Atherosclerosis 153 (2000) 161 – 168
The methylenetetrahydrofolate reductase gene is associated with
increased cardiovascular risk in Japan, but not in other
populations
Sun Ha Jee
a,*, Terri H. Beaty
b, Il Suh
c, Young-sup Yoon
d, Lawrence J. Appel
baDepartment of Epidemiology and Disease Control,Yonsei Uni
6ersity Graduate School of Health Science and Management,CPO POB8044,
Seoul,South Korea bDepartment of Epidemiology,Johns Hopkins Uni
6ersity School of Hygiene and Public Health,Baltimore,MD,USA
cDepartment of Pre6enti6e Medicine and Public Health,Yonsei Uni6ersity College of Medicine,Seoul,South Korea dDi6ision of Cardiology,Yonsei Cardio6ascular Center,Yonsei Uni6ersity College of Medicine,Seoul,South Korea
Received 7 June 1999; received in revised form 13 December 1999; accepted 14 January 2000
Abstract
The methylenetetrahydrofolate reductase (MTHFR) gene has been associated with increased risk for cardiovascular disease in some, but not all studies. Our data sources included a MEDLINE search of the literature published before December 1998, a bibliography review, and expert consultation. Of 23 studies initially identified, 18 (9855 persons) met the inclusion criteria. Information on sample size, study design, Hardy – Weinberg equilibrium, method of genotype determination, plasma folate and homocysteine were abstracted by two reviewers using a standardized protocol. The overall odds ratio of the MTHFR gene on cardiovascular disease was estimated using the Mantel – Haenzel method. From 12 studies with angiographically-confirmed coronary artery disease (CAD), the overall odds ratio (OR) for CAD among those with heterozygous (V/A) was 1.3 (95% CI, 1.1 – 1.5), while it was 1.4 (1.2 – 1.6) for the homozygous mutant (V/V) compared to those with homozygous normal (A/A). However, the overall odds ratio for CAD among those with the V/V genotype versus A/A genotype was not statistically significant (OR: 1.1; 95% CI: 0.9 – 1.3) after excluding three Japanese studies. The corresponding OR for the three Japanese studies was 2.0 (1.6 – 2.7). For six studies with myocardial infarction (MI), the overall OR of MI was 1.0 (0.8 – 1.1) for those with the V/A genotype and 0.9 (0.7 – 1.1) for those with the V/V genotype, respectively; none of these ORs for MI was statistically significant. The MTHFR gene is associated with increased risk for CAD in Japan, but not in other populations. © 2000 Elsevier Science Ireland Ltd. All rights reserved.
Keywords:Genotype; MTHFR; Coronary artery disease; Myocardial infarction; Racial difference
www.elsevier.com/locate/atherosclerosis
1. Introduction
Over the past decade, mild hyperhomocysteinemia has been recognized as a risk factor for occlusive arte-rial disease and thrombosis [1 – 5]. A meta-analysis of 27 studies investigating the relation between fasting total homocysteine (tHcy) levels and coronary artery disease (CAD) yielded an odds ratio of 1.6 for men and 1.8 for women for every 5 mmol/l increase in tHcy plasma
levels [6]. Homocysteine levels are influenced by
envi-ronmental (folate, vitamin B6, and vitamin B12 intake) as well as genetic factors [2,3,7 – 9].
A common mutation of enzyme methylenetetrahy-drofolate reductase (MTHFR) has been implicated in the development of hyperhomocysteinemia [10,11] and CAD. A missense mutation in the gene encoding MTHFR has recently been described as the molecular basis for this defect. MTHFR activity in the V/V genotype has been found to be reduced [12] and tHcy significantly elevated [12,13] compared with the A/A and V/A genotypes.
One [24], but not the other [40], meta-analysis has identified a positive association between the MTHFR gene and increased risk for CAD. Results of these
* Corresponding author. Tel.: +82-2-3615095; fax: + 82-2-3655118.
E-mail address:[email protected] (S.H. Jee).
studies have been inconsistent, perhaps because of racial differences, small sample sizes or other design features. Pooling the results of case-control studies provided a means of exploring the basis for heterogene-ity in study outcomes.
The objective of this study was to quantitatively summarize the evidence for a relationship between the MTHFR gene, coronary artery disease and myocardial infarction.
2. Methods
2.1. Study selection
Relevant studies in humans were identified by search-ing the MEDLINE database for the years 1988 through 1998. The search combined the following terms; methylenetetrahydrofolate reductase (MTHFR), homo-cysteine and cardiovascular disease. In addition, refer-ence lists of identified studies and review articles were examined for other relevant studies. Finally, experts in the field were surveyed to identify additional studies. Twenty-three case-control studies [14 – 35] were iden-tified. These articles were reviewed by two authors (Jee and Yoon) to determine whether they met predeter-mined criteria for inclusion in our subsequent analysis. Areas of disagreement or uncertainty were adjudicated by consensus. To be included, a study must have in-cluded the following criteria: [1] been conducted in humans; [2] assessed MTHFR genotype determination as an exposure using standardized laboratory methods; [12] and [3] reported the number of cases and controls with the different MTHFR genotypes. Studies [15,18 – 21,23 – 27] of CAD used angiographically — confirmed occlusion as outcomes, while studies of myocardial infarction (MI) used WHO criteria for MI [16 – 18] which relies on symptoms, enzyme elevations or electro-cardiographic changes, and clinical/past history [14,22,23].
2.2. Data abstraction
Information was abstracted on general characteristics (name of first author, year of publication, country of origin, sample size, mean age, gender of participants and body mass index), study design (case and control selection criteria), folate intake, plasma folate and tHcy in the study population, method of MTHFR genotype determination including information on the polymerase chain reaction and enzyme used, and information on Hardy – Weinberg equilibrium. If different outcomes were employed in the same report, they were analyzed as separate studies [18,23]. When cases of CAD or MI could not be separated, these studies were not included [27 – 34].
2.3. Statistical analysis
Alleic and genotypic frequencies were determined from observed genotype counts, and the expectations of the Hardy – Weinberg equilibrium were evaluated byx2 analysis. Comparisons between genotypic frequencies were done using x2 analysis.
A meta-analysis consists of tests for association and homogeneity. Homogeneity testing assesses the homo-geneity of the different odds ratios determined in re-spective studies. The overall test for association then assesses the significance of the association between the C677T mutation and CAD or MI for all studies com-bined. To calculate the pooled effects of the MTHFR gene, each study was assigned a weight consisting of the reciprocal of variance of its odds-ratio estimate [36]. Estimations of the mean odds ratio of cardiovascular disease associated with those having the MTHFR gene and those with 95% confidence intervals were calculated using the Mantel – Haenszel method [26].
3. Results
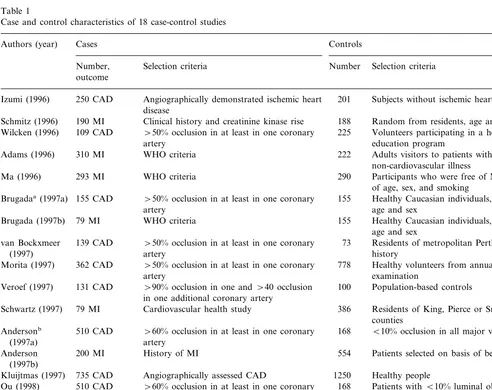
Table 1 shows the definition or criteria of cases and controls. The case-control studies were conducted be-tween 1996 and 1998, and varied in size from 84 to 735 cases and from 73 to 1250 controls. The total number of participants was 9855 (4594 cases and 5261 controls). In 16 studies, healthy individuals were used as controls. In the remaining two studies, those subjects angio-graphically-confirmed as normal were used as controls. CAD patients with stenosis greater than 50% in at least one coronary artery were used as the CAD case group in eight studies [15] [18 – 21,23,25] and [27] whereas the four remaining studies [14,26,32,33] did not indicate which criteria of angiography was employed. In two studies for CAD and three studies for MI, the control group was matched for age and sex, whereas the re-maining 13 studies did not indicate whether controls were matched for age and sex. All of the studies used a standardized laboratory method [12] to determine the MTHFR genotype. Three studies were conducted in Japan [20,25,33]; the others were conducted in Aus-tralia [15,19], the United States [18,23], Netherlands [21,24], Italy [26,32], and Germany [27].
S.H.Jee et al./Atherosclerosis153 (2000) 161 – 168 163
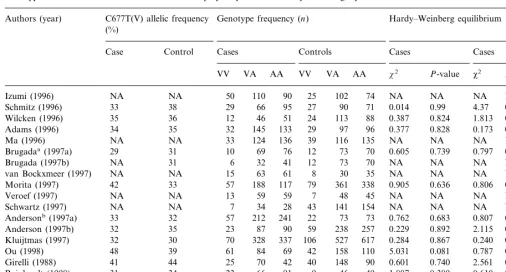
Table 3 shows the genotypic and allelic distribution of the MTHFR gene and its Hardy – Weinberg equilibrium. In all the studies included, the distributions of the genotypes were in the Hardy – Weinberg equilibrium for both patients and controls. From 12 studies with angio-graphically-confirmed coronary artery disease (CAD), the frequency of the three genotypes among controls was A/A (homozygous normal), 43.6%; V/A (heterozygous), 45.2%; and V/V (homozygous mutant, 677 CT), 11.2%. These frequencies in cases were 42.1% for A/A, 43.9% for V/A, and 13.2% for V/V.
3.1. Coronary artery disease
Twelve case-control studies examined the risk of CAD associated with those having the MTHFR gene.
Geno-type VV increased in the risk of CAD compared to the corresponding AA group in eight (66.7%) of the 12 studies. In just two of these studies, the relationship was statistically significant.
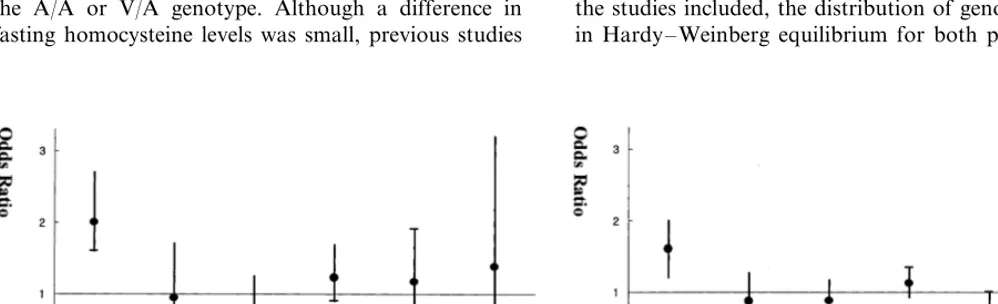
Table 4 provides the overall odds ratios of CAD across these 12 case-control studies. Compared to the A/A genotype, the overall odds ratio for CAD was 1.3 (95% CI. 1.1 – 1.5) for the V/A genotype and 1.4 (1.2 – 1.6) for the V/V genotype. However, after excluding the three Japanese studies [20,25,33] which had a strong associa-tion between MTHFR and CAD [20,25], the correspond-ing CAD odds ratios for the V/V or V/A genotypes were not statistically significant (1.1, 0.9 – 1.3) and (1.1, 0.9 – 1.3), respectively. For the three Japanese studies [20,25,33] the overall odds ratios were 1.6 (1.2 – 2.0) for the VA genotype and 2.0 (1.6 – 2.7) for the VV genotype.
Table 1
Case and control characteristics of 18 case-control studies
Authors (year) Cases Controls
Selection criteria Number Selection criteria Number,
outcome
250 CAD 201 Subjects without ischemic heart disease
Izumi (1996) Angiographically demonstrated ischemic heart disease
Clinical history and creatinine kinase rise
190 MI 188 Random from residents, age and sex matched
Schmitz (1996)
\50% occlusion in at least in one coronary 225 Volunteers participating in a heart health 109 CAD
Wilcken (1996)
education program artery
310 MI
Adams (1996) WHO criteria 222 Adults visitors to patients with
non-cardiovascular illness 293 MI WHO criteria
Ma (1996) 290 Participants who were free of MI; matching
of age, sex, and smoking \50% occlusion in at least in one coronary
155 CAD 155 Healthy Caucasian individuals, matched for
Brugadaa(1997a)
artery age and sex
79 MI
Brugada (1997b) WHO criteria 155 Healthy Caucasian individuals, matched for age and sex
Residents of metropolitan Perth, with no 73
139 CAD \50% occlusion in at least in one coronary van Bockxmeer
history
(1997) artery
362 CAD
Morita (1997) \50% occlusion in at least in one coronary 778 Healthy volunteers from annual health examination
artery 131 CAD
Veroef (1997) \90% occlusion in one and\40 occlusion 100 Population-based controls in one additional coronary artery
79 MI Cardiovascular health study
Schwartz (1997) 386 Residents of King, Pierce or Snohomish
counties 168
510 CAD
Andersonb \60% occlusion in at least in one coronary B10% occlusion in all major vessels
(1997a) artery
Patients selected on basis of being healthy
Anderson 200 MI History of MI 554
(1997b)
Angiographically assessed CAD
735 CAD Healthy people
Kluijtmas (1997) 1250
168 Patients withB10% luminal obstruction 510 CAD
Ou (1998) \60% occlusion in at least in one coronary artery
Girelli (1998) 278 CAD Angiographically documented multivessel 137 Angiographically documented normal coronary arteries
CAD
\50% occlusion in at least in one coronary
180 CAD 105
Reinhardt (1998) Healthy volunteers
artery
84 CAD Angiographically documented CAD
Abbate (1998) 106 Healthy subjects in the same age bracket
S
.
H
.
Jee
et
al
.
/
Atherosclerosis
153
(2000)
161
–
168
Table 2
Participants and study design characteristics of 18 case-control studies
Plasma folate (nmol/l) Body mass index (kg/m2)
Author (year) Country Age (years) Percentage of males
Range Mean
Case Control Case Control Case Control
Control
Case Control Case
Japan 60 59 43–76 74 54 NA NA 23.3 23.3
Izumi (1996) 41–80
USA 58 59 B76 NA NA 9.1 9.8 25.9 25.7
Schmitz (1996) B76
72 46 NA NA 28.2
18–65 28.2c
42 65
Wilcken (1996) Australia NA
NA
UK 65 56 NA 65 64 NA NA 25.8 25.4
Adams (1996)
100 NA 3.3 3.7 NA
Ma (1996) USA 62 NA 40–84 NA NA
72 72 NA NA NA
NA NA
USA
Brugadaa(1997a) 60 60 NA
NA
USA NA 60 NA NA 72 NA NA NA NA
Brugada (1997b)
van Bockxmeer (1997) Australia NA NA B50 B50 NA NA 6.4 7.6 NA NA
100 100 NA NA 23.8
26–86 23.8c
NA
Morita (1997) Japan 62 48
85 60 NA NA 26.6
Veroef (1997) Netherlands 53 50 NA NA 26.0
NA NA NA NA NA
18–44 NA
Schwartz (1997) USA NA NA 18–44
17–89
USA 63 65 31–89 79 67 NA NA NA NA
Anderson (1997a)b
79 48 NA NA NA
17–84 NA
USA
Anderson (1997b) 65 62 36–89
NA
Netherlands NA NA NA 100 100 NA NA NA NA
Kluijtmas (1997)
NA NA 86 87 NA NA 23.2 23.9
Japan
Ou (1998) 55 55
87 58 11.3 14.4 26.3
NA 24.9
61 NA
Girelli (1988) Italy 59
NA
Germany 61 52 NA 75 67 NA NA 26.9 27.0
Reinhardt (1998)
NA
Abbate (1998) Italy NA NA NA NA NA NA NA NA NA
83 69 7.5 8.9 25.5 25.4
17–89 18–89
56
Overall 60
aBrugada (1997): a, outcome was a CAD; b, outcome was a MI.
S.H.Jee et al./Atherosclerosis153 (2000) 161 – 168 165
Table 3
Genotypic and alleic distributions of the MTHFR polymorphism and Hardy–Weinberg equilibrium of 18 case-control studies
C677T(V) allelic frequency
Authors (year) Genotype frequency (n) Hardy–Weinberg equilibrium (%)
Control Cases Controls
Case Cases Cases
VV VA AA VV VA AA x2 P-value x2 P-value
NA 50 110 90 25
Izumi (1996) NA 102 74 NA NA NA NA
38 29 66 95 27
Schmitz (1996) 33 90 71 0.014 0.99 4.37 0.11
36 12 46 51 24 113
35 88
Wilcken (1996) 0.387 0.824 1.813 0.404
34
Adams (1996) 35 32 145 133 29 97 96 0.377 0.828 0.173 0.917
NA 33 124 136 39 116
NA 135
Ma (1996) NA NA NA NA
31 10 69 76 12 73
Brugadaa(1997a) 29 70 0.605 0.739 0.797 0.671
31 6 32 41 12 73
NA 70
Brugada (1997b) NA NA NA NA
NA
van Bockxmeer (1997) NA 15 63 61 8 30 35 NA NA NA NA
33 57 188 117 79 361
42 338
Morita (1997) 0.905 0.636 0.806 0.668
NA
Veroef (1997) NA 13 59 59 7 48 45 NA NA NA NA
NA 7 34 28 43 141 154 NA NA
Schwartz (1997) NA NA NA
32 57 212 241 22 73
33 73
Andersonb(1997a) 0.762 0.683 0.807 0.668
35 23 87 90 59 238
Anderson (1997b) 32 257 0.229 0.892 2.115 0.347
30 70 328 337 106 527
32 617
Kluijtmas (1997) 0.284 0.867 0.240 0.887
39 61 84 69 42 158
Ou (1998) 48 110 5.031 0.081 0.787 0.675
44 25 70 42 40 148
41 90
Girelli (1988) 0.601 0.740 2.561 0.278
34 23 66 91 9 46
Reinhardt (1998) 31 49 1.887 0.389 0.610 0.737
53 25 38 21 30 52
53 24
Abbate (1998) 0.022 0.989 0.052 0.951
33
Overall 33 473 1673 1667 558 2332 2298 2.134 0.344 0.080 0.961
aBrugada (1997): a, outcome was a CAD; b, outcome was a MI.
bAnderson (1997): a, outcome was a CAD; b, outcome was a MI. Note: NA means data was not available.
Table 4
Odds ratios (OR) of coronary artery disease associated with MTHFR gene in 12 case-control studies
VV vs AAa
No. of studies VA vs AAa
OR 95% CI P-value OR 95% CI P-value
1.4 1.2–1.6 0.0002
Overall 12 1.3 1.1–1.5 0.0056
Japanese studies [20,25,33] excluded 9 1.1 0.9–1.3 0.42 1.1 0.9–1.3 0.49 Japanese studies [20,25,33] only 3 2.0 1.6–2.7 B0.00001 1.6 1.2–2.0 0.00047
aVV, homozygous for the mutant; VA, heterozygous; AA, homozygous normal.
3.2. Myocardial infarction
Six studies examined the relationship between the risk of myocardial infarction (MI) and the MTHFR gene. Genotype VV increased the risk of MI compared to the corresponding AA group in four (66.7%) of six studies; while in none of these studies was the lower bound of the 95% confidence interval greater than one. Compared to those with the A/A genotype, the overall odds ratio for MI associated with the MTHFR gene was 1.0 (0.8 – 1.1) among those with the V/A genotype and 0.9 (0.7 – 1.2) among those with the V/V genotype (eachP\0.05).
3.3. Coronary artery disease or myocardial infarction
Using data from all 18 studies, the occurrence of
CAD or MI was assessed by the MTHFR genotype. Compared to the A/A genotype, the overall odds ratio for the combination of CAD and MI was 1.1 (95% CI, 1.0 – 1.2) for the V/A genotype and 1.2 (1.1 – 1.4) for the V/V genotype. After excluding the three Japanese stud-ies, the corresponding odds ratios were 1.0 (0.9 – 1.2) and 1.0 (0.9 – 1.1).
4. Discussion
MTHFR gene on the combination of CAD and MI, and CAD disappeared after excluding three Japanese studies. The discrepancy between the results from the Japanese studies and other populations is unclear and may be due to differences in nutritional intake of co-factors required for the MTHFR pathway, such as vitamin B12 or folate, or to other ethnic differences (e.g. weight, BMI).
Over the past decade, a consensus has emerged that elevated plasma homocysteine is an independent risk factor for coronary heart disease [37 – 39]. MTHFR, a folate-dependent enzyme, is essential for the remethyla-tion of homocysteine. Frosst et al. [12] identified a single common mutation of the MTHFR gene that causes reduced activity and thermolability of the en-zyme; while homozygosity for this mutant allele results in significantly-elevated plasma tHcy levels. Therefore, in the Japanese studies, the finding of an increased risk of CAD associated with MTHFR is not surprising because MTHFR is strongly associated with an in-creased risk of plasma homocysteine. In particular, Morita [20] reported that the frequency of this mutation was correlated with the severity of stenotic lesions (the presence of \=99% stenosis) and the number of stenotic coronary arteries, suggesting that this mutation is closely associated with the severity of CAD. In our meta-analysis, the V allele has a codomi-nant effect on coronary arterial risk among the Japanese studies.
Previously, Kang et al. [10] reported that thermo-labile MTHFR, which is thought to be correlated with the V/V genotype, can be an inherited risk factor for CAD. They documented a prevalence of thermolabile MTHFR of 17% in patients with CAD and of 5% in control subjects. Frosst et al. [12] documented that plasma homocysteine levels were significantly higher in patients with the V/V genotype than in patients with the A/A or V/A genotype. Although a difference in fasting homocysteine levels was small, previous studies
have suggested that plasma homocysteine levels after dietary methionine loading are greatly affected by this mutation. These findings suggested that the V/V geno-type of MTHFR, which can cause a predisposition to increased plasma homocysteine levels, may itself be an independent genetic risk factor for CAD.
However, our meta analysis does not support the hypothesis that the MTHFR gene is an independent predictor of developing CAD. Still, the MTHFR gene was a strong predictor for CAD in Japanese individu-als, who have a low fat diet and are generally lean. According to the present study, when BMI is high, the MTHFR gene has little effect on CAD (Fig. 1). Wilcken et al. [15] reported that the MTHFR mutation was associated with increased BMI, although MTHFR was still not associated with the number of diseased vessels. Therefore, it will be of great interest to deter-mine the impact of this polymorphism in other lean populations, e.g. other East-Asian countries, and sub-samples of other populations, e.g. populations with a lower intake of folate. In this study, unfortunately, we could not examine this hypothesis because only four studies reported the plasma intake level. According to the Frosst study [15], when folate consumption is high, this common genetic variant has little effect on tHcy levels. It will be of great interest to determine the impact of this polymorphism in a general population sample with a lower intake of folate, as well as among younger individuals.
Overall, we found no association between the MTHFR genotype and the risk of MI. However, be-cause only six studies included this outcome, these analyses have relatively little power to test for an association between the risk of MI and MTHFR gene mutation. Schmitz (1996) [14] reported that only the survivors of MI entered their study, and this group may not be representative of all MI cases. However, in all the studies included, the distribution of genotypes were in Hardy – Weinberg equilibrium for both patients and
S.H.Jee et al./Atherosclerosis153 (2000) 161 – 168 167
controls, which meant that there was no evidence of selected samples of cases and controls.
Our results raise several important research ques-tions. Specifically, what factors, genetic and environ-mental, account for the apparent association of MTHFR genotypes with CAD in Japan? In that coun-try, the population tends to be leaner and to have lower levels of serum cholesterol than Western countries. Does leanness or lower cholesterol modify the risk of a relationship between MTHFR and CAD? Additional research, e.g. prospective observational studies in lean populations and those with a low intake of folate, is warranted.
Acknowledgements
This study was supported by a grant (No. HMP-98-M-1-0004) of the 1998 Good Health R&D Project, Ministry of Health and Welfare, Korea.
References
[1] Wilcken DEL, Wilcken B. The pathogenesis of coronary artery disease: a possible role for methionine metabolism. J Clin Invest 1976;57:1079 – 82.
[2] Boers GHJ, Smals AGH, Trijbels JMF, Fowler B, Bakkeren JAJM, Schoonderwaldt HC, Kleijer WT, et al. Heterozygosity for homocystinuria in premature peripheral and cerebral occlu-sive arterial disease. New Engl J Med 1985;313:709 – 15. [3] Kang S-S, Wong PWK, Malinow MR. Hyperhomocyst(e)inemia
as a risk factor for occlusive vascular disease. Annu Rev Nutr 1992;12:279 – 98.
[4] Malinow MR. Homocyst(e)ine and arterial occlusive disease. J Intern Med 1994;236:603 – 17.
[5] Den Heijer M, Blom HJ, Gerrits WBJ, Rosendaal FR, Haak HL, Wijermans PW, Bos GMJ. Is hyperhomocysteinaemia a risk factor for recurrent venous thrombosis? Lancet 1995;345:882 – 5. [6] Boushey CJ, Bereford SA, Omenn GS, Motulsky AG. A quanti-tative assessment of plasma homocysteine as a risk factor for vascular disease: probable benefits of increasing folic acid in-takes. J Am Med Assoc 1995;274:1049 – 57.
[7] Miller JW, Ribaya-Mercado JD, Russell RM, Shepard DC, Morrow FD, Cochary EF, Sadowski JA, et al. Effect of vitamin B-6 deficiency on fasting homocysteine concentrations. Am J Clin Nutr 1992;55:1154 – 60.
[8] Daly L, Robinson K, Tan KS, Graham IM. Hyperhomocys-teinemia: a metabolic risk factor for coronary heart disease determined by both genetic and environmental influences? Quart J Med 1993;86:685 – 9.
[9] Selhub J, Jacques PF, Wilson PWF, Rush D, Rosenberg IH. Vitamin status and intake as primary determinants of homocys-teinemia in an elderly population. J Am Med Assoc 1993;270:2693 – 8.
[10] Kang SS, Wong PW, Susmano A, Sora J, Norusis M, Ruggie N. Thermolabile methylenetetrahydrofolate reductase: an inherited risk factor for coronary artery disease. Am J Hum Genet 1991;48:536 – 45.
[11] Engbersen AM, Franken DG, Boers GH, Stevens EM, Trijbels FJ, Blom HJ. Thermolabile 5,10-methylenetetrahydrofolate re-ductase: as a cause of mild hyperhomocysteinemia. Am J Hum Genet 1995;56:142 – 50.
[12] Frosst P, Blom HJ, Milos R, Goyette P, Sheppard CA, Mathewes RG, Boers GJ, den Heijer M, Kluijtmans LA, van den Heuvel LP, Rozen R. A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofo-late reductase. Nat Gene 1995;10:111 – 3.
[13] van der Put NM, Steegers-Theunissen RP, Frosst P, Trijbels FJ, Eskes TK, van den Heuvel LP, Mariman EC, den Heyer M, Rozen R, Blom HJ. Mutated methylenetetrahydrofolate reduc-tase as a risk factor for spina bifida. Lancet 1995;346:1070 – 1. [14] Schmitz C, Lindpaintner K, Petra Verhoef P, Gaziano JM,
Buring J. Genetic polymorphism of methylenetetrahydrofolate reductase and myocardial infarction. A case-control study. Cir-culation 1996;94:1812 – 4.
[15] Wilcken DEL, Wang XL, Sim AS, McCredie RM. Distribution in healthy and coronary population of the methylenetetrahydro-folate reductase (MTHFR) C677T mutation. Arterioscler Thromb Vas Biol 1996;16:878 – 82.
[16] Adams M, Smith PD, Martin D, Thompson JR, Lodwick D, Samani NJ. Genetic analysis of thermolabile methylenetetrahy-drofolate reductase as a risk factor for myocardial infarction. Quart J Med 1996;89:437 – 44.
[17] Ma J, Stampfer MJ, Hennekens CH, Frosst P, Selhub J, Hors-ford J, Malinow MR, Willett WC, Rozen R. Methylenetetrahy-drofolate reductase polymorphism, plasma folate, homocysteine, and risk of myocardial infarction in US physicians. Circulation 1996;94:2410 – 6.
[18] Brugada R, Marian AJ. A common mutation in methylenete-trahydrofolate reductase gene is not a major risk of coronary artery disease or myocardial infarction. Atheriosclerosis 1997;128:107 – 12.
[19] van Bockxmeer FM, Mamotte CDS, Vasikaran SD, Taylor RR. Methylenetetrahydrofolate reductase gene and coronary artery disease. Circulation 1997;95:21 – 3.
[20] Morita H, Taguchi J, Kurihara H, Kitaoka M, Kaneda H, Kurihara Y, Maemura K, Shindo T, Minamino T, Ohno M, Yamaoki K, Ogasawara K, Aizawa T, Suzuki S, Yazaki Y. Genetic polymorphism of 5,10-methylenetetrahydrofolate reduc-tase (MTHFR) as a risk factor for coronary artery disease. Circulation 1997;95:2032 – 6.
[21] Verhoef P, Kok FJ, Kluijtmans LAJ, Blom HJ, Refsum H, Ueland PM, Kruyssen DACM. The 677CT mutation in the methylenetetrahydrofolate reductase gene: associations with plasma total homocysteine levels and risk of coronary atherosclerotic disease. Atherosclerosis 1997;132:105 – 13. [22] Schwartz SM, Siscovick DS, Malinow MR, et al. Myocardial
infarction in young women in relation to plasma total homocys-teine, folate, and a common variant in the methylenetetrahydro-folate reductase gene. Circulation 1997;96:412 – 7.
[23] Anderson JL, King GJ, Thomson MJ, et al. A mutation in the methylenetetrahydrofolate reductase gene is not associated with increased risk for coronary artery disease or myocardial infarc-tion. J Am Coll Cardiol 1997;30:1206 – 11.
[24] Kluijtmans LAJ, Kastelein JJP, Lindemans J, et al. Thermolabile methylenetetrahydrofolate reductase in coronary artery disease. Circulation 1997;96:2573 – 7.
[25] Ou T, Kobayashi Y, Arinami T, et al. Methylenetetrahydrofo-late reductase and apolipoprotein E polymorphisms are indepen-dent risk factors for coronary heart disease in Japanese: a case-control study. Atheroslcerosis 1998;137:23 – 8.
[26] Girelli D, Friso S, Trabetti E, et al. Methylenetetrahydrofolate reductase C677T mutation, plasma homocysteine, and folate in subjects from Northern Italy with or without angiographically documented severe coronary atherosclerotic disease: evidence for an important genetic-environment interaction. Blood 1998;91(11):4158 – 63.
methylenetetrahydrofo-late reductase gene and the risk of coronary artery disease. Eur J Clin Invest 1998;28:20 – 3.
[28] Kluijtmans LAJ, van den Heuvel LPWJ, Boers GHJ, Frosst P, Stevens EMB, van Oost BA, den Heijer M, Trijbels FJM. Molecular genetic analysis in mild hyperhomocysteinemia: a common mutation in the methylenetetrahydrofolate reductase gene is a genetic risk factor for cardiovascular disease. Am J Hum Genet 1996;58:35 – 41.
[29] Gallagher PM, Meleady R, Shields DC, Tan KS, McMaster D, Rozen R, Evans A, Graham IM, Whitehead AS. Homocysteine and risk of premature coronary heart disease; evidence for a common gene mutation. Circulation 1996;94:2154 – 8.
[30] Deloughery TG, Evans A, Sadeghi A, Mcwilliams J, Henner WD, Taylor LM, Press RD. Common mutation in methylenete-trahydrofolate reductase; correlation with homocysteine metabolism and late-onset vascular disease. Circulation 1996;94:3074 – 8.
[31] Christensen B, Frosst P, Lussier-Cacan S, Selhub J, Goyette P, Rosenblatt DS, Genest J, Rozen R. Correlation of a common mutation in the methylenetetrahydrofolate reductase gene with plasma homocysteine in patient with premature coronary artery disease. Arterioslcer Thromb Vasc Biol 1997;17:569 – 73. [32] Abbate R, Sardi I, Pepe G, et al. The high prevalence of
thermolabile 5,10-methylenetetrahydrofolate reductase (MTHFR) in Italians is not associated to an increased risk for coronary artery disease (CAD). Thromb Haemost 1998;79:727 – 30.
[33] Izumi M, Iwai N, Ohmichi N, et al. Molecular variant of 5,10-methylenetetrahydrofolate reductase is a risk factor of is-chemic heart disease in the Japanese population. Atheroslcerosis 1998;121:293 – 4.
[34] Dunn J, Title LM, Bata I, et al. Relation of a common mutation in methylenetetrahydrofolate reductase to plasma homocysteine and early onset coronary artery disease. Clin Biochem 1998;31(2):95 – 100.
[35] Kostulas K, Crisby M, Huang WX, et al. A methylenetetrahy-drofolate reductase gene polymorphism in ischaemic stroke and in carotid artery stenosis. Eur J Clin Invest 1998;28(4):285 – 9. [36] Cooper H, Hedges LV. The Handbook of Research Synthesis.
New York: Russell Sagge Foundation, 1994.
[37] Clarke R, Daly L, Robinson K, Cahalane S, Fowler B, Graham I. Hyperhomocysteinemia: an independent risk factor for vascu-lar disease. New Engl J Med 1991;324:1149 – 55.
[38] Kang SS, Wong PWK, Cook HY, Norusis M, Messer JV. Protein-bound homocyst(e)ine: a possible risk factor for coro-nary artery disease. J Clin Invest 1986;77:1482 – 6.
[39] Arnesen E, Refsum H, Bonaa K, Ueland PM, Forde OH, Nordrehaug JE. Serum total homocysteine and coronary heart disease. Int J Epidemiol 1995;24:704 – 9.
[40] Brattstrom L, Wilcken DEL, Ohrvik J, Brudin L. Common methylenetetrahydrofolate reductase gene mutation leads to hy-perhomocysteinemia but not to vascular disease: the results of a meta-analysis. Circulation 1998;98:2520 – 6.