IDENTIFIKASI OTOMATIS GALUR MENCIT DAN TIKUS MENGGUNAKAN
SEQUENCE DNA DENGAN PENDEKATAN DYNAMIC PROGRAMMING
SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Komputer
Jurusan Teknik Informatika
Oleh :
Tulus Wardoyo
NIM : 065314073
PROGRAM STUDI TEKNIK INFORMATIKA
JURUSAN TEKNIK INFORMATIKA
FAKULTAS SAINS DAN TEKNOLOGI
UNIVERSITAS SANATA DHARMA
YOGYAKARTA
AUTOMATIC IDENTIFICATION OF STRAINS OF MICE AND RATS BY DNA SEQUENCES USING DYNAMIC PROGRAMMING APPROACH
A THESIS
Presented as Partial Fulfillment of the Requirements
To Obtain the Sarjana Komputer Degree
In Department of Informatics Engineering
By :
Tulus Wardoyo
NIM : 065314073
INFORMATICS ENGINEERING STUDY PROGRAM
INFORMATICS ENGINEERING DEPARTMENT
FACULTY OF SCIENCE AND TECHNOLOGY
SANATA DHARMA UNIVERSITY
YOGYAKARTA
“Hidup bukanlah perjuangan menghadapi badai, tapi bagaimana tetap menari di tengah hujan.”
PERNYATAAN KEASLIAN KARYA
Saya menyatakan sesungguhnya bahwa skripsi yang saya tulis ini tidak memuat
karya orang lain kecuali telah disebutkan dalam kutipan atau daftar pustaka,
sebagaimana layaknya karya ilmiah.
Yogyakarta, 9 April 2012,
Penulis
PERNYATAAN PERSETUJUAN
PUBLIKASI KARYA ILMIAH UNTUK KEPENTINGAN AKADEMIS Yang bertanda tangan di bawah ini, saya mahasiswa Universitas Sanata Dharma :
Nama : Tulus Wardoyo
NIM : 065314073
Demi pengembangan ilmu pengetahuan,saya memberikan kepada perpustakaan
Universitas Sanata Dharma karya ilmiah saya yang berjudul :
IDENTIFIKASI OTOMATIS GALUR MENCIT DAN TIKUS MENGGUNAKAN SEQUENCE DNA DENGAN PENDEKATAN
DYNAMIC PROGRAMMING
Beserta perangkat yang diperlukan (bila ada). Dengan demikian saya memberikan
kepada Perpustakaan Universitas Sanata Dharma hak untuk menyimpan,
mengalihkan dalam bentuk media lain, mengelolanya dalam bentuk pangkalan
data, mendistribusikannya secara terbatas dan mempublikasikannya di internet
atau media lain untuk kepentingan akademis tanpa perlu meminta ijin dari saya
maupun memberikan royalty kepada saya selama tetap mencantumkan nama saya
sebagai penulis. Demikian pernyataan ini yang saya buat dengan sebenarnya.
Yogyakarta, 9 April 2012,
Penulis,
ABSTRAK
Dalam pengembangan bahan obat diperlukan uji praklinik yang
membutuhkan hewan percobaan bergalur murni. Penelitian ini membangun suatu
sistem indentifikasi secara otomatis tikus dan mencit bergalur murni
menggunakan pendekatan Dynamic Programming. Penelitian ini menggunakan
data sequence DNA 3 mencit dengan berbeda galur, tikus, dan tikus Lemur,
dengan jumlah total 300 sequence DNA mencit dan tikus. Berdasarkan hasil
penelitian yang dilakukan dengan kombinasi dari 3 metode multiple sequence
alignment dan 2 metode consensus, diperoleh akurasi bervariasi dari yang
terendah yaitu 38,000 % sampai dengan yang tertinggi 81,667 % .
Kata kunci : sequence DNA, Bioinformatika, galur mencit, Dynamic
Programming, sequence alignment, sequence consensus, multiple sequence
ABSTRACT
In the development of pharmaceuticals needed preclinic trials that require
animal experiments fluted pure. This research builds a system to automatically
identify rats and mice using fluted pure Dynamic Programming approach. This
research uses DNA sequence of data with three different strains of mice, rats, and
Lemurs, with a total of 300 sequence the DNA of mice and rats. Based on the
results of the research conducted with combination of three multiple sequence
alignment method and two method sequence consensus, obtained accuracy varies
from the lowest i.e. 38,000% up to the highest 81,667%.
Keywords : DNA sequence, Bioinformatics, a strain of mice, Dynamic
Programming, sequence alignment, sequence consensus, multiple sequence
KATA PENGANTAR
Puji syukur kepada Tuhan Yang Maha Esa yang telah memberikan segala
karunia-Nya sehingga penulis dapat menyelesaikan skripsi dengan judul “Identifikasi
Otomatis Galur Mencit dan Tikus Menggunakan Sequence DNA dengan Pendekatan Dynamic Programming”. Dalam kesempatan ini, penulis ingin mengucapkan terima kasih yangs ebesar-besarnya kepada semua pihak yang turut
memberikan dukungan, semangat dan bantuan hingga selesainya skripsi ini:
1. Romo Dr. Cyprianus Kuntoro Adi, S.J., M.A., M.Sc. selaku dosen
pembimbing, terimakasih atas segala bimbingan dan kesabaran dalam
mengarahkan dan membimbing penulis dalam menyelesaikan tugas akhir
ini.
2. Ibu P.H. Prima Rosa, S.Si., M.Sc selaku Dekan Fakultas Sains dan
Teknologi Universitas Sanata Dharma Yogyakarta.
3. Ibu Ridowati Gunawan, S.Kom., M.T. selaku kaprodi Teknik Informatika.
4. Bapak J. Eka Priyatma, M.Sc., Ph.D. dan Bapak Puspaningtyas Sanjoyo
Adi, S.T., M.T. selaku dosen penguji.
5. Seluruh staff pengajar Prodi Teknik Informatika Fakultas Sains dan
Teknologi Universitas Sanata Dharma.
6. Kedua orang tua saya yang tercinta, yang telah memberi dukungan kepada
7. Eka Permatasari, Siwi Febrianti, Widiyo Sutoto, dan Ferdinandus Andaru
Prima Yudha yang sudah banyak membantu dalam proses pembuatan
skripsi ini, atas dukungan, semangat, dan bantuan.
8. Teman-teman prodi Teknik Informatika angkatan 2006, atas
kebersamaanya selama penulis menjalani masa studi.
9. Serta semua pihak yang tidak dapat disebutkan satu-persatu yang telah
membantu penulis dalam menyelesaikan skripsi ini.
Penulis menyadari bahwa tugas akhir ini jauh dari sempurna, oleh karena itu
kritik dan saran yang sifatnya membangun sangat penulis harapkan.
Akhir kata, penulis berharap semoga skripsi ini dapat bermanfaat bagi
pembaca dan pihak lain yang membutuhkannya.
Yogyakarta, 23 April 2012,
DAFTAR ISI
HALAMAN JUDUL ... i
HALAMAN JUDUL (INGGRIS) ... ii
HALAMAN PERSETUJUAN ... iii
HALAMAN PENGESAHAN ... iv
PERNYATAAN KEASLIAN KARYA ... vi
PERNYATAAN PERSETUJUAN ... vii
ABSTRAK ... viii
ABSTRACT ... ix
KATA PENGANTAR ... x
DAFTAR GAMBAR ... xv
DAFTAR TABEL ... xix
Glosarium ... xx
BAB I : PENDAHULUAN ... 1
1.1. Latar Belakang... 1
1.2. Rumusan Masalah. ... 2
1.3. Tujuan Penelitian. ... 3
1.4. Batasan Masalah. ... 3
1.5. Metodologi Penelitian. ... 3
BAB II : LANDASAN TEORI ... 6
2.1. Pengenalan Pola Dalam Bioinformatika. ... 6
2.2. Pengertian DNA. ... 7
2.3. Format Fasta dan Format Stockholm. ... 10
2.4. Sequence Alignment.... 13
2.5. Multiple Sequence Alignment. ... 14
2.6. Sequence Consensus. ... 37
2.7. Substitution Matrix. ... 49
2.8. Dynamic Programming. ... 49
2.9. Komplesitas Waktu. ... 59
BAB III : METODOLOGI PENELITIAN ... 61
3.1. Data Mencit Dan Tikus. ... 61
3.2. 5 Fold Cross-Validation. ... 62
3.3. Proses Training. ... 64
3.4. Proses Testing. ... 65
3.5. Perancangan Sistem. ... 67
3.6. Spesifikasi Hardware. ... 71
BAB IV : IMPLEMENTASI DAN ANALISA HASIL ... 72
4.1. Hasil dan Analisis. ... 72
4.3. Kompleksitas Waktu Asimptotik. ... 89
BAB V : PENUTUP ... 97
5.1. Kesimpulan ... 97
5.2. Saran ... 98
DAFTAR PUSTAKA ... 99
LAMPIRAN ... 103
A. Lampiran Proses Preprosesing. ... 104
DAFTAR GAMBAR
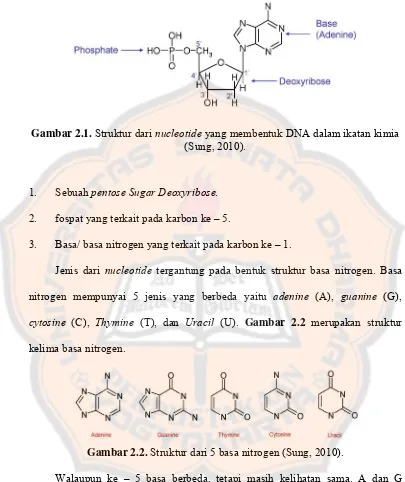
Gambar 2.1. Struktur dari nucleotide yang membentuk DNA dalam ikatan kimia
(Sung, 2010). ... 8
Gambar 2.2. Struktur dari 5 basa nitrogen (Sung, 2010). ... 8
Gambar 2.3. Contoh untaian DNA yang dibentuk oleh 5 nucleotide yang saling berikatan, dan itu terdiri dari sebuah backbone phosphate-sugar dan 5 basa (Sung, 2010). ... 9
Gambar 2.4. Sequence DNA dalam Format FASTA. ... 10
Gambar 2.5. Contoh Format Stockholm yang paling sederhana (Eddy, 2005). .. 11
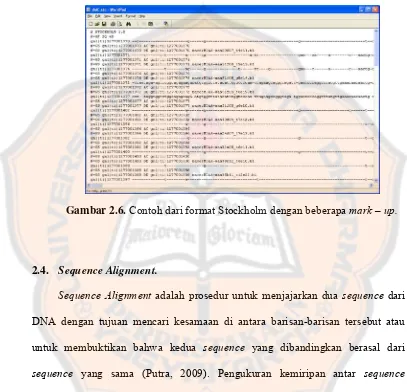
Gambar 2.6. Contoh dari format Stockholm dengan beberapa mark – up. ... 13
Gambar 2.7. Sequence DNA untuk global alignment ... 14
Gambar 2.8. Sequence DNA untuk Local alignment. ... 14
Gambar 2.9. Contoh perhitungan jarak dari P – Distance... 15
Gambar 2.10. Contoh penentuan letak root dari sebuah tree unrooted (Stewart, 2004). ... 17
Gambar 2.11. Tree dari hasil proses neighbor – join dan midpoint (Singh, 2000). ... 18
Gambar 2.12. Sebuah sequence alignment dengan gap. ... 19
Gambar 2.13. Konversi sequence DNA ke sequence profile ... 20
Gambar 2.14. Gambaran proses Multidimensional dynamic programming. ... 21
Gambar 2.15. Gambaran Algoritma Muscle (Edgar, 2004b). ... 22
Gambar 2.16. Gambaran dari subsequencek -mer dengan k = 6. ... 23
Gambar 2.18. Bagian A menunjukan proses progressive alignment. Sedangkan
bagian B menunjukan proses iterative refinement (Katoh, 2008)... 28
Gambar 2.19. Konversi sequence DNA ke urutan vector 4 dimensi. ... 30
Gambar 2.20. Sebuah contoh dari optimasi segment sequence homolog dengan DP (Katoh, 2002). ... 32
Gambar 2.21. Perhitungan dari Position Weight Matrix (Sung, 2010). ... 37
Gambar 2.22. Markov chains untuk DNA (Attaluri, 2007). ... 38
Gambar 2.23. Model HMM dari kedua koin (Attaluri, 2007). ... 40
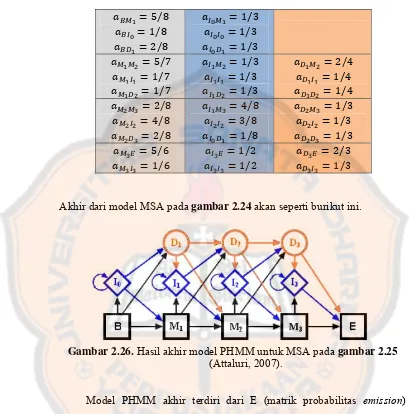
Gambar 2.24. Struktur model PHMM (Attaluri, 2007). ... 42
Gambar 2.25. MSA dari alignment DNA (Attaluri, 2007). ... 43
Gambar 2.26. Hasil akhir model PHMM untuk MSA pada gambar 2.25 (Attaluri, 2007). ... 45
Gambar 2.27. Peta rute perjalanan (Wahyujati, 2005). ... 50
Gambar 2.28. Tahapan pencarian rute terpendek (Wahyujati, 2005). ... 51
Gambar 2.29. Solusi terakhir yang merupakan jalur terpendek ... 53
Gambar 2.30. Contoh alignment dari global alignment. ... 54
Gambar 2.31. Contoh alignment dari local alignment... 54
Gambar 2.32. Pengisian gap pinalti dalam matrik. ... 55
Gambar 2.33. Matrik nilai yang sudah terisi. ... 57
Gambar 2.34. Pointer nilai optimal. ... 58
Gambar 2.35. Sequencealignment optimal. ... 59
Gambar 3.2. Pembagian data 5 fold cross-validation. ... 62
Gambar 3.3. Pembagian data training dan testing pada setiap modelnya. ... 63
Gambar 3.4. Proses Training untuk setiap model. ... 64
Gambar 3.5. Contoh proses pembentukan model/ consensus. ... 65
Gambar 3.6. Proses testing /proses Pengenalan dalam setiap model. ... 66
Gambar 3.7. Contoh proses pengenalan dengan setiap model. ... 66
Gambar 3.8. Halaman Depan /Halaman Menu Utama. ... 67
Gambar 3.9. Halaman Proses Multiple Sequence Alignment. ... 68
Gambar 3.10. Halaman pembentukan sequence consensus. ... 68
Gambar 3.11. Halaman konversi file fasta ke Stockholm... 69
Gambar 3.12. Halaman Pengenalan Tunggal galur mencit dan Tikus. ... 69
Gambar 3.13. Halaman Bantuan Program. ... 70
Gambar 4.1. Grafik akurasi ke – 6 skenario yang sudah dilakukan. ... 76
Gambar 4.2. Implementasi Halaman Depan. ... 79
Gambar 4.3. Implementasi Halaman MSA. ... 80
Gambar 4.4. Implementasi Halaman Consensus. ... 81
Gambar 4.5. Implementasi Halaman Fasta to Stockholm. ... 82
Gambar 4.6. Implementasi Halaman Pengenalan Tikus dan Mencit. ... 83
Gambar 4.7. Implementasi Halaman Bantuan Program. ... 83
Gambar 4.8. Implementasi Halaman open file. ... 84
Gambar 4.9. Implementasi Halaman Save File. ... 84
Gambar 4.10. Eror – Handling Save 1. ... 85
Gambar 4.12. Eror – Handling Save 3. ... 86
Gambar 4.13. Eror Handling Peringatan pada MSA. ... 86
Gambar 4.14. Eror Handling pada Halaman Consensus 1... 87
Gambar 4.15. Eror – Handling pada Halaman Consensus 2... 87
Gambar 4.16. Progress Bar Perhitungan Jarak. ... 88
Gambar 4.17. Progress Bar Progressive Alignment. ... 88
DAFTAR TABEL
Tabel 2.1. Kompleksitas Waktu MSA Phylogenetic. ... 21
Tabel 2.2. Kompleksitas Waktu MSA Muscle. ... 27
Tabel 2.3. Probabilitas emission dari profile HMM pada gambar 2.25. ... 44
Tabel 2.4. Probabilitas transisi dari PHMM pada gambar 2.24. ... 45
Tabel 2.5. Jarak tiap node. ... 51
Tabel 2.6. Gambaran pemecahan pada stage 1 ... 52
Tabel 2.7. Gamabaran pemecahan solusi stage 2. ... 52
Tabel 2.8. Perhitungan pencarian solusi stage 3. ... 53
Tabel 2.9. Perbandingan pertumbuhan T(n) dengan n2 ... 60
Tabel 3.1. Confusion Matrix. ... 63
Tabel 3.2. Enam skenario pemodelan /pembentukan consensus. ... 65
Tabel 4.1. Enam skenario kombinasi metode preprosessing. ... 72
Tabel 4.2. Tabel hasil akurasi dari ke 6 skanario. ... 73
Tabel 4.3. Confusion matrix MAFFT – HMMER. ... 77
Glosarium
Efikasi = Kemampuan obat untuk menghasilkan efek
terapeutik yang diinginkan.
Farmakokinetik = Kerja obat dalam tubuh selama periode waktu,
termasuk proses penyerapan distribusi, lokalisasi,
dalam jariangan biotransformasi dan ekskresi.
Homolog = Gen yang sama dalam struktur dan asal usul
evolusi dengan gen spesies lain.
Isolasi = Extraksi kimia pada subtansi yang tidak diketahui
dalam bentuk dari jaringan.
Kultur = Perkembang biakan mikroorganisme atau jaringan
sel hidup pada media yuang menyebabkan
pertumbuhanya.
Kultur sel = Pertumbuhan sel,
Reseptor = Molekul dipermukaan atau didalam sitoplasma sel
yang mengenal dan mengikat molekul spesifik,
menghasilkan efek khusus dalam sel.
Sintesis = terbentuknya senyawa dengan penyatuan elemen
penyusunnya, dilakukan secara buatan atau
sebagaian hasil proses alami.
Toksis = bersifat racun.
Toksisitas = kualitas bersifat racun, khususnya derajat virulensi mikroba toksis atau racun.
BAB I
PENDAHULUAN
1.1. Latar Belakang.
Pengembangan suatu obat berawal dari sintesis atau isolasi berbagai bahan
untuk memunculkan calon obat. Sebelum calon obat resmi menjadi obat,
diperlukan uji yang sering memakan waktu yang panjang dan biaya yang tidak
sedikit (Sukandar, 2004). Uji yang harus ditempuh oleh calon obat ada dua uji
prakinik dan uji klinik.
1. Uji Praklinik
Uji praklinik merupakan persyaratan uji untuk calon obat. Dari uji ini
diperoleh informasi tentang efikasi ( efek farmakologi), profil farmakokinetik dan
toksisitas calon obat (Sukandar, 2004). Pada uji ini diperlukan hewan yang
bergalur murni /hewan utuh seperti mencit, tikus, kelinci, marmot, hamster,
anjing atau beberapa uji menggunakan primata.
2. Uji Klinik
Uji klinik merupakan kelajutan dari uji praklinik, setelah calon obat
dinyatakan mempunyai kemanfaatan dan aman pada hewan percobaan. Uji klinik
pada manusia perlu melewati proses yang dilakukan oleh komite etik mengikuti
Deklarasi Helsinki. (Sukandar, 2004).
Uji praklinik memerlukan hewan percobaan salah satunya tikus atau mencit.
Melalui tikus bergalur murni dapat diketahui apakah obat tersebut aman, ataukah
obat dapat dievaluasi. Hasil pengamatan pada hewan menentukan apakah dapat
diteruskan dengan uji pada manusia. Oleh karena itu sangat penting untuk
mengetahui tikus yang dipakai bergalur murni.
Genom atau cetak biru informasi genetik menentukan sifat setiap makhluk
hidup yang disandi dalam bentuk pita molekul DNA. Melalui perhitungan
sequence alignment informasi makhluk hidup bisa ditentukan. Oleh karena itu
DNA dapat digunakan untuk membedakan galur seekor tikus. Namun analisis
molekul DNA yang dilakukan secara manual seringkali memerlukan waktu yang
lama karena panjangnya sequence DNA. Sebagai contoh DNA mikro organisme
mempunyai panjang sequence DNA 106 karakter (Lesk, 2005). Maka dari itu,
perlu dikembangkan sebuah sistem yang mampu mengidentifikasi sequence DNA
tikus secara otomatis. Penelitian ini akan membangun sistem pengenalan otomatis
berdasar pendekatan dynamic programming. Berdasarkan penelitian yang ada,
metode ini mampu memberikan hasil optimal dalam analisis sequence DNA
(Lesk, 2005).
1.2. Rumusan Masalah.
Berdasarkan latar belakang diatas, masalah dapat dirumuskan berikut :
1. Bagaimana cara merancang sistem yang dapat mengenali sequence
DNA tikus dengan cepat ?
2. Bagaimana cara mengukur kemiripan sequence DNA mencit galur
murni ?
1.3. Tujuan Penelitian.
Tujuan dari penelitian adalah membangun suatu sistem yang secara otomatis
mampu mengidentifikasi sequence DNA dari 2 jenis tikus dan 3 jenis mencit galur
murni.
1.4. Batasan Masalah.
Sistem yang akan dikerjakan, mempunyai beberapa batasan sebagai berikut :
1. Data yang dibandingkan hanya berupa sequence DNA tikus, dan
mempunyai format data dengan extensi *.fasta atau*. fa.
2. Inputan data dari sistem ini hanya DNA yang telah mengalami proses
sequencing atau sequence DNA.
3. Sistem ini hanya dapat mengidentifikasi atau mengukur kemiripan
sequence DNA tikus saja.
1.5. Metodologi Penelitian.
Metodologi penelitian yang dilakukan adalah sebagai berikut :
1. Melakukan studi literature, dengan tujuan sebagai berikut :
a. Mempelajari teori sequence DNA analisis.
b. Memahami algoritma – algoritma sequence DNA.
2. Perancangan Sistem.
Setelah memulai tahap literature, selanjutnya dilakukan perancangan
3. Implementasi.
Tahap ini adalah penerapan disain kedalam bentuk program dengan
memanfaatkan bahasa pemrograman yang ada berdasarkan perancangan
sistem.
4. Pengujian dan Evaluasi.
Menganalisa hasil dari proses indetifikasi atau mengukur kemiripan dari
sistem yang telah dibuat sebelum menentukan kesimpulan.
1.6. Sistematika Penulisan.
Untuk memudahkan dalam penyusunan dan pemahaman isi dari skripsi ini,
maka digunakan sistematika penulisan sebagai berikut :
BAB I : PENDAHULUAN.
Bab ini berisi tentang latar belakang masalah, perumusan masalah, batasan
masalah, tujuan, metode penulisan dan keterangan mengenai sistematika
penulisan.
BAB II : LADASAN TEORI.
Bab ini berisikan berisikan tentang landasan teori yang digunakan dalam
analisis, perancangan dan implementasi program serta penulisan isi dari
pembahasan dan evaluasi hasil penelitian.
BAB III : METODOLOGI.
Bab ini berisi tentang data tikus dan mencit, alur proses training dan
BAB IV : IMPLEMENTASI DAN ANALISA HASIL.
Bab ini berisi implementasi antarmuka sistem dan analisa hasil pengujian
sistem.
BAB V : PENUTUP
Bab ini berisi kesimpulan dan saran-saran yang dapat dipertimbangkan
BAB II
LANDASAN TEORI
Pada landasan teori ini akan dijelaskan akan dijelaskan secara singkat hal –
hal yang berkaitan dengan identifikasi sequence DNA, metode – metode pre –
prosesing dan metode pengenalan kemiripan sequence DNA dengan algoritma
Needlemen Wunsch.
2.1. Pengenalan Pola Dalam Bioinformatika.
Pengenalan pola adalah disiplin ilmu yang mengklasifikasikan object
berdasar image, berat atau parameter-parameter yang telah ditentukan ke dalam
sejumlah kategori atau kelas (Priatama, 2010). Pengenalan pola meliputi berbagai
aplikasi dan implementasi dalam kasus-kasus di dunia nyata. Salah satu contoh
penerapan dari pengenalan pola yaitu bioinformatika.
Bioinformatika adalah penggunaan matematika, statistik dan metode
komputer untuk menganalisis biologi, biokimia, dan data biofisik (Priatama,
2010). Ilmu ini mengajarkan aplikasi, analisis, dan mengorganisir miliaran bit
informasi genetik dalam sel mahluk hidup. Studi bioinformatika merupakan
perpaduan oleh studi genomik, biologi komputasi, dan teknologi komputer.
Genomik adalah studi yang berhubungan dengan pemetaan, sekuen, dan analisis
genom. Walaupun belum jelas, secara umum Genomik bisa diartikan sebagai
penggunaan informasi genom secara sistematis, dengan data eksperimental baru
Bioinformatika dapat membantu menjawab pertanyaan seperti apakah gen
baru dianalisis adalah serupa dengan gen apapun sebelumnya dikenal, apakah
urutan protein yang dapat menunjukkan bagaimana fungsi protein, dan apakah gen
diaktifkan pada sel kanker berbeda dari yang diaktifkan dalam yang sehat sel.
2.2. Pengertian DNA.
Asam deoksiribonukleat (deoxyribonucleic acid), atau biasa disebut DNA,
adalah biomolekul yang berupa asam nucleotide (terdapat dalam inti sel atau
nucleus), yang berfungsi untuk menyimpan informasi genetik suatu organisme
(Putra, 2009). DNA dapat memberi informasi tentang sifat – sifat fisik suatu
organism. Pada manusia DNA mampu memberi informasi mengenai tinggi badan,
warna kulit, golongan darah, ataupun bentuk wajah. DNA tidak hanya
menentukan oleh sifat-sifat fisik saja, ia juga mengendalikan ribuan operasi dan
sistem lainnya yang berjalan di dalam sel dan tubuh. Tinggi rendah atau tekanan
darah yang normal seseorang misalnya tergantung pada informasi yang tersimpan
di dalam DNA ( Yahya,2005). DNA terdiri dari dua untaian yang terjalin bersama,
dan membentuk helix ganda. Dalam setiap untaian terdiri dari molekul kecil yang
dinamakan nucleotide (Sung, 2010).
a. Nucleotide.
Nucleotide adalah komponen yang membangun semua molekul asam
nukleat (Sung, 2010). Struktur nucleotide digambarkan pada gambar 2.1. Setiap
Gambar 2.1. Struktur dari nucleotide yang membentuk DNA dalam ikatan kimia (Sung, 2010).
1. Sebuah pentose Sugar Deoxyribose.
2. fospat yang terkait pada karbon ke – 5.
3. Basa/ basa nitrogen yang terkait pada karbon ke – 1.
Jenis dari nucleotide tergantung pada bentuk struktur basa nitrogen. Basa
nitrogen mempunyai 5 jenis yang berbeda yaitu adenine (A), guanine (G),
cytosine (C), Thymine (T), dan Uracil (U). Gambar 2.2 merupakan struktur kelima basa nitrogen.
Gambar 2.2. Struktur dari 5 basa nitrogen (Sung, 2010).
Walaupun ke – 5 basa berbeda, tetapi masih kelihatan sama. A dan G
disebut purine mempunyai 2 cincin dalam strukturnya, sedangkan C, T, dan U
disebut pyrimidine dan mempunyai satu cincin dalam strukturnya. Dalam DNA
Nucleotide dihubungkan dengan ikatan glukosa fosfat, sambungan fosfat
terdapat pada sebuah nucleotide yang terikat karbon 5’ ke ikatan karbon 3’ pada
nucleotide lainnya. Hubungan antar nucleotide dinamakan polynucleotide atau
sering disebut sequence DNA. Gambar 2.3 menunjukan sebuah contoh sequence
rantai DNA yang terdiri dari 5 nucleotide ACGTA.
Gambar 2.3. Contoh untaian DNA yang dibentuk oleh 5 nucleotide yang saling berikatan, dan itu terdiri dari sebuah backbone phosphate-sugar dan 5 basa (Sung,
2010).
b. Struktur DNA.
Bentuk struktur DNA diawali dari dua penelitian yaitu sebagai berikut :
1. E. Chargaff mengemukakan bahwa konsentrasi dari thymine selalu
sama dengan konsentrasi adenine dan konsentrasi cytosine selalu sama
dengan konsentrasi guanine. Penelitaian ini mendukung kuat untuk
menetapkan bahwa A selalu berpasangan dengan T dan C selalu
berpasangan dengan G (Sung, 2010).
2. X – Ray Diffraction pattern oleh R. Franklin, M. H. F. Wilkins, dan
co - workers. Dari data menunjukan bahwa DNA sangat teratur,
dengan struktur beruntai ganda dengan pengulangan untuk setiap
Dari dua penelitian tersebut, Watson dan Crick mengemukakan bahwa
struktur DNA berbentuk helix ganda yang didalamnya terdapat rantai
polynucleotide yang terdiri dari sebuah sequence dari nucleotide – nucleotide
yang diikat oleh ikatan sugar-phosphate. Untuk setiap pasangan basa mempunyai
ketetapan bahwa A selalu berpasangan dengan T dan C selalu berpasangan dengan
G (Sung, 2010).
2.3. Format Fasta dan Format Stockholm.
Dalam Bioinformatika terdapat berbagai format data penyimpanan untuk
sequence DNA, RNA, dan Protein. Dalam penelitian ini penulis hanya
menggunakan dua format data sequence yaitu format FASTA dan Stockholm.
1. Format Fasta.
Format FASTA merupakan format yang sangat umum untuk data sequence
dalam bioinformatika. Format ini berasal dari ketetapan dari FASTA, sebuah
program dari algoritma FAST oleh W. R. Pearson (Lesk, 2005). Penulisan
sequence dari format FASTA terdapat dua bagian. Di awali dengan baris diskripsi,
penulisan dalam baris ini harus diawali dengan tanda lebih dari (>). Untuk
penulisan baris diskripsi ini bebas, tetapi harus informatif dengan data
sequencenya dan baris berikutnya berisi sequence DNA. Gambar 2.4 merupakan sebuah contoh dari sequence DNA yang disimpan dalam format FASTA.
2. Format Stockholm.
Format Stockholm merupakan salah satu format file untuk multiple sequence
alignment. Format ini dipakai oleh HMMER sebagai format standar untuk
multiple sequence aligment. Format Stockholm memiliki dokumentasi secara
detail tentang data yang disimpan didalamnya (Eddy, 2005). Contoh penulisan
format Stockholm ditunjukan pada gambar 2.5.
Gambar 2.5. Contoh Format Stockholm yang paling sederhana (Eddy, 2005).
Terdapat dua bagian utama dalam file Stockholm yang pertama bagian
header yang harus berisi nama format file dan versinya, dan sekarang format
Stockholm baru sampai versi Stockholm 1.0. Bagian kedua dinamakan bagian
sequence alignment. Bagian ini berisi nama sequence, lalu diikuti dengan data
sequence yang telah mengalami proses multiple sequence alignment. Pada baris
terbawah terdapat tanda “ // ” merupakan akhir dari sequencealignment.
Dalam Stockholm yang lengkap terdapat mark – up untuk keterangan
tambahan mengenai sequence alignment. Ada empat macam tipe mark – up dari
a. #=GF merupakan tanda keterangan untuk tiap file, dalam GF terdapat
beberapa tag untuk memperjelas keterangan yang akan ditulis. Berikut ini
tag dari #=GF :
1 #=GF ID
Merupakan tag identitas alignment yang disimpan. Dalam penulisannya harus satu kata dan unik.
2 #=GF AC
Merupakan tag tambahan dukumentasi yang berupa
angka, sebagai kode dari alignment.
3 #=GF DE
Merupakan tag diskripsi tentang alignment yang disimpan.
4 #=GF AU Merupakan tag untuk menunjukan pembuat alignment.
5 #=GF SQ
Merupakan tag untuk menunjukkan berapa jumlah
sequence yang dsimpan dalam satu file.
b. #=GC merupakan tanda catatan untuk tiap kolom, dan dibawah ini tag dari
#=GC :
1 #=GC RF
Merupakan tag untuk acuan untuk menunjukan consensus dari tiap kolom.
c. #=GS merupakan tanda keterangan untuk tiap sequence. Dalam #=GS
terdapat beberapa tag yaitu sebagai berikut :
1 #=GS AC Merupakan tag accession untuk setiap sequencenya
yang berisi dengan kode angka.
2 #=GS DE Merupakan tag diskripsi, berisi keterangan untuk
menjelaskan untuk setiap sequence.
d. #=GR merupakan tanda keterangan untuk tiap residu atau karakternya. Tag
untuk menunjukan label consensus dari suatu sequence. Gambar 2.6 merupakan contoh dari format Stockholm dengan beberapa mark – up.
Contoh :
Gambar 2.6. Contoh dari format Stockholm dengan beberapa mark – up.
2.4. Sequence Alignment.
Sequence Alignment adalah prosedur untuk menjajarkan dua sequence dari
DNA dengan tujuan mencari kesamaan di antara barisan-barisan tersebut atau
untuk membuktikan bahwa kedua sequence yang dibandingkan berasal dari
sequence yang sama (Putra, 2009). Pengukuran kemiripan antar sequence
umumnya menggunakan levenshtein distance/edit distance. Levenshtein distance
adalah menghitung dua string dengan panjang yang berbeda, dan untuk
menemukan kemiripan yang optimal dilakukan “edit operations” (Lesk, 2005).
karakter dalam kedua sequence. Ada dua macam metode sequence aligment,
yaitu :
1. Global alignment
Global alignment adalah penjajaran dua sequence untuk mencari kemiripan
yang optimal antar sequence pada seluruh sequence (Lesk ,2005).
Contoh :
Gambar 2.7. Sequence DNA untuk global alignment
2. Local alignment
Local alignment adalah pencarian alignment terbaik dari beberapa
subsequence dengan subsequence yang lain dari sequence yang berbeda.
(Lesk, 2005).
Contoh :
Gambar 2.8. Sequence DNA untuk Local alignment. 2.5. Multiple Sequence Alignment.
Multiple Sequence Alignment (MSA) adalah kumpulan 3 atau lebih
sequence yang disejajarkan sehingga membentuk matrik persegi panjang
(Edgar,2006) .MSA berfungsi untuk membantu menemukan struktur dan
karakteristik dari protein atau DNA, selain itu juga berfungsi untuk menemukan
sequence leluhur dan itu sering disebut sequence consensus (Sung, 2010). Dalam
pembentukan multiple sequence alignment terdapat beberapa metode. Dalam
penelitian ini, mengunakan 3 metode pembentukan multiple sequence alignment
yaitu MSA Phylogenetic, MSA Muscle, dan MSA MAFFT.
1. MSA Phylogenetic.
MSA Plylogenetic merupakan metode MSA yang digunakan didalam fungsi
di MATLAB. Berikut ini merupakan langkah – langkah algoritma MSA
phylogenetic :
a. Menghitung Jarak antar sequence dengan algoritma p – distance. P –
distance adalah perhitungan ketidaksamaan antara dua sequence yang sudah
mengalami proses pesejajaran dengan algoritma Needleman-wunsch(2.6.3),
dibagi jumlah panjang alignmentnya dengan menghilangkan alignment yang
mempunyai pasangan gap (Lavrov,2011). Berikut merupakan rumus dari p
– distance.
(2 – 1)
Contoh :
Hasil dari perhitungan dari gambar 2.9 dimisalkan D. Karena sequence
yang berpasangan dengan gap tidak diperhitungkan maka sequence yang
tidak cocok sebanyak 2 sedangkan sequence totalnya sebanyak 5. Sehingga
nilai D adalah 2/5 yaitu 0.4.
b. Membentuk guide tree dengan mencari jarak terdekat antar sequence
dengan algoritma neighbor – join ( NJ ) merupakan algortima pencarian
jarak terdekat dengan mencari nilai minimum dalam distance matrix untuk
membentuk sebuah tree biner tanpa root (Studier, 1988). Algoritma ini
memiliki tahap perhitungan yang pertama mencari jarak terdekat dengan
rumus dibawah ini :
(2 – 2)
Dimana D adalah matrik yang menyimpan jarak antar sequence yang
dihitung dengan p – distance. N adalah banyaknya sequence yang dalam
MSA. S adalah sebuah matrik perhitungan untuk mencari jarak terpendek
dari matrik D. Ri dan Rj adalah jumlah jarak antar sequence dalam satu
kolom i dan j. Berikut ini merupakan rumus untuk mencari nilai R.
( 2 – 3 )
D merupakan matrik dari hasil p – distance, i adalah kolom dari D dan k
adalah banyaknya baris dari matrik D. Membuat node baru u dari dua
sequence yang mempunyai jarak paling pendek, dan setelah itu menghitung
jarak dari node u ke – kedua sequence tersebut. Berikut rumus untuk
( 2 – 4a )
Dan
( 2 – 4b )
Dui dan Duj merupakan jarak sequence i dan sequence j dengan node u.
seqeuence i dan sequence j merupakan sequence dengan jarak paling dekat.
Dan untuk jarak sequence yang lain dengan node u dirumuskan sebagai
berikut.
(2 – 5 )
Dku merupakan jarak sequence i ke node u, Dik adalah jarak sequence i
dengan sequence k dan Djk adalah jarak sequence j dengan sequence k.
Setelah itu diulang ke persamaan ke (2 – 2) sampai dengan tidak ada node
yang akan digabungkan lagi. Setelah tree terbentuk langkah berikutnya
menentukan root dengan metode midpoint. Midpoint merupakan metode
pencarian root dengan mencari nilai tengah dari kedua cabang yang
jaraknya paling jauh(Stewart, 2004). Gambar 2.10 merupakan gambaran
metode midpoint untuk menentukan root.
Gambar 2.10. Contoh penentuan letak root dari sebuah tree unrooted (Stewart, 2004).
c. Langkah terakhir membentuk sequence dengan algoritma progressive
alignment. Dalam progressive alignment mempunyai 2 tahap yaitu :
c.1. Sequence weight
Sequence weight merupakan pembobotan untuk setiap sequence. Berat
dihitung dari jarak sequence dengan root, dan bila terdapat dua atau lebih
sequence yang berbagi cabang, maka panjang cabang dibagi sebanyak
jumlah sequence yang didalam cabang. Setelah itu dilakukan normalisasi
berat sehingga maksimum berat adalah 1 (Singh, 2000). Gambar 2.11
merupakan contoh perhitungan dari sequence weight.
Gambar 2.11. Tree dari hasil proses neighbor – join dan midpoint (Singh, 2000).
Untuk menghitung berat sequence yang memiliki sebagai berikut :
Missal hitung berat Hbb_human :
0.81+(0.226/2)+(0.016/4)+(0.015/5)+(0.062/6) = 0.9403
c.2. Multidimensional dynamic Programming.
Multidimensional dynamic programming merupakan proses pencarian
dynamic programming ini menggunakan algoritma Needleman
Wunsch(2.6.3) yang dimodifikasi dengan affine gap. Dengan catatan untuk
sequence DNA diubah menjadi sequence profile. Pengisian matrik
kemiripan memakai sequence profile dilakukan dengan cara mengalikan
kedua sequence profile dengan substitution matrix(2.5), dimana salah satu
sequence profile harus di transpose lebih dulu. Sebelum proses dynamic
programming sequence profile dikalikan dengan berat/weight masing –
masing sequence (Thompson,1994). Affine gap adalah penentuan nilai gap
pada perhitungan pencarian sequence alignment optimal (Roshan, 2007).
Affine gap dapat dirumuskan sebagai berikut :
1 (2 – 6)
y merupakan gap pinalti, d merupakan gap pinalti dari pembukaan gap (gap
open), e adalah gap pinalti untuk gap yang ditemukan setelah gap pertama
(extend gap), dan n merupakan panjang dari sequence (Roshan, 2007).
Gambar 2.12 merupakan gambaran dari affine gap.
Gambar 2.12. Sebuah sequence alignment dengan gap.
Sequence profile adalah model matetatis yang mewakili dari satu sequence
DNA atau lebih (Kuznetsov,2010). Gambar 2.13 merupakan contoh dari
Gambar 2.13. Konversi sequence DNA ke sequence profile
Proses progressive alignment secara keseluruhan dapat dijelas sebagai
berikut:
a. Mencari berat setiap sequence dari tree dengan metode sequence
weight.
b. Mengubah sequence data menjadi sequence profile, lalu dikalikan
dengan berat setiap sequence.
c. Membentuk sequence alignment dengan algoritma Needleman
Wunsch(2.6.3) dari sequence profile yang telah dikalikan.
Hasil dari algoritma Needleman Wunsch(2.6.3) membentuk sequence
profile baru. Setelah itu sequence profile baru akan dihitung lagi dengan
sequence profile yang lain dan begitu seterusnya sampai semua sequence
dihitung. Urutan perhitungan dimulai dari sequence terdekat berdasarkan
Gambar 2.14. Gambaran proses Multidimensional dynamic programming.
Kompleksitas waktu untuk algoritma multiple alignment ini
ditunjukan pada tabel berikut :
Tabel 2.1. Kompleksitas Waktu MSA Phylogenetic.
Step O(Time)
Distance Matrix N2L2
Neighbor Join N4
Progressive alignment(satu
iterasi) NLp+Lp
2 = N2+L2
Progressive alignment (total) N3+NL
TOTAL N4+L2
Dimana L adalah panjang sequence dan N merupakan jumlah
Gambar 2.16. Gambaran dari subsequencek -mer dengan k = 6.
Sedangkan k – mer distance adalah jarak antar sequence dengan
perhitungan k – mer. Untuk memperjelas k – mer distance ditunjukan
dalam persamaan berikut :
, ∑ min , ⁄min , 1 (2-7)
F(X,Y) adalah Jarak antara sequenceX dan sequenceY, τ merupakan k – mer,
misalkan pada gambar 2.16 τ = GAGAAG, nX(τ) dan nY(τ) adalah jumlah
kejadian dari τ dalam sequence X dan sequence Y. Lalu LX dan LY adalah
panjang sequence X dan sequenceY sedangkan k merupakan nilai k dari k –
mer.
b. Matrix D1 merupakan klastering dari UPGMA, menghasilkan biner tree
Tree1. UPGMA atau Unweighted Pair Group Method with Arithmetic
means merupakan algoritma clastering untuk pembentuk tree untuk muscle
(Lavrov, 2011). Berikut ini merupakan langkah dari algoritma tersebut.
b.1. Menentukan nilai ni dan nj, dimana untuk menampung jumlah
sequence dalam sebuah node. Untuk awal nilai ni dan nj sama yaitu 1.
b.2. Mencari nilai minimum dari matrik jarak D1, misal jarak minimum
D(i,j) .
b.4. Menganti i dan j menjadi u,mengupdate jarak matrik yang sudah
disatukan. Persamaan update jarak sebagai berikut :
, , , (2 - 8)
Dimana k≠i,j
b.5. Set nu = ni +njuntuk nilai n baru.
b.6. Mengulangi langkah b.1 sampai tidak ada sequence yang digabungkan
lagi.
c. Progressive alignment dibangun dari urutan percabangan Tree1 seperti
progressive alingment di MSA phylogenetic (2.4.1.c).
2.2. Improved Progressive.
Pada tahap draft progressive tree yang dibentuk masih belum optimal. Oleh
karena itu Muscle mnggunakan kimura distance untuk menghitung jarak antar
sequence dan langkah ini dimungkinkan terjadi iterasi. Berikut langkah –
langkahnya.
a. Menghitung jarak sequence dengan kimura distance dari MSA1, yang akan
menghasilkan matrix D2. Kimura distance merupakan perhitungan jarak
antar sequence DNA dengan mempertimbangkan mutasi transition dan
transversion (Mount,2001). Transition lebih jarang terjadi dari pada
transversion. Untuk transition mempunyai 4 cara untuk mutasi yaitu : A
ÅÆ G dan C ÅÆ T, sedangkan transvertion menpunyai 8 cari mutasi
yaitu AÅÆC, AÅÆT, GÅÆT, dan GÅÆC. untuk lebih jelasnya
Gambar 2.17. Mutasi DNA menurut kimura.
Dalam gambar 2.17 transition = α, dan transvertion = β. Jika Dtransition =
banyaknya frekuensi mutasi transition antara sequence A dan sequence B,
dan Dtransversion = banyaknya frekuensi mutasi transversion antara sequence
A dan sequence B. Maka α dan β mempunyai persamaan sebagai berikut:
1/ 1 2 (2 – 9a)
dan
1 1 2⁄ (2 -9b)
Lalu untuk mengukur jarak dari kimura sebagai berikut:
, log log (2 – 9c)
b. Matrix D2 diklasifikasikan dengan UPGMA, menghasilkan tree biner yang
bernama Tree2.
c. Membandingkan Tree1 dengan Tree2, bila node dalam urutan berbeda maka
akan mengulangi langkah a.
d. Langkah ini sama dengan langkah c pada draft progressive, yang akan
2.3. Refinement.
Langkah – langkah refinement sebagai berikut :
a. Memisahkan Tree2 menjadi 2 tree dengan cara mencari panjang cabang
yang terdekat dengan root.
b. Menghitung profile untuk setiap sub multiple alignment, dan membuang
kolom yang tidak beri isi karakter DNA.
c. Membentuk multiplealignment baru dari hasil dua sub multiple alignment.
Menggunakan profile – profile alignment. Profile – profile alignment di
muscle menggunakan profile sum-of-pairs (PSP) merupakan penjumalahan
sequence weight dengan substitution matrix skor untuk setiap pasangan
karakter DNA untuk setiap kolomnya (Edgar, 2004). Berikut ini persamaan
untuk menghitung PSP.
∑ ∑ (2 – 10a)
Dimana
log (2 – 10b)
i dan j adalah karakter dari DNA, dan merupakan frekuensi dari i dan
j untuk colom x untuk profile 1 dan y untuk profile 2. Sij merupakan
substitutionmatrix score, dimana pijadalah probabilitas gabungan dari i dan
j yang disejajarkan. Sedangkan pi dan pj adalah probabilitas dasar dari i dan
j. Dalam Persamaan diatas belum menyertakan gap untuk perhitungannya.
1 1 log ∑ ∑ ⁄ (2 - 11)
Dimana dan merupakan frekuensi gap dari kolom x untuk profile 1
dan y untuk profile 2.
d. Jika nilai Sum Pair Score meningkat, alignment baru akan disimpan, selain
itu akan dibuang.
Langkah a – d diulangi sampai konvergensi atau sampai semua cabang dari
tree tidak mengalami perubahan lagi. Dan untuk kompleksitas waktu dari Muscle
ditunjukan pada tabel dibawah ini, dimana L adalah panjang sequence dan N
merupakan jumlah sequence.
Tabel 2.2. Kompleksitas Waktu MSA Muscle.
Step O(time)
k – mer Distance Matrix N2L
UPGMA N2
Progressive (satu iterasi) N2 + L2
Progressive (root) N2 log N + NL log N
Progressive (N iterasi + root ) N3 + NL2
Refinement (satu edge) N3 + L2
Refinement (N edge) N4 + NL2
3. MAFFT
MAFFT merupakan kepanjangan dari Multiple Alignment Fast Fourier
Transform. Pendekatan Fast Fourier Transform digunakan saat proses
progressive alignment berlangsung (Katoh, 2002). Berikut ini langkah – langkah
proses MAFFT terlihat pada gambar 2.18 :
Gambar 2.18. Bagian A menunjukan proses progressive alignment. Sedangkan bagian B menunjukan proses iterative refinement (Katoh, 2008).
Berikut ini merupakan penjelasan langkah diatas :
3.1. Menghitung jarak kemiripan antara dua sequence dengan perhitungan
berdasar jumlah dari pembagian 6 – tuples antara kedua sequence. Untuk
persamaan perhitungan jarak kemiripan antara dua sequence sebagai berikut
:
Dimana Dij adalah jarak kemiripan antar sequence i dsn sequence j. Tij
adalah jumlah 6 tuples antara sequence i dan j. Tii adalah jumlah 6 tuples
pada sequence i, sedangkan Tjj adalah jumlah 6 tuples pada sequence j
(Katoh, 2002).
3.2. Pembentukan guide tree mengunakan algoritma UPGMA yang
dimodifikasi untuk perhitungan pada pengupdate jarak antar sequence pada
persamaan (2 - 8), diganti dengan persamaan berikut :
, 0,5 , , 1 , , (2 -13)
Dimana x =0,1 nilai tersebut berkerja baik untuk menangani perhitungan
sequence yang terpisah (Katoh,2002).
3.3. Progressive Alignment 1
Dalam proses ini mengunakan pendekatan untuk menghitung group to
group alignment dengan pendekatan fast fourier transform (FFT)
correlation untuk pencarian segment sequence homolog (Katoh, 2002).
Berikut ini merupakan langkah perhitungan groupto groupalignment untuk
DNA:
a. Menemukan segment sequence homolog dengan FFT correlation .
Pertama, mengubah sequence DNA menjadi urutan vector 4 dimensi
dari frekuensi A, C, G, dan T dengan setiap nucleotide yang muncul
Gambar 2.19. Konversi sequence DNA ke urutan vector 4 dimensi.
Setelah itu menghitung correlation untuk setiap nucleotide. Berikut
perhitungan correlation tiap nucleotide.
∑ (2 – 14)
Dimana CA(k) merupakan correlation dari nucleotide A, persamaan
diatas juga berlaku untuk nucleotide lainya. x merupakan sequence x
dengan panjang nx dan y merupakan sequence y dengan panjang ny.
Sedangkan n = (nx + ny) -1, dan k= 0,.., n. Dan x × y merupakan
perhitungan dari Discrete Fourirer Transfroms (DFT) dengan
persamaan sebagai berikut :
(2 – 15)
Persaman diatas merupakan sebuah perkalian discrete fourier
transform dari x dengan inverse discrete fourier transform (Wu,
2007). Berikut ini merupakan persamaan DFT dengan jumlah
sequence sebanyak ak dan panjang sequence dimisalkan n.
∑
, 0
1
(2 – 16a)
∑
, 0
1
(2 – 16b)
Persamaan diatas merupakan perhitungan inverse DFT dari hasil DFT
dengan sequence sebanyak Ak dan mempunyai panjang n. Dan berikut
ini persamaan keseluruhan correlation antara dua sequence DNA:
(2 - 17)
C(k) adalah correlation antara dua sequence DNA, sedangkan CA(k),
CC(k), CG(k), dan CT(k) merupakan correlation dari nucleotide A,C,G,
dan T.
b. Membagi menjadi sebuah Homology Matrix. Untuk menghasilkan
alignment yang mempunyai segment homolog yang konsisten dari
antara kedua sequence. Sebuah matrik Sij(1 ≤i, j ≤n), n adalah jumlah
dari segment homolog yang telah terbentuk. Jika segment homolog ke
– i pada sequence 1, sama dengan segment homolog ke – j dari
sequence 2, maka matrik Sij diisi dengan nilai dari perhitungan
pencarian segmentsequence homolog, dan sebaliknya jika tidak sama
akan diisi dengan nilai 0. Dan dengan Dynamic programming
digunakan untuk membentuk sequence homolog yang optimal dari
segment – segment homolog. Gambar 2.20 merupakan gambaran
pencarian sequence homolog yang optimal dari 5 segment sequence
Gambar 2.20. Sebuah contoh dari optimasi segment sequence homolog dengan DP (Katoh, 2002).
Dimana anak panah yang tebal menunjukan optimal dari kelima
segment sequence yang homolog.
3.4. Mengukur jarak kemiripan antar sequence dengan kimura distance dengan
rasio transition/transversion 2.0. Untuk menaikan efesiensi dari alignment.
Nilai similarity matrix (kimura distance) dan gap pinalti mengalami
modifikasi sebagai berikut:
(2 -18a)
Dimana
1 ∑ (2 – 18b)
2 ∑ , (2 – 18c)
Dimana adalah similarity matrix dari nucleotide yang sudah
dimodifikasi dan a, b adalah karakter nucleotide. Mab adalah similarity
matrix dari kimura distance. Untuk fa dan fb merupakan frekuensi kejadian
sequence random. Untuk DNA fa dan fb bernilai sama di set dengan nilai
0.25, sedangkan untuk Sa = 0.06.
3.5. Pembentukan guide tree dengan algoritma UPGMA yang telah
dimodifikasi.
3.6. Progressivealignment 2
Dalam progressive yang kedua ini untuk menghitung group to group
dengan homolog matrik, dengan persamaan sebagai berikut :
, ∑ , , , , (2 – 19)
Dimana i dan j merupakan posisi dari sequence n dan sequence m. wn dan
wm merupakan berat sequence ke – n dari group1 dan berat sequence ke – m
dari group2. A(n,i) adalah posisi ke – i dari sequence ke – n dalam group1
dan B(m,j) adalah posisi ke – j dari sequence ke – m dalam group2.
Sedangkan adalah normalisasi dari distance matrik kimura 2 paramater
yang terdapat pada persamaan (2 – 18). Untuk alignment optimal dalam
kedua group dihitung sebagai berikut :
, , , 1 1,, 11 1
1, , 1 1
(2 – 20a)
Dimana P(i,j) merupakan nilai akumulasi dari bagian optimal (1,1) ke (i,j),
sedangkan G1(i,x), dan G2(j,y) merupakan gap pinalti yang didefinisikan
dibawah ini :
Dimana Sop adalah gap open pinalti, (x) adalah jumlah gap yang
dimulai dari posisi ke – x, dan adalah jumlah gap yang berakhir
pada posisi ke – i. Dan Berikut persamaan perhitungannya :
∑ 1 (2 – 20b)
Dan,
∑ 1 (2 – 20c)
Dimana zm(i) = 1 dan am(i)=0, jika posisi ke – i dalam sequence m adalah
gap, jika tidak maka zm(i)= 0 dan am(i)=1, sedangkan wm merupakan berat
sequencem. G2(j,y) dihitung dengan cara yang sama.
3.7. Iterative refinement dengan WSP ( Weight Sum of Pair ).
Pada langkah ini bertujuan untuk menaikan akurasi jarak hubungan antar
sequence dari multiple alignment dengan cara mengevaluasi efek dari jumlah
homolog yang dimasukan dalam alignment (Katoh, 2004). Berikut ini merupakan
langkah – langkah iterativerefinement :
Untuk setiap pasangan alignment di bagi menjadi gap – free segment, dan
banyaknya n untuk menentukan setiap bagian. Informasi dari setiap bagian
disimpan dalam sebuah array, skor S(s,t,n) yang mewakili skor alignment dari
gap – free segment ke – n antara sequence s dan t, L(s,t,n) merupakan panjang
dari segment alignment (s,t,n). P(s,t,n) adalah posisi dalam setiap sequence s dan
t, dan E(s,t,n) adalah importance value yang dihitung seperti persamaan (2 – 21b),
segment. Misalkan (s,t,p,q) Є P(s,t,n), jika posisi ke – p dari sequence s, disejajarkan posisi ke – q dari sequence t dalam segment alignment (s,t,n).
Nilai frekuensi f(s, p) yang mewakili frekuensi posisi ke – p dalam sequence
s yang terdapat dalam gap – free segment, dihitung sebagai berikut :
, ∑ , ,, , , , , (2 – 21a)
Dimana wt adalah berat dari sequence t. Untuk importance valueE(s, t, n) untuk
segment alignment dihitung sebagai berikut :
, , ∑,, , , , , , , , , , , (2 – 21b)
Lalu menetapkan matrik importanceI(s, t, p, q) antara posisi ke – p dari sequence
s dan posisi ke – q dari sequence t, seperti berikut :
, , , ∑ 0, , (2 – 21c)
Dengan catatan jika (s, t, p, q) ЄP(s,t.n) atau sebaliknya.
Sebuah alignment dari beberapa sequence yang dihasilkan selama prosedur
progressive dan iterative refinement ditunjukan sebagai ‘group’. Untuk align
normalisasi. wst merupakan berat sequence antara sequence s dan t, dengan
perhitungan sebagai berikut :
, . . , (2 - 22)
Dimana wp = wjk = wst untuk jarak yang tidak langsung berhubungan w(j,x)
adalah jumlah berat sequence j dan x, dimana sequence j dan x langsung
berhubungan. we adalah jumlah berat antara sequence, yang menghubungkan
sequence x dan y. w(y,k) adalah jumlah berat sequence y dan k, dimana sequence y
dan k langsung berhubungan. Sedangkan WI adalah faktor berat yang di set
dengan nilai 2.7.
Untuk mendapatkan align yang optimal, kedua group dihitung dengan
Needleman Wunsch untuk matrik H(…) pada setiap langkah progressive alignment.
Setelah itu alignment yang terbentuk dicari nilai optimalnya, dengan menghitung
Weight Sum of Pairs (WSP) dengan persamaan sebagai berikut :
∑ ∑ , , (2 – 23)
Dimana WSP(A) adalalah berat dari alignment A, N adalah panjang
alignment A. j dan k adalah sequence dalam alignment A. wj,k adalah berat dari
sequence j dan k, sedangkan Sj,k adalah skor dari perhitungan dynamic
programming Needleman Wunsch antara sequence j dan k. Proses iterative
refinement akan diulangi sampai nilai WSP tidak berubah atau konvergen. Untuk
kompleksitas waktu dari MAFFT secara keseluruhan adalah O(NL)+O(N3) =
O(NL+N3). Dimana L adalah panjang sequence dan N merupakan jumlah
2.6. Sequence Consensus.
Sequenceconsensus atau juga sering disebut dengan sequence Motif adalah
fitur dari efisiensi pengabungan dari sebuah posisi karakter dari beberapa
sequence dengan panjang yang sama. Sequenceconsensus digunakan sebagai pola
yang mewakili dari beberapa sequence (Sung, 2010). Dalam penelitian ini untuk
pembentukan sequence consensus mengunakan dua metode yaitu Positional
Weight Matrix (PWM) dan Profile Hidden Markov Model.
1. Position Weight Matrix (PWM).
Position Weight Matrix merupakan pembentukan sequence consensus
dengan cara menghitung frekuensi dalam setiap posisi dari 4 basa DNA atau
protein yang sudah disejajarkan.Umumnya PWM berukuran matrik 4 × l atau 5 ×
l, dimana l adalah panjang dari sequence alignment. nilai 4 adalah basa DNA
yang terdiri dari A, C, G, T dan berdimensi 5 × l adalah untuk gap bila ada .
Dalam setiap kolom dihitung frekuensi dari A, C, G, T dengan total jumlah setiap
kolom sama dengan 1 (Sung, 2010). Untuk memperjelas proses perhitungan PWM
ditunjukan pada gambar 2.21.
Kompleksitas waktu dari algoritma PWM adalah O(NL) dimana N adalah
jumlah dari sequence dalam MSA dan L adalah panjang dari sequence MSA.
2. Profile Hidden markov model.
a. Markov chains.
Markov chains adalah serangkain state dengan probabilitas yang
berhubungan untuk setiap taransisi antara state. Probabilitas transisi
dihitung dari arus state yang terdapat pada state sebelumnya. Markov chains
pada sequence DNA ditunjukan pada gambar 2.22, dengan 4 basa DNA
yaitu adenine (A), cytosine (C), guanine (G), dan thymine (T).
Gambar 2.22. Markov chains untuk DNA (Attaluri, 2007).
Setiap anak panah dalam gambar 2.22 mewakili dari probalitas
transisi dari keempat basa nucleotide. Probabilitas transisi dihitung setelah
mengamati beberapa sequence DNA. DNA markov model adalah first order
markov model saat setiap kejadian tergantung pada kejadian sebelumnya.
Probabilitas transisi ast ( probabilitas transisi dari state sebelumnya dengan
simbol s, dan untuk state sekarang dengan simbol t ) dihitung sebagai
berikut :
Penjumlahan dari probabilitas transisi untuk setiap statenya sama
dengan 1. Karena terdapat pengabungan probabilitas dalam setiap langkah,
model ini disebut sebagai probabilistic Markov model. Bila sebuah
sequence diketahui probabilitasnya maka untuk sebuah model dihitung
sebagai berikut :
P(x1) adalah probabilitas dari permulaan state dengan simbol x1. P(x1) dapat
dihitung dengan menambahkan begin state dan end state untuk menampung
simbol pertama dan akhir dari urutan.
b. Hidden markov model.
Hidden markov models adalah sebuah model statistik dengan Markov
model yang state – state (transisi state) tidak dapat diamati secara langsung
atau dengan kata lain parameter – parameter tersebut tersembunyi (hidden).
Notasi umum dalam HMM sebagai berikut :
O – Sequence observasi.
T – Jumlah total simbol dari sequence obsevasi.
N – Jumlah total sate.
α – Alphabet dari model.
M – Jumlah total dari simbol alphabet.
A – Matrik probabilitas taransisi state.
aij– Probabilitas transisi dari statei ke j.
B – Simbol matrik probabilitas distribusi.
bi(k) – Probabilitas distribusi dari k dalam statei.
λ– Model HMM.
Untuk lebih memahami lebih HMM akan ditunjukan dalam sebuah
kasus dua koin yang terdiri dari satu koin normal dan satu koin bias. Kedua
koin dilempar secara begantian sehingga membentuk sequence observasi O
= {HTHTHH} dimana H mengantikan kepala (head), dan T mengantikan
ekor (tail). Untuk nilai T = 6, N = 2 terdiri dari koin normal dan bias, α =
{HT}, M=2 yang terdiri dari H dan T. Gambar 2.23 menunjukan model
HMM dari kasus ini.
Gambar 2.23. Model HMM dari kedua koin (Attaluri, 2007).
Matrik probabilitas transisi koin normal sebagai state 1 dan bias sebagai
state 2 sebagai berikut :
0.95 0.05
Dengan kata lain a12 = 0.05 mewakili probabilitas transisi dari state 1
ke state 2. Untuk matrik probabilitas distribusi (B) dari H dan T dari kedua
koin adalah sebagai berikut :
0.5 0.5 0.7 0.3
Pada baris pertama dalam matrik merupakan probabilitas distribusi
dari (H,T) dalam koin normal, dan baris kedua merupakn probabilitas
ditribusi koin bias. Untuk b1(H) melambangkan probabilitas distribusi dari
H dalam kasus koin normal. Dalam kasus pengambilan secara random
inisialisasi distribusi koin ditentukan sebagai berikut :
0.5 0.5
c. ProfileHidden markov model.
Pendekatan untuk modeling consensus sequence akan membentuk
sebuah probabilitas model, sehingga dalam pengembangaannya tipe hidden
markov model sangat cocok untuk modeling multiple alignment. Profile
HMM merupakan alipkasi HMM yang terkenal dalam biologi molekuler
saat ini (Eddy, 1996). Profile hidden markov model mempunyai sturktur
Gambar 2.24. Struktur model PHMM (Attaluri, 2007).
Pada gambar 2.24 struktur model PHMM mempunyai 3 macam state
yaitu match state dilambangkan dengan persegi, insertstate dilambangkan
dalam bentuk diamond dan delete state berbentuk lingkaran. delete State
digunakan untuk null transisi, insert state digunakan untuk gap dari
alignment, dan match state digunakan untuk perhitungan kemiripan dari
state. Match dan insert state merupakan emission state dari PHMM.
Probabilitas emission dihitung tergantung pada frekuensi simbol yang
dipancarkan. Delete state boleh dilewati oleh gap yang ditemukan dalam
MSA dan diberikan pada emissionstate lainnya. Anak panah dalam gambar
menunjukan transisi yang mungkin pada state sekarang ke state selanjutnya.
Dan itu kemudian disebut dengan probabilitas transisi yang menentukan
likelihood dari state berikutnya yang diambil. Berikut ini persamaan
perhitungan probabilitas emission dan probabilitas transisi :
∑ (2 -25a) dan ∑ (2 – 25b)
akl merupakan probabilitas transisi dari state k ke state l, Akl adalah jumlah
transisi dari state k ke state l. dan ∑ adalah jumlah total transisi dari state k ke semua state. ek(a) merupakan probabilitas emission dari state k
untuk simbol a. Ek(a) = jumlah kejadian simbol a dalam state k, dan
∑ = jumlah total semua simbol dalam state k. Untuk lebih
Gambar 2.25. MSA dari alignment DNA (Attaluri, 2007).
Sebuah MSA yang terdiri dari 5 sequence DNA, langkah pertama
yaitu membangun model PHMM dengan menentukan match state dan
insertstate. Pada gambar 2.25 matchstate terdapat pada kolom 1,2 dan 6,
sedangkan insert state terdapat pada kolom 3, 4, dan 5, untuk insert state
ditentukan bila jumlah gap lebih banyak dari pada karakter dalam tiap
kolomya. Probabilitas emission pada kolom pertama sebagai berikut :
(A) = 4/4, C) = 0/4, G) = 0/4, (T) = 0/4
Dalam perhitungan probabilitas emission diatas terdapat banyak nilai
nol. Untuk menggabungkan semua kemungkinan yang muncul perhitungan
harus ditambah probabilitas kecil, sehingga dalam perhitungan tidak
ditemukan nilai nol. Penambahan probabilitas itu disebut “add-one rule”
dimana pembilang untuk probabilitas emission ditambah dengan nilai 1 dan
untuk penyebut ditambah dengan jumlah karakter DNA yaitu 4 (Attaluri,
2007). Sebagai contoh (A) = (4+1)/(4+4)=5/8. Dan untuk keseluruhan
perhitungan probabilitas emission dari kasus diatas ditunjukan pada tabel
Tabel 2.3.Probabilitas emission dari profile HMM pada gambar 2.25.
Lalu untuk pehitungan probabilitas transisi mengunakan perhitungan
pada persamaan (2 – 25a), pada begin state ke matchstate pertama dihitung
seperti dibawah ini.
BM BM ⁄ BM BI BD = 4/(4+0+1) = 4/5
Untuk seluruh kemungkinan dari perhitungan probabilitas transisi juga
mengunakan “add-one rule”. Dalam probabilitas transisi pembilang
ditambah dengan 1, dan penyebut ditambah dengan 3, karena dalam PHMM
terdapat 3 jenis state yang melakukan transisi yaitu matchstate, insertstate,
dan delete state. Sebagai contoh untuk nilai BM 5/8. Dan hasil
perhitungan probabilitas transisi ditunjukan pada tabel 2.2.
Tabel 2.4.Probabilitas transisi dari PHMM padagambar 2.24.
Akhir dari model MSA pada gambar 2.24 akan seperti burikut ini.
Gambar 2.26. Hasil akhir model PHMM untuk MSA pada gambar 2.25 (Attaluri, 2007).
Model PHMM akhir terdiri dari E (matrik probabilitas emission)
dengan probabilitas emission match dan insert state yang ditunjukan pada
tabel 2.1, dan A (matrik probabilitas transisi) yang berisi transisi dari setiap match, insert, dan delete state yang ditunjukan pada tabel 2.2. Serta jumlah
state dari begin state sampai ke end state (N) adalah 4.
Dalam HMM terdapat 3 masalah yang dibicarakan yaitu :