could reveal an inhibition of VCAM-1 expression by prolonged preincubation of cells with HDL. Indeed, a modest induction (7%, PB0.01) above the cytokine-stimulated level of VCAM-1 was seen after pretreat-ment with HDL (1.5 mg protein/ml), whereas an inhibition of about 90% had been anticipated [19]. This inability of HDL to downregulate VCAM-1 was also seen with suboptimal doses of TNF-a when only lim-ited induction occurred.
Our discrepant data were not due to a general poor responsiveness of the HUVECs. Induction of CAM expression by IL-1b or TNF-a was consistent with other reports [19,20], while positive controls established that this expression was subject to downregulation; addition of the nitric oxide donor, GSNO or of 17b -estradiol suppressed TNF-a-induced VCAM-1 by
20.993.3 and 60.493.0%, respectively (both PB
0.001) [24]. Furthermore, we recently reported VCAM-1 suppression in HUVECs by cell-derived, minimally-lipidated apo E particles [38]. One possible explanation, that could be explored further, is that other groups have supplemented culture media with supra-physiological doses of heparin [19,27,39,40]. However, as excess heparin has the ability to modulate ICAM-1 transcription [41] and to bind TNF-a, thus limiting its availability to interact with endothelial cells [42], heparin supplementation was omitted from our experiments.
It is also unlikely that our HDL lacked biological potency; previous preparations, using identical method-ology, have shown characteristic functional properties towards several cell types, including those cultured in vitro [4,25,43] or used ex vivo [6,44,45]. Donor varia-tion, which is reported to influence inhibitory activity [27], was excluded since we sampled HDL from 14 healthy individuals, both male and female with ages ranging from 20 to 50 years, while procedural changes to reduce isolation time also failed to yield HDL capa-ble of inhibiting CAM expression. In addition, all HDL preparations had a minimal effect on basal VCAM-1 expression (Fig. 3A); an important observation, as con-tamination of HDL with endotoxin or oxidation prod-ucts might have stimulated VCAM-1 and E-selectin expression and hence counteracted any downregulatory effect of the HDL. Finally, none of our experimental conditions caused cytotoxic effects, as judged by micro-scopic examination of cells, and by assays for cell viability and loss of cellular protein.
In agreement with our findings, there is one prelimi-nary report that apo A-I fails to prevent monocyte binding to human aortic endothelial cell monolayers [46]. However, although we are unaware of additional studies of HDL effects on CAM expression in HU-VECs, several groups have studied reconstituted HDL particles (rHDL), mainly apo A-I – phospholipid com-plexes. These studies have been somewhat inconsistent, possibly because procedures used to isolate and purify apolipoproteins may result in a variable biological ac-tivity [47]. Thus, while the original authors suggested that downregulatory effects of rHDL on CAMs were comparable to native HDL [19], there are other reports that rHDL only partially reduces (30%) VCAM-1 in HUVECs [39,40], while even phospholipid alone has mild inhibitory effects [39]; in addition, suppression of ICAM-1 and E-selectin is marginal (B10%) [48].
overex-pressing apo A-I, atheroma formation was inhibited
after endothelial activation and monocyte adherence [49]. By contrast, others report that elevating HDL levels, by either apo A-I overexpression [50] or rHDL infusion [51], does suppress VCAM-1 in apo E-deficient mice, although it was not possible to distinguish be-tween direct and indirect effects. Thus, the reduction of VCAM-1 expression could reflect the antioxidant prop-erties of HDL, as this diminishes both oxidative stress and production of pro-inflammatory lipoproteins [50,51].
In summary, although the hypothesis that HDL di-minishes expression of CAMs in arterial endothelium, a prerequisite for capture of circulating leukocytes, is an attractive explanation for HDL antiatherogenicity, we found no evidence for such a regulatory effect in our model of human arterial endothelial cells in vitro. Thus, although further studies are needed to understand HDL interactions with vascular endothelial cells, we tenta-tively conclude that attenuation of CAM induction in arterial endothelium is unlikely to contribute to protec-tive actions of HDL in vivo.
Acknowledgements
We thank the British Heart Foundation for support-ing A.K. Stannard with a Ph.D. Studentship (FS/
95051) and A. Graham with an Intermediate Fellowship (FS/95026). We are grateful to Dr D.G. Hassall (Glaxo Wellcome Research and Development, Stevenage, UK) for help with flow cytometry measure-ments of HUVECs.
References
[1] Gordon DJ, Probstfield JL, Garrison RJ, et al. High-density lipoprotein cholesterol and cardiovascular disease. Four prospec-tive American studies. Circulation 1989;79:8 – 15.
[2] Fielding CJ, Fielding PE. Molecular physiology of reverse cholesterol transport. J Lipid Res 1995;36:211 – 28.
[3] Gillotte KL, Davidson WS, Lund-Katz S, Rothblat GH, Phillips MC. Removal of cellular cholesterol by pre-beta-HDL involves plasma membrane microsolubilization. J Lipid Res 1998;39:1918 – 28.
[4] Graham A, Hassall DG, Rafique S, Owen JS. Evidence for a paraoxonase-independent inhibition of low-density lipoprotein oxidation by high-density lipoprotein. Atherosclerosis 1997;135:193 – 204.
[5] Mackness MI, Durrington PN. HDL, its enzymes and its poten-tial to influence lipid peroxidation. Atherosclerosis 1995;115:243 – 53.
[6] Desai K, Bruckdorfer KR, Hutton RA, Owen JS. Binding of apoE-rich high density lipoprotein particles by saturable sites on human blood platelets inhibits agonist-induced platelet aggrega-tion. J Lipid Res 1989;30:831 – 40.
[7] Nofer JR, Walter M, Kehrel B, Wierwille S, Tepel M, Seedorf U, Assmann G. HDL3-mediated inhibition of thrombin-induced
platelet aggregation and fibrinogen binding occurs via decreased production of phosphoinositide-derived second messengers 1,2-diacylglycerol and inositol 1,4,5-tris-phosphate. Arterioscler Thromb Vasc Biol 1998;18:861 – 9.
[8] Rubin EM, Krauss RM, Spangler EA, Verstuyft JG, Clift SM. Inhibition of early atherogenesis in transgenic mice by human apolipoprotein AI. Nature 1991;353:265 – 7.
[9] Plump AS, Scott CJ, Breslow JL. Human apolipoprotein A-I gene expression increases high density lipoprotein and suppresses atherosclerosis in the apolipoprotein E-deficient mouse. Proc Natl Acad Sci USA 1994;91:9607 – 11.
[10] Liu AC, Lawn RM, Verstuyft JG, Rubin EM. Human protein A-I prevents atherosclerosis associated with apolipo-protein[a] in transgenic mice. J Lipid Res 1994;35:2263 – 7. [11] Badimon JJ, Badimon L, Fuster V. Regression of atherosclerotic
lesions by high density lipoprotein plasma fraction in the choles-terol-fed rabbit. J Clin Invest 1990;85:1234 – 41.
[12] Imhof BA, Dunon D. Leukocyte migration and adhesion. Adv Immunol 1995;58:345 – 416.
[13] van der Wal AC, Das PK, Tigges AJ, Becker AE. Adhesion molecules on the endothelium and mononuclear cells in human atherosclerotic lesions. Am J Pathol 1992;141:1427 – 33. [14] O’Brien KD, Allen MD, McDonald TO, et al. Vascular cell
adhesion molecule-1 is expressed in human coronary atheroscle-rotic plaques. Implications for the mode of progression of ad-vanced coronary atherosclerosis. J Clin Invest 1993;92:945 – 51. [15] Johnson-Tidey RR, McGregor JL, Taylor PR, Poston RN.
Increase in the adhesion molecule P-selectin in endothelium overlying atherosclerotic plaques. Coexpression with intercellular adhesion molecule-1. Am J Pathol 1994;144:952 – 61.
[16] Osborn L, Hession C, Tizard R, Vassallo C, Luhowskyj S, Chi-Rosso G, Lobb R. Direct expression cloning of vascular cell adhesion molecule 1, a cytokine-induced endothelial protein that binds to lymphocytes. Cell 1989;59:1203 – 11.
[17] Elices MJ, Osborn L, Takada Y, Crouse C, Luhowskyj S, Hemler ME, Lobb RR. VCAM-1 on activated endothelium interacts with the leukocyte integrin VLA-4 at a site distinct from the VLA-4/fibronectin binding site. Cell 1990;60:577 – 84. [18] Nakashima Y, Raines EW, Plump AS, Breslow JL, Ross R.
Upregulation of VCAM-1 and ICAM-1 at atherosclerosis-prone sites on the endothelium in the ApoE-deficient mouse. Arte-rioscler Thromb Vasc Biol 1998;18:842 – 51.
[19] Cockerill GW, Rye KA, Gamble JR, Vadas MA, Barter PJ. High-density lipoproteins inhibit cytokine-induced expression of endothelial cell adhesion molecules. Arterioscler Thromb Vasc Biol 1995;15:1987 – 94.
[20] Klein CL, Ko¨hler H, Bittinger F, Wagner M, Hermanns I, Grant K, Lewis JC, Kirkpatrick CJ. Comparative studies on vascular endothelium in vitro. I. Cytokine effects on the expres-sion of adheexpres-sion molecules by human umbilical vein, saphenous vein and femoral artery endothelial cells. Pathobiology 1994;62:199 – 208.
[21] Fina L, Molgaard HV, Robertson D, et al. Expression of the CD34 gene in vascular endothelial cells. Blood 1990;75:2417 – 26. [22] Allen S, Khan S, Al-Mohanna F, Batten P, Yacoub M. Native low density lipoprotein-induced calcium transients trigger VCAM-1 and E-selectin expression in cultured human vascular endothelial cells. J Clin Invest 1998;101:1064 – 75.
[23] Allen S, Khan S, Tam S, Koschinsky M, Taylor P, Yacoub M. Expression of adhesion molecules by Lp(a): a potential novel mechanism for its atherogenicity. FASEB J 1998;12:1765 – 76. [24] Stannard AK, Riddell DR, Bradley NJ, Hassall DG, Graham A,
Owen JS. Apolipoprotein E and regulation of cytokine-induced cell adhesion molecule expression in endothelial cells. Atheroscle-rosis 1998;139:57 – 64.
disease regulate cholesterol metabolism in cultured human skin fibroblasts. J Lipid Res 1984;25:919 – 31.
[26] Desai K, Mistry P, Bagget C, Burroughs AK, Bellamy MF, Owen JS. Inhibition of platelet aggregation by abnormal high density lipoprotein particles in plasma from patients with hepatic cirrhosis. Lancet 1989;1:693 – 5.
[27] Ashby DT, Rye KA, Clay MA, Vadas MA, Gamble JR, Barter PJ. Factors influencing the ability of HDL to inhibit expression of vascular cell adhesion molecule-1 in endothelial cells. Arte-rioscler Thromb Vasc Biol 1998;18:1450 – 5.
[28] Caulin-Glaser T, Watson CA, Pardi R, Bender JR. Effects of 17beta-estradiol on cytokine-induced endothelial cell adhesion molecule expression. J Clin Invest 1996;98:36 – 42.
[29] Nakai K, Itoh C, Hotta K, Itoh T, Yoshizumi M, Hiramori K. Estradiol-17 beta regulates the induction of VCAM-1 mRNA expression by interleukin-1 beta in human umbilical vein en-dothelial cells. Life Sci 1994;54:L221 – 7.
[30] De Caterina R, Libby P, Peng HB, Thannickal VJ, Rajavashisth TB, Gimbrone MA, Shin WS, Liao JK. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest 1995;96:60 – 8. [31] Khan BV, Harrison DG, Olbrych MT, Alexander RW, Medford
RM. Nitric oxide regulates vascular cell adhesion molecule 1 gene expression and redox-sensitive transcriptional events in human vascular endothelial cells. Proc Natl Acad Sci USA 1996;93:9114 – 9.
[32] Quyyumi AA. Endothelial function in health and disease: new insights into the genesis of cardiovascular disease. Am J Med 1998;105:32S.
[33] Liao JK. Endothelium and acute coronary syndromes. Clin Chem 1998;44:1799 – 808.
[34] Fang X, Weintraub NL, Rios CD, et al. Overexpression of human superoxide dismutase inhibits oxidation of low-density lipoprotein by endothelial cells. Circ Res 1998;82:1289 – 97. [35] Sakai A, Kume N, Nishi E, Tanoue K, Miyasaka M, Kita T.
P-selectin and vascular cell adhesion molecule-1 are focally ex-pressed in aortas of hypercholesterolemic rabbits before intimal accumulation of macrophages and T lymphocytes. Arterioscler Thromb Vasc Biol 1997;17:310 – 6.
[36] Barter PJ, Rye KA. High density lipoproteins and coronary heart disease. Atherosclerosis 1996;121:1 – 12.
[37] Garlanda C, Dejana E. Heterogeneity of endothelial cells. Spe-cific markers. Arterioscler Thromb Vasc Biol 1997;17:1193 – 202. [38] Stannard AK, Riddell DR, Tagalakis AD, Athanasopoulos T, Dickson JG, Owen JS. Locally secreted apolipoprotein (apo)E inhibits vascular cell adhesion molecule-1 (VCAM-1) expression in human endothelial cells. Atherosclerosis 1999;144:86. [39] Calabresi L, Franceschini G, Sirtori CR, De Palma A, Saresella
M, Ferrante P, Taramelli D. Inhibition of VCAM-1 expression in endothelial cells by reconstituted high density lipoproteins.
Biochem Biophys Res Commun 1997;238:61 – 5.
[40] Baker PW, Rye KA, Gamble JR, Vadas MA, Barter PJ. Ability of reconstituted high density lipoproteins to inhibit cytokine-in-duced expression of vascular cell adhesion molecule-1 in human umbilical vein endothelial cells. J Lipid Res 1999;40:345 – 53. [41] Miller SJ, Hoggatt AM, Faulk WP. Heparin regulates ICAM-1
expression in human endothelial cells: an example of non-cy-tokine-mediated endothelial activation. Thromb Haemost 1998;80:481 – 7.
[42] Lantz M, Thysell H, Nilsson E, Olsson I. On the binding of tumor necrosis factor (TNF) to heparin and the release in vivo of the TNF-binding protein I by heparin. J Clin Invest 1991;88:2026 – 31.
[43] Ong AC, Jowett TP, Moorhead JF, Owen JS. Human high density lipoproteins stimulate endothelin-1 release by cultured human renal proximal tubular cells. Kidney Int 1994;46:1315 – 21.
[44] Owen JS, Brown DJ, Harry DS, McIntyre N, Beaven GH, Isenberg H, Gratzer WB. Erythrocyte echinocytosis in liver disease. Role of abnormal plasma high density lipoproteins. J Clin Invest 1985;76:2275 – 85.
[45] Gillett MP, Owen JS. Comparison of the cytolytic effects in vitro on Trypanosoma brucei brucei of plasma, high density lipo-proteins, and apolipoprotein A-I from hosts both susceptible (cattle and sheep) and resistant (human and baboon) to infec-tion. J Lipid Res 1992;33:513 – 23.
[46] Hama SY, Cardinez CJ, Hough GP, Castellani LW, Lusis AJ, Laks H, Fogelman AM. Incubation with apolipoprotein A-II markedly induces monocyte adhesion to human aortic endothe-lial cells. FASEB J 1998;12:2807.
[47] LaDu MJ, Pederson TM, Frail DE, Reardon CA, Getz GS, Falduto MT. Purification of apolipoprotein E attenuates iso-form-specific binding to beta-amyloid. J Biol Chem 1995;270:9039 – 42.
[48] Moudry R, Spycher MO, Doran JE. Reconstituted high density lipoprotein modulates adherence of polymorphonuclear leuko-cytes to human endothelial cells. Shock 1997;7:175 – 81. [49] Dansky HM, Charlton SA, Barlow CB, Tamminen M, Smith
JD, Frank JS, Breslow JL. Apo A-I inhibits foam cell formation in apo E-deficient mice after monocyte adherence to endothe-lium. J Clin Invest 1999;104:31 – 9.
[50] Theilmeier G, Van Veldhoven PP, Michiels C, Collen D, Him-pens B, Holvoet P. Human-like HDL in vivo reduces oxidative stress, Ca2+ transients, adhesion molecule expression and
macrophage homing in apoE deficient mice. Circulation (Suppl.) I 100:189.
[51] Dimayuga P, Zhu J, Oguchi S, et al. Reconstituted HDL con-taining human apolipoprotein A-1 reduces VCAM-1 expression and neointima formation following periadventitial cuff-induced carotid injury in apoE null mice. Biochem Biophys Res Commun 1999;264:465 – 8.