Ascorbic acid enhances 17
b
-estradiol-mediated inhibition of
oxidized low density lipoprotein formation
Juliana Hwang
a,b,*, Hazel Peterson
a, Howard N. Hodis
a,b,c, Bune Choi
c,
Alex Sevanian
a,baDepartment of Molecular Pharmacology and Toxicology,School of Pharmacy,Uni6ersity of Southern California,1985Zonal A6enue,PSC612, Los Angeles,CA90033,USA
bUni6ersity of Southern California,School of Medicine,Atherosclerosis Research Unit,Di6ision of Cardiology,Los Angeles,CA,USA cDepartment of Pre6enti6e Medicine,School of Medicine,Uni6ersity of Southern California,Los Angeles,CA,USA
Received 20 April 1999; received in revised form 15 June 1999; accepted 20 August 1999
Abstract
Postmenopausal women who use estrogen appear to be protected from coronary heart disease (CHD). Studies have demonstrated that estrogen can lower low-density lipoprotein (LDL) levels and the antioxidant activity of 17 b-estradiol can prevent the oxidation of this LDL. Ascorbic acid is regarded as a major hydrophylic antioxidant, however, its impact on the prevention of CHD has yet to be clearly demonstrated. Modified low density lipoprotein (LDL−) is an important marker of LDL
oxidation in vivo, since it contributes to the oxidative susceptibility of low density lipoprotein, and at physiological levels displays pro-inflammatory and cytotoxic properties. Previously we showed that women taking estrogen replacement therapy have lower LDL− levels along with lower predisposition of the LDL to oxidize. In this study, we evaluated the potential action of 17
b-estradiol (E2) in combination with ascorbic acid (AA) measured on the basis of LDL oxidative susceptibility in vitro and in the
presence of cultured cells. High concentrations of E2were able to inhibit LDL oxidation, whereas in the presence of ascorbic acid
nano- to picomolar levels of E2were sufficient to suppress LDL oxidation (PB0.05). Preconditioning male aortic endothelial cells
(RAEC) with 5 ng/ml of E2(E2RAEC) reduced the formation of LDL−(PB0.005), and a more extensive inhibition was found
in the presence of AA (PB0.0001). Interestingly, E2enhanced the uptake of LDL in the absence or presence of AA, however,
this was not seen for the uptake of LDL−. These results provide the first evidence that ascorbic acid can enhance the antioxidant
effect of E2by preventing LDL oxidation by copper ions or cells. The cytoprotective and antiatherogenic effect of E2appears to
involve a reduction in the extent of oxidized LDL formation and uptake. The enhanced activity of E2in the presence of ascorbate
indicates that the antioxidant and antiatherosclerosis activity of E2may occur at concentrations within the physiological range.
© 2000 Elsevier Science Ireland Ltd. All rights reserved.
Keywords:Estrogen; Coronary heart disease; Low-density lipoprotein; 17b-estradiol; Male aortic endothelial cells; Ascorbic acid
www.elsevier.com/locate/atherosclerosis
1. Introduction
Coronary heart disease (CHD) is the leading cause of death in men and women in the United States and in developing countries. The progression of CHD is deter-mined by several factors among which gender is a striking example. The risk for CHD is less in pre-menopausal women versus age-matched men, however, this ‘protective’ effect is lost with menopause and
po-tentially regained with estrogen replacement therapy (ERT). The potentially protective effect of estrogen has been described in animal and epidemiological studies and is now being investigated by several randomized controlled clinical trials.
The mechanisms by which estrogen inhibits atheroge-nesis are not known, but part of this beneficial effect may be explained by alteration of the plasma lipo-protein profile [1 – 4]. Changes in the lipid profile do not fully account for the cardioprotective effect afforded by estrogen, indicating that other mechanisms are likely to be involved [5,6]. One mechanism may include estro-gen’s antioxidant activity [7]. Estrogen exhibits natural * Corresponding author. Tel.: +1-323-4423355; fax: +
1-323-2247473.
E-mail address:[email protected] (J. Hwang)
antioxidant activity during membrane phospholipid peroxidation [8], influences agonist-mediated calcium signaling responses [9], potentiates endothelium-derived relaxing factor in coronary arteries [10], and inhibits LDL modification [11,12]. Estrogen also facilitates prostracyclin production [13,14], limits cholesterol ester influx [15], reduces apoptosis of human endothelial cells [16], and suppresses TNF-a, IL-1 and NFkB activation as induced by lipopolysacharides [17]. Many of these processes are linked to the production of reactive oxy-gen species (ROS) and hence the modulation of ROS production and consequent effects on cell redox status may be a mechanism by which estrogen exerts its ‘antioxidant/atheroprotective’ activity.
It is widely held that oxidative modification of low density lipoprotein (LDL) contributes to the develop-ment of atherosclerosis [18,19]. Oxidized LDL has been found in human plasma as a more electronegatively charged lipoprotein (LDL−) that contains a number of oxidized components [12,20 – 22] similar to other forms of oxidized LDL produced in vitro [11,23,24]. LDL− exists mainly in the denser LDL subclass (\1.055 g/ml), is more susceptible to oxidation and is capable of facilitating the oxidation of other lipoproteins [25]. Individuals with higher levels of small dense LDL (1.050 – 1.063 g/ml) are more prone to CHD [26], sug-gesting that the risk factor(s) may reside in an increased proportion of oxidatively modified LDL subclasses.
Reports have been inconsistent in postmenopausal women taking ERT in regards to the susceptibility of LDL to oxidize in vitro. Some reports have shown that ERT can inhibit LDL and HDL oxidation in the presence of copper [27,28] whereas others have shown that there was no effect on LDL oxidative susceptibility in postmenopausal women receiving ERT [2,29].
Inhibition of LDL oxidative modification in vivo has been approached in large measure through the use of dietary antioxidants. Studies have often included ascor-bic acid, a central component of the antioxidant de-fense system [30]. Ascorbate intake and dietary demand varies with lifestyle and age, and in the latter instance plasma levels were shown to decrease during aging at a rate of approximately 0.06 mg/dl per decade [31]. Men tend to have lower levels of ascorbic acid than women [32] and require larger doses of ascorbic acid to reach the same serum concentration [33]. This gender related effect seems to begin in adolescence and is thought to be hormonally mediated [34].
Epidemiological studies suggest that increased ascor-bic acid intake may afford protection against cardiovas-cular disease based on the postulated inhibition of lipoprotein oxidation, regulation of cholesterol homeostasis, and lowering of blood pressure [34]. Mar-tin et al. [35] reported recently that both intracellular and extracellular ascorbic acid inhibited endothelial cell-mediated LDL oxidative modification. Surprisingly,
there is very little information concerning the interac-tion between estradiol and ascorbic acid with respect to their effects on oxidatively modified LDL formation. In this report, we describe an antioxidant action of estro-gen that is facilitated by ascorbic acid.
2. Methods
2.1. Chemical and reagents
The following reagents were obtained from Sigma Chemical Co. (St. Louis, MO): EDTA, BHT, 17 b -estradiol (E2), progesterone, L-ascorbic acid (Vitamin C), 2,4,6, tripyridyl-s-triazine, ferric chloride, sodium acetate, NaBr, NaCl, TRIS, trichloroacetic acid, metaphosphoric acid, triethanolamine, 2-vinylpyridine. 1,1%-dioctadecyl-3,3,3%,3%-tetramethylindocarbocyanine perchlorate (DiI), was purchased from Molecular Probes (Eugene, OR). Buffers, media, and cell culture supplies were obtained from Life Science (New York, NY) and male serum from Omega Scientific (Van Nuys, CA). All organic solvents and copper sulfate were HLPC grade and purchased from T.J. Baker Chemical Co. (Phillipsburg, NJ).
2.2. Lipoprotein separation
Venous blood was obtained from fasting adult hu-man volunteers and plasma was immediately separated by centrifugation at 1500×g for 10 min at 4°C. LDL (d=1.019 – 1.063 g/ml) was isolated from freshly sepa-rated plasma by preparative ultracentrifugation using a Beckman L8-55 ultracentrifuge and a SW-41 rotor. The technique used for separating LDL was similar to that described previously [20]. The isolated LDL was exten-sively dialyzed against argon-sparged 0.01 mol/l TRIS buffer, pH 7.2, containing 10 mmol/l EDTA, sterilized by filtration (0.2mm Millipore membrane) and stored at 4°C under nitrogen.
2.3. LDL−
preparation
2.4. In 6itro oxidation of LDL
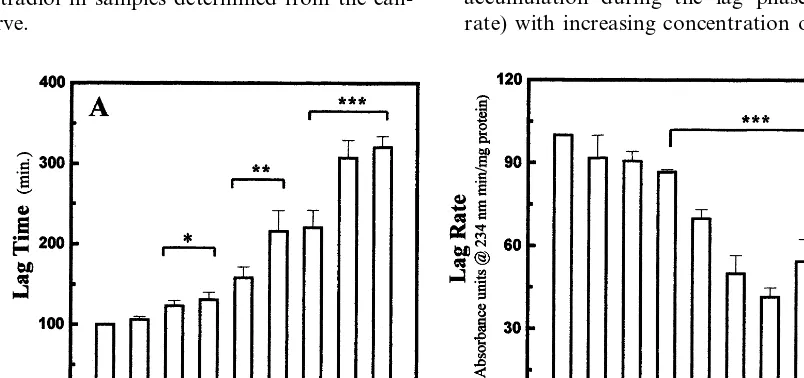
The kinetics of LDL oxidation were analyzed by adding 10 mmol/l CuSO4 to 200 mg/ml LDL protein. Formation of conjugated dienes was monitored contin-uously at 234 nm for up to 5 h using a Beckman DU-650 spectrophotometer equipped with a six posi-tion automated sample changer. Oxidaposi-tion kinetics were analyzed in phosphate buffer on the basis of: (1) the oxidation lag time which was defined as the interval between initiation of oxidation and the intercept of the tangent for the slope of the absorbance curve during the propagation phase; (2) the rate of oxidation during the lag time, defined as the initial oxidation rate before the onset of the propagation phase; (3) the rate of oxidation during the propagation phase, i.e. thelog rate which was defined as the maximal rate of oxidation calculated from the slope of the absorbance curve dur-ing the propagation phase.
The kinetics of LDL oxidation were analyzed in the presence or absence of E2, progesterone, a-tocopherol (added in ethanol), and ascorbic acid. The amounts of ethanol vehicle were the same for all the experiments (0.2% v/v).
2.5. Vitamin E measurements
LDL fractions were collected during the kinetic assay and extracted for vitamin E analysis. A modified method of Bui [36] was used to measure vitamin E levels. 500 ml of LDL was mixed with 50ml of internal standard (a-tocopherol acetate) and extracted twice with 500 ml of hexane containing BHT. The pooled phases were evaporated under nitrogen, the residue dissolved in 200 ml of ethanol, and injected into a Perkin Elmer Series 4 HPLC equipped with a BioRad reverse-phase column Bio-Sil ODS-5S, 250×4 mm (BioRad Instruments, CA). Samples were eluted with acetronitrile-tetrahydrofuran-H2O at 0.9 ml/min, the eluent monitored using a UV/Vis monitor model 1706 (BioRad instruments, CA), and the peak areas inte-grated with Axxi-chrom 747 analytical chromatography software. The amounts of vitamin E (a and g -toco-pherol) were measured using established calibration curves.
2.6. Ascorbic acid measurement
Total ascorbic acid levels were determined by a modified colorimetric method of Day et al. [37] based on the reduction of ferric chloride by ascorbic acid. Samples were added to 0.615 M of trichloroacetic acid, mixed, and centrifuged at 1500 rpm for 10 min and the supernatant mixed with the color reagent consisting of 2,4,6 tripyridyl-s-triazine and ferric chloride in sodium acetate buffer.
2.7. Cell culture
Male New Zealand White rabbit aortic endothelial cells (RAEC) were used between passages 19 and 21. Cells were passaged using a 1:2 split ratio, allowed to grow to confluency, and transferred by trypsinization. The doubling time of the cells was approximately 38 h in complete media (DMEM without phenol red con-taining 15% male serum, 20 mg/ml ECGS, 50 mg/ml gentamicin and 20mg/ml heparin). This is referred to as control medium. Prior (5 days) to the experiments, cells were split in complete media in the presence (E2RAEC) or absence (RAEC) of 5 ng/ml of E2.
2.8. Cytotoxicity assays
Cells were seeded into 24-well dishes 1 day prior to measurements of cell survival and plating efficiency which were used as estimates of cytotoxicity following various treatments. The cytotoxicity produced by LDL and LDL− was determined using nearly confluent cul-tures grown in the presence of 2% male serum and cell numbers measured after 24 h incubation using a Coul-ter counCoul-ter (Model ZB). Treatments typically used 1× 104
cells/cm2
. The parameters related to the measurement of cytotoxicity included: plating efficiency (PE) and growth curves based on the surviving fraction (SF) measured at 24 h after treatment with lipoprotein.
2.9. Measurement of cell-mediated oxidation
Formation of LDL−by endothelial cells was used as a measure of cell-mediated LDL oxidation. LDL− for-mation was determined after adding 100mg/ml of fresh LDL protein and 0.5 mM CuSO4 to endothelial cell cultures comprised of 2×105 cells/10 cm2. Ratios of LDL− formation relative to total LDL were measured over a 24 h interval for control cells with and without 100 mM of ascorbic acid, and E2RAEC cultures with and without ascorbic acid in serum-free control media. At selected time points, aliquots of the media were removed and centrifuged at 800 rpm for 5 min to remove dead cells and cell debris. The supernatant was mixed with 15 ml PBS and concentrated using a Centri-con 30 000 molecular weight cut-off microCentri-concentrator to adjust LDL concentrations and remove low molecu-lar weight components from the medium. These brief reconstituting procedures and adjustments of LDL con-centrations did not affect the amounts of LDL− forma-tion [25] [20]. Samples were concentrated to the volume of the initial aliquot removed from the cell culture medium and then analyzed by HLPC for LDL− con-tent. LDL− levels were calculated on the basis of the cell number and corrected for LDL−
2.10. DiI LDL uptake
Sub-confluent cultures (4×104
cells) were treated with DiI-labled lipoproteins at 10mg/5×104 cells for 2 h in complete medium in the presence of 2% male serum. The incorporated DiI was measured fluoromet-rically (523 nm Excitation/563 nm Emition) after ex-traction with isopropanol. Cells were stained with Giemsa and protein concentrations determined on the basis of absorbance at 590 nm with parallel measure-ments of cell number using a Coulter counter. Confocal microscopy (Zeiss LSM-1) was used to confirm the internalization of LDL particles [38] relative to the amount of fluorescence associated with the cell mem-brane. Measurements of relative electorphoretic mobil-ity (REM) of the LDL samples after labeling with DiI indicated that the lipoproteins were unaltered by this labeling procedure. The extent of uptake and concen-tration of lipoprotein fluorescence was estimated on the basis of relative fluorescence intensity per mg LDL protein. The assumption was made that the amount of cell-associated fluorescence represented the same fluorescence concentration as in the original LDL. Re-sults were expressed as mg uptake of LDL as mg cell protein in 2 h.
2.11. 17 b-estradiol measurement
Estradiol levels were measured using a Pantex RIA assay (Santa Monica, CA), where estradiol calibrators and fixed amounts of radiolabeled estradiol (tracer) compete for binding sites in fixed volumes of antiserum. Binding values are plotted versus concentrations, and levels of estradiol in samples determined from the cali-bration curve.
2.12. Glutathione measurements
Total glutathione (GSH) levels were measured using a standard kit (Cayman, MI), Briefly, cells were re-moved by scrapping in cold PBS, the pellets were washed once with PBS and deproteinized with 10% of metaphosphoric acid. The assay utilizes glutathione reductase for the quantification of GSSG and GSH as total GSH. Concentrations of samples were determined using established calibration curves that were run with each batch of samples.
2.13. Statistics
All results are expressed as mean and standard errors determined from at least five independent experiments with all measurements performed in duplicate. Determi-nations of statistical significance between various treat-ment groups were made using the paired two-tailed student t-test.
3. Results
3.1. In 6itro oxidation of LDL is inhibited by E2 and ascorbic acid
Fig. 1 shows an antioxidant effect of E2 that is typical of the suppression of radical chain reactions in LDL lipids by various antioxidants. The inhibitory effect of E2 was manifested by a prolongation of the oxidation lag time Fig. 1A, and inhibition of peroxide accumulation during the lag phase (a decreased lag rate) with increasing concentration of E2. In Fig. 1, E2
Fig. 2. Kinetics of LDL oxidation measured by the formation of conjugated dienes (OD234) in the presence of 17 b-estradiol. Freshly isolated human LDL (200 mg protein/ml) was incubated with 10 mmol/l of CuSO4 in the absence and presence (50 ng/ml) of 17 b-estradiol as described in Section 2. Symbols are for LDL (); LDL in the presence 50 ng/ml of 17b-estradiol ( ); LDL in the presence of 100mM of ascorbic acid ();and LDL in the presence of 100mM of ascorbic acid and 50 ng/ml of 17 b-estradiol (). Vitamin E con-sumption under the same treatment conditions is shown as open symbols.
presence of E2as compared to LDL alone (Fig. 2). This suggested that the antioxidant effect of E2 was not due to direct interaction with vitamin E, and that the presence or content of vitamin E in the LDL samples had little effect on the oxidation kinetics as influenced by E2. Also shown in Fig. 2 are the effects of ascorbic acid on LDL vitamin E levels during oxidation. The levels of vitamin E were determined during the oxida-tion of LDL alone, LDL in the presence of 50 ng/ml of E2, LDL in the presence of 100 mM of ascorbic acid and LDL in the presence of 100 mM of ascorbic acid and 50 ng/ml of E2. Relatively small decreases in vita-min E levels were found when 100mM of ascorbic acid or 100 mM of ascorbic acid and 50 ng/ml of E2 were added to LDL, indicating a preservation of vitamin E by ascorbate but not by E2 alone. When 100 mM ascorbic acid was added to the LDL samples after addition of E2, a strong antioxidant effect was evident and the amounts of added E2 required to significantly inhibit LDL oxidation was reduced to the range of 1 – 5 ng/ml (Fig. 3). The combination of ascorbic acid and E2 was 4 times more potent than E2 alone and more than twice as potent as ascorbic acid alone (Fig. 4). Vitamin E did not show the same effect as ascorbic acid (Fig. 4) when 200 mg/ml of LDL protein was incubated with either E2 and ascorbic acid or E2 and vitamin E.
Notably, progesterone added at 200 mg/ml, had no significant effect on LDL oxidation (data not shown), indicating that the observed antioxidant activity was not a general property of steroids hormones.
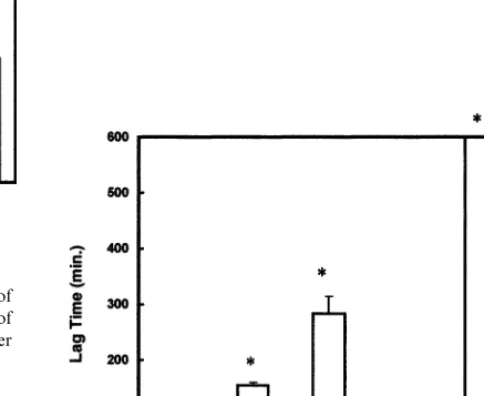
Fig. 3. Oxidation lag times of freshly isolated LDL in the presence of 10mM of CuSO4, 100mM of ascorbic acid and increasing amounts of 17b-estradiol as described in Methods. Significant differences over control values are indicated by *PB0.05.
Fig. 4. Oxidation lag times of freshly isolated LDL in the presence of 10mM of CuSO4. Comparisons are shown for 5 ng/ml of 17b -estra-diol; 100 mM of ascorbic acid; 10mM of vitamin E; 5 ng/ml of 17 b-estradiol and 100mM of ascorbic acid and 17b-estradiol plus 10 mM of vitamin E. Significant differences over control values are indicated by *PB0.05.
was added to cuvettes containing 200 mg/ml of LDL protein over a concentration range 1 – 200 ng/ml. The inhibitory effect of E2 was significant (PB0.05) at concentrations greater than 5 ng/ml. Fig. 1B shows that the oxidation lag rates were also inhibited in the pres-ence of E2 with significant inhibition occurring at con-centrations \10 ng/ml.
Table 1
Determinations of cytotoxicity after 24 h treatmentsa
Surviving fraction Treatment
RAEC+AA E2RAEC
RAEC E2RAEC+AA
0.94390.071 0.99290.044 0.98090.046 Control media 1
0.70690.080 0.80890.051
0.76890.081 0.79290.034
10mg/ml of LDL−
0.74490.076*
20mg/ml of LDL− 0.67490.088 0.96990.058* 0.82590.093
250mg/ml of LDL 0.72590.070 0.60990.095 0.79590.067 0.72190.067 0.60790.049+ 0.81090.056 **,++ 0.68990.049++
0.55190.041**,+
500mg/ml of LDL
a10 and 20mg/ml of LDL−, 250 and 500mg/ml of LDL in control cells (RAEC), control cells in the presence of 100mM of ascorbic acid
(RAEC+AA), 17b-estradiol pre-treated cells for 5 days (E2RAEC) and 17b-estradiol pre-treated cells for 5 days in the presence of 100mM of ascorbic acid (E2RAEC+AA).
*PB0.001. **PB0.0005.
+PB0.0001. ++PB0.05.
3.2. E2 cells are more resistant to LDL and LDL−
Male rabbit endothelial cells pre-incubated with 5 ng/ml of E2 (E2RAEC) for 5 days were more resistant to the cytotoxic effects of high concentrations of LDL or LDL−, as shown in Table 1. Treatments with 10 and 20 mg/ml of LDL− produced significant toxicity (PB 0.01) to control cells but no toxicity to E2RAEC under the same treatment conditions. Treatment of E2RAEC with 500 mg/ml LDL also produced no increase in cytotoxicity whereas these high levels of LDL were toxic to RAEC (PB0.001). Pre-incubation with E2was sufficient to confer protection, and the presence of E2 was not required during the treatment periods with LDL or LDL−. This indicates that protection afforded by E2 did not require interaction with lipoproteins in the cell culture medium but was related to a conferred protection or induction of resistance of the cells to normal or modified LDL.
Table 1 also shows the effects of LDL and LDL− treatments in the presence of 100 mM of ascorbic acid. There was no cytoprotective effect of ascorbic acid on LDL-mediated toxicity in RAEC or E2RAEC, but a protective effect was found when the cells were treated with 500 mg/ml of LDL (PB0.05).
3.3. Cell-mediated LDL− formation
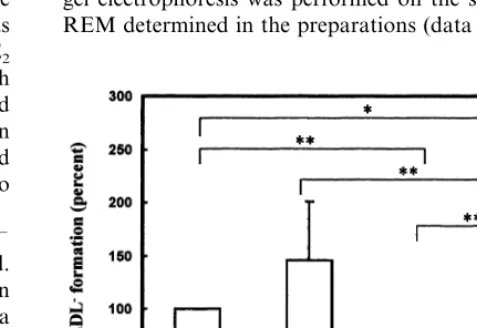
Control RAEC and E2RAEC were incubated with 100 mg/ml of LDL protein for 24 h and the amount of LDL− formation determined by HPLC analysis of total LDL recovered from the medium. Fig. 5 shows that less LDL was converted to LDL− in the presence of E2 (E2RAEC) than RAEC (PB0.005). The results are expressed as a percent of the rate of LDL− forma-tion by RAEC under the standard culture condiforma-tions. The SF following 24 h incubation with 100mg/ml LDL was not significantly different from untreated (control)
cultures. Moreover, the presence of ascorbic acid in E2RAEC cultures further inhibited LDL− formation (PB0.005) as compared to E2RAEC without ascor-bate. By contrast, addition of ascorbic acid to RAEC tended to increase the rate of LDL− formation. The effect of ascorbic acid was clearly related to increased cell-mediated activity on LDL since no effects of ascor-bic acid were found under cell-free conditions (data not shown). To confirm the oxidation status of LDL and LDL−
before addition to RAEC and E2RAEC, agarose gel electrophoresis was performed on the samples, and REM determined in the preparations (data not shown).
Fig. 5. LDL−formation mediated by cells after addition of LDL at
100mg protein/ml. Cells were then incubated for 24 h under standard conditions-control (C), and the following treatment conditions: 100 mM of ascorbic acid (C/AA), after pre-conditioning with 17b -estra-diol (E2), and after pre-conditioning with 17b-estradiol and 100mM of ascorbic acid (E2/AA). Control cells produced 1.425% of LDL−, the formation of LDL−being calculated as a percent of total LDL.
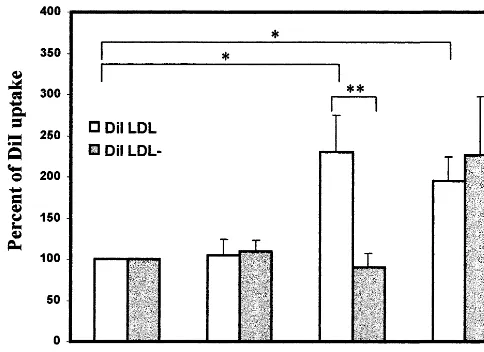
Fig. 6. Uptake of LDL and LDL−by control cells, cells
pre-incu-bated with 17b-estradiol, ascorbic acid (100 mM), or ascorbic acid and 17b-estradiol as described in methods. Symbols are for DiI-LDL uptake () and for DiI-LDL−uptake (). Significant differences
over control values are indicated by *PB0.005, and between E2RAEC DiI LDL and DiI LDL−uptake by **PB0.05.
3.5. 17 b-estradiol le6els are increased in E2RAEC
Levels of 17b-estradiol were measured in the RAEC and E2RAEC incubated in the presence and absence of ascorbic acid. The measurements were performed in media and cells. Table 2 shows that E2RAEC cells and E2RAEC incubated with ascorbic acid have increased 17b-estradiol levels relative to RAEC or RAEC treated with ascorbic acid. Incubation of E2RAEC with ascor-bic acid did not increase the levels of 17 b-estradiol in cells as compared to E2RAEC. The levels of 17b -estra-diol in the medium from E2RAEC or E2RAEC incu-bated with ascorbic acid were not different.
3.6. Total glutathione le6els are increased in E2RAEC
Table 2 shows that RAEC and RAEC in the pres-ence of ascorbic acid have similar levels of total GSH while E2RAEC and E2RAEC in the presence of ascor-bic acid have increased GSH levels as compared to RAEC. The increase in GSH in E2RAEC is essentially attributable to the effects of E2since ascorbate addition had little effect on GSH levels in E2RAEC.
4. Discussion
A novel antioxidant interaction between estradiol and ascorbic acid has been found that is highly effective in preventing LDL oxidation. The marked reduction of the rates of conjugated diene formation during the lag phase (i.e. lag rate) in the presence of E2 indicates a suppression of lipid peroxide formation, typical of the antioxidant protected phase for radical chain propaga-tion during lipid peroxidapropaga-tion. The enhanced protecpropaga-tion in the presence of ascorbic acid suggests an interaction between E2and ascorbic acid and/or other LDL antiox-idants. This interaction may be similar to that described for vitamin E and ascorbate where the former is re-duced by the latter and its antioxidant activity pre-served [39]. Conservation of vitamin E in LDL is
3.4. LDL but not LDL− uptake is enhanced in E2RAEC
As shown in Fig. 6, uptake of LDL was greater in E2RAEC and E2RAEC in the presence of ascorbic acid than in RAEC or RAEC in the presence of ascorbic acid when cells were grown in standard medium con-taining 15% of male serum, PB0.05. The extent of uptake is shown relative to LDL or LDL− uptake in control cultures (RAEC), which was set as 100%. DiI LDL uptake in RAEC was 0.74390.169 mg/105 cells while DiI-LDL−
uptake in RAEC was 0.30490.034 mg/105 cells. Although there was apparently greater LDL− uptake in E2RAEC treated with ascorbic acid, the effect was not significant compared to E2 alone or to control RAEC. The tendency for E2RAEC to incor-porate more LDL− in the presence of ascorbic acid may be due to an antioxidant effect such that less LDL is degraded to a form that is not assimilated by LDL receptors.
Table 2
Estradiol levels in cells and mediaaand total glutathione levels in control cells (RAEC)b
RAEC RAEC+AA E2RAEC E2RAEC+AA
Estradiol le6els
Media (pg/ml) 14.790.33 17.091.53 42009925 57009706
25.394.66**,++
Cell (pg/mg protein) 12.191.00*,** 11.290.97+,++ 23.893.26*,+ Glutatione le6els
1.4590.032*,** 1.4490.048+,++ 1.7290.118*,+
Cell (Nm/mg/cell protein) 1.8190.071**,++
aRAEC (control cell), RAEC+AA (control cells in the presence of 100mM ascorbic acid), E
2RAEC (cells incubated with 17b-estradiol for 5 days) and E2RAEC+AA (E2RAEC in the presence of 100mM of ascorbic acid).
bControl cells in the presence of 100mM of ascorbic acid (RAEC+AA), 17b-estradiol pre-conditioned cells (E
considered as a primary antioxidant action for ascor-bate in terms of suppressing LDL oxidation [40]. It can not be determined at this time whether the protective effect of E2 was related to the amount of incorporation or association with LDL. It is possible that the physico-chemical characteristics of LDL could be altered by E2 to a form more resistant to oxidation. Moreover, incu-bating LDL with E2 for up to 4 h did not confer more resistance to oxidation (data not shown), indicating that the extent of uptake or association with LDL is not influenced by the time of incubation but is related to E2treatment concentrations. It should be noted that Shwaery et al. [11] reported that an antioxidant effect of E2at low concentrations was only seen when E2and LDL were incubated in plasma. The extent of E2 incor-poration into LDL may be facilitated by plasma en-zymes or factors, however, the extent of incorporation is difficult to measure given the amounts of LDL available for analysis of E2 content. The apparent syn-ergistic interaction between ascorbic acid and E2 may also be related to the recently described inhibition by ascorbic acid of apoB-100 oxidation by copper [41]. The ability of ascorbic acid to inhibit copper binding via oxidation of histidine to oxohistidine may reduce the extent of LDL oxidation, thus decreasing the de-mand for antioxidant activity from E2 and other antioxidants.
The antioxidant activity of E2is in one aspect atypi-cal of other LDL antioxidants since there is no inhibi-tion of vitamin E consumpinhibi-tion during LDL oxidainhibi-tion. This suggests that E2 is not interacting directly with vitamin E or preventing its reaction with peroxyl radi-cals, as described for other antioxidants [42]. Estrogen is a weak chain breaking antioxidant based on the rate constant for reactions with peroxyl radicals and high concentrations are typically required to effectively in-hibit LDL lipid peroxidation [43]. The antioxidant pro-tective action may be due to its phenolic structure, which can donate hydrogens after E2 is incorporated into the lipid core of LDL. Direct reactions with per-oxyl radicals are possible, as described thermodynami-cally for reactions between phenolic compounds (including estrogen) and peroxyl radicals [43]. This is supported by the clear suppression of conjugated diene formation during the prolonged lag phase. The con-sumption of vitamin E during this phase may reflect the facile reaction of a-tocopherol with copper [44]. The kinetic profiles suggest, but do not prove, that E2 may be inhibiting tocopherol-mediated peroxidation [45], and one may speculate that E2 suppresses interactions between tocopheroxyl radical and LDL core lipids. Since the levels of vitamin E and rates of vitamin E consumption are unaffected by E2, it is likely that peroxidation reactions subsequent to initiation of toco-pheroxyl radical are being inhibited by E2. Although a direct antioxidant activity for E2 is not found using
concentrations comparable to those found in human blood, (which in pre-menopausal women can reach 443 pg/ml and is B59 pg/ml in postmenopausal women), inhibition of LDL oxidation was found at concentra-tions as low as 5 ng/ml. This represents levels that are more than 10 times lower than reported previously [11]. More importantly, our findings show that significant antioxidant activity can be demonstrated by E2 in the presence of physiological concentrations of ascorbic acid and under these conditions marked antioxidant activity for E2 is found even at physiological concentrations.
Since LDL is less oxidized in the presence of E2, it is possible that suppressed formation of LDL−
, or other modified forms of LDL, may contribute to E2-mediated protection. This may also account for the cytoprotec-tion afforded after treatments with high levels of LDL. Treatments with 500 mg/ml of LDL are likely to be accompanied by substantial amounts of contaminating LDL−, present either in the isolated LDL or formed by cell-mediated oxidation. In either case, if the levels of LDL− were comparable to that reported previously for LDL from human plasma (i.e. 2 – 6%) [25], then LDL− levels could be as high as 20 mg/ml which are sufficient to produce cytotoxicity [25]. Surprisingly, the presence of ascorbic acid did not afford a protection against LDL-mediated toxicity to RAEC or E2RAEC. The protective effect E2 appears to be due to altered cell properties (conferred resistance) elicited only after pre-treatment or pre-conditioning of cells. Since cytotoxic-ity resulting from high doses of LDL or LDL−may be due to factors other than oxidative stress, ascorbic acid could alter the response of cells to E2even though there were no apparent effects on uptake (Table 2). However, the combination of E2 and ascorbic acid may increase the amount of LDL− uptake as compared to E2 alone (Fig. 6) producing the greater cytotoxicity described in Table 1.
During cell-mediated oxidation a variety of events contribute to the formation of LDL−. Two important processes include: (1) extracellular modification of LDL, and (2) uptake of LDL and/or LDL−. Inhibition of extracellular modification of LDL by E2 could be explained by the increased levels of GSH found in E2 pre treated cells, either in the presence or absence of ascorbic acid. The latter could plausibly influence the degree of LDL modification. Although E2RAEC assim-ilated LDL more readily than RAEC, E2RAEC was less susceptible to LDL or LDL− -induced toxicity. This suggests that E2 protects endothelial cells by as-similating LDL more readily, but not LDL−.
stimulating monocyte/macrophage adherence to en-dothelium and influx into the intima [46] followed by uptake of oxidized LDL-cholesterol [18]. E2 may not only inhibit LDL accumulation by vascular cells but also suppress LDL modification and, in turn, the conse-quent pro-inflammatory effects of modified LDL [47]. Estrogens have been shown to inhibit LDL oxidation as catalyzed by cupric ions, vascular smooth muscle cells, and endothelial cells [24,48 – 51]. E2 and ascorbic acid may prevent this process by reducing LDL−formation and its potential vascular cell effects. Increased uptake of LDL may contribute to the lower levels of plasma LDL in women taking estrogen replacement therapy. Our findings suggest three possible properties of E2that can contribute to its anti-atherogenic effect: (1) reduced formation of LDL−; (2) less uptake of LDL−; (3) higher uptake and recycling of LDL, making it less prone to oxidation.
These results provide the first evidence that ascorbic acid can enhance the antioxidant effect of E2 by pre-venting LDL oxidation by copper ions or cells. The cytoprotective and possibly anti-atherogenic effect ap-pears to be manifested by reducing the extent of oxi-dized LDL formation and uptake. The enhanced activity of E2in the presence of ascorbate indicates that the antioxidant and anti-atherosclerosis activity of E2 may occur at concentrations within the physiological range in the presence of ascorbate.
Acknowledgements
This project was supported by grant HL 50350 from National Institutes of Health and the National Phar-macy Cholesterol Council. The authors appreciate the excellent assistance of Dr Frank Stancyzk for the mea-surement of 17 b-estradiol levels.
References
[1] Godsland IF, Wynn V, Crook D, Miller NE. Sex plasma lipo-proteins and atherosclerosis: prevailing assumptions and out-standing question. Am Heart J 1987;114:1467 – 503.
[2] McManus J, Mc Eneny J, Thompson W, Young IS. The effect of hormone replacement therapy on the oxidation of low density lipoprotein in postmenopausal women. Atherosclerosis 1997;135:73 – 81.
[3] Manson JE. Postmenopausal hormone therapy and atheroscle-rotic disease. Am Heart J 1994;128:1337 – 43.
[4] Gruchow HW, Anderson AJ, Barboriak JJ, Sobocisnki KA. Postmenopausal use estrogen and occlusion of coronary arteries. Am Heart J 1988;115:954 – 63.
[5] Clarkson TB, Anthony MS, Klein KP. Effects of estrogen on arterial wall structure and function. Drugs 1994;47:42 – 51. [6] Mendelsohn ME, Faras RH. Estrogen and the blood vessel wall.
Curr Opin Cardiol 1994;9:626 – 91.
[7] Behl C, Skutella T, Lezoualc’h F, et al. Neuroprotection against oxidative stress by estrogens: structure-activity relationship. Mol Pharmacol 1997;51:535 – 41.
[8] Akishita M, Ouchi Y, Miyoshi H, et al. Estrogen inhibits endothelin-1 production and c-fos gene expression in rat aorta. Atherosclerosis 1996;125:27 – 38.
[9] Wells KE, Miguel R, Alexander JJ. Sex hormones affect the calcium signaling response of human adterial cells to LDL. J Surg Res 1996;63:64 – 72.
[10] Cicinelli E, Ignarro LJ, Lograno M, Matteo G, Falco N, Schonauer LM. Acute effects of transdermal estradiol adminis-tration on plasma levels of nitric oxide in postmenopausal women. Fertil Steril 1997;67:63 – 6.
[11] Shwaery GT, Vita JA, Keaney JF. Antioxidant protection of LDL by physiological concentrations of 17b-estradiol require-ment for estradiol modification. Circulation 1997;95:1378 – 85. [12] Wilcox JG, Hwang J, Hodis HN, Sevanian A, Stanczky FZ,
Lobo RA. Cardioprotective effects of individual conjugated equine estrogens through their possible modulation of insulin resistance and oxidation of low-density lipoprotein. Fertil Steril 1997;67:57 – 62.
[13] Myers SI, Turnage RH, Bartula L, Kalley B, Meng Y. Estrogen increases male rat aortic endothelial cell (RAEC) PGI2 release. Prostaglandins Leukot Essent Fatty Acids 1996;54:403 – 9. [14] Mikkola T, Ranta V, Orpana A, Viinikka L, Ylikorkala O.
Hormone replacement therapy modifies the capacity of plasma and serum to regulate prostacyclin and endothelin-1 production in human vascular endothelial cells. Fertil Steril 1996;66:389 – 93. [15] Sulistiyani ASJ, St. Clair RW. Effect of 17 b-estradiol on metabolism of acetylated low-density lipoprotein by THP-1 macrophages in culture. Arterioscler Thromb Vasc Biol 1997;17:1691 – 700.
[16] Spyridopoulos, I, Sullivan AB, Kearney BA, Isner JM, Losordo DW. Estrogen-receptor-mediated inhibition of human endothe-lial apoptosis estradiol as a survival factor. Circulation 1997;95:1505 – 14.
[17] Deshpande R, Khalili H, Pergolizzi RG, Michael SD, Chang MY. Estradiol down regulated LPS-induced cytokine production and NFkB activation in murine macrophages. Am J Reprod Immunol 1997;38:46 – 54.
[18] Steinberg D. A critical look at the evidence for the oxidation of LDL in atherogenesis. Atherosclerosis 1997;131:S5 – 7.
[19] Berliner JA, Heineicke JW. The role of oxidized lipoproteins in atherogenesis. Free Rad Biol Med 1996;20:707 – 27.
[20] Hodis HN, Kramsch DM, Avogaro P, et al. Biochemical and cytotoxic characteristics of an in vivo circulating oxidized low density lipoprotein (LDL−). J Lipid Res 1994;35:669 – 77.
[21] Keaney JF, Shwaery GT, Xu A, Nicolosi RJ, Loscalzo J, Foxall TL, Vita JA. 17(-estradiol preserves endothelial vasodilator func-tion and limits low-density lipoprotein oxidafunc-tion in hypercholes-terolemic swine. Circulation 1994;89:2251 – 9.
[22] Samsioe G. Cardioprotection by estrogens: mechanisms of ac-tion- the lipids. Int J Fertil 1994;39:43 – 9.
[23] Rifici VA, Khachadurian AK. The inhibition of low-density lipoprotein oxidation by 17-b estradiol. Metabolism 1992;41:1110 – 4.
[24] Neugarten J, Ghossein C, Silbiger S. Estradiol inhibits mesangial cell-mediated oxidation of low-density lipoprotein. J Lab Clin Med 1995;126:385 – 91.
[25] Sevanian A, Hwang J, Hodis HN, Cazzolato G, Avogaro P, Bittolo-Bon G. Contribution of an in vivo oxidized LDL to LDL oxidation and its association with dense LDL subpopula-tion. Arterioscler Thromb Vasc Biol 1996;16:784 – 93.
[26] Tribble DL, Holl LG, wood PD, Krauss RM. Variations in oxidative susceptibility among six low-density lipoprotein sub-fractions of differing density and particle size. Atherosclerosis 1992;93:189 – 99.
[28] Sack MN, Rafer DJ, Cannon III RO. Oestrogen and inhibition of oxidation of low-density lipoprotein in postmenopausal women. Lancet 1994;343:269 – 70.
[29] Brussaard HE, Leuven JAG, Kluft C, Krans MJ, van Duyven-voorde W, Buytenhek R, van der Laarse A, Princen HMG. Effect of 17b-estradiol on plasma lipids and LDL oxidation in postmenopausal women with type II diabetes mellitus. Arte-rioscler Thromb Vasc Biol 1997;17:324 – 30.
[30] Rose RC, Bode AM. Biology of free radical scavengers: an evaluation of ascorbate. Faseb J 1993;7:1135 – 42.
[31] Burr MT, Sweetman PT, Hurley RJ, Powell GH. Effects of age and intake on plasma ascorbic acid levels. Lancet 1974;1:163 – 4. [32] Morgan AF, Fillum HL, Williams RI. Nutritional status of aging. III Serum ascorbic acid intake. J Nutr 1955;55:431 – 48. [33] Itoh R, Yamada K, Oka J, Echizen H, Murakami K. Sex as a
factor in levels of serum ascorbic acid in healthy elderly popula-tion. Int J Vitamin Nutr Res 1989;59:365 – 572.
[34] Barja G. Ascorbic acid and aging. In: Harris JR, editor. Subcel-lular Biochemistry 25. New York: Plenum, 1996:157 – 87. [35] Martin A, Frei B. Both intracellular and extracellular vitamin C
inhibit atherogenesic modification of LDL by human vascular endothelial cells. Arterioscler Thromb Vasc Biol 1997;17:1583 – 90.
[36] Bui MH. Simple determination of retinol, a-tocopherol and carotenoids (lutein, all trans-lycopene, a and b carotenes) in human plasma by isocratic liquid chromatography. J Chro-matogr B: Biomed Appl 1994;654:129 – 33.
[37] Day BR, Williams DR, Marsh CA. A rapid manual method for routine assay of ascorbic acid in serum and plasma. Clin Biochem 1979;12:22 – 6.
[38] Reynolds G, St. Clair R. A comparative microscopic and bio-chemical study of the uptake of fluorescent and 125I-labeled lipoproteins by skin fibroblasts, smooth muscle cells, and peri-toneal macrophages in culture. Am J Pathol 1985;121:200 – 11. [39] Niki E. Action of ascorbic acid as a scavenger of active and
stable oxygen radicals. Am J Clin Nutr 1991;54:1119S – 24S. [40] Frei B. Ascorbic acid protects lipids in humnan plasma low
densisty lipoprotein against oxidative damage. Am J Clin Nutr 1991;54:1113S – 8S.
[41] Retsky KL, Chen K, Zeind J, Frei B. Inhibition of copper-in-duced LDL oxidation by vitamin C is associated with decreased
copper-binding to LDL and 2-oxo-histidine formation. Free Rad Biol Med 1999;26:90 – 8.
[42] Ingold KU, Bowry VW, Stocker R, Walling C. Autoxidation of lipids and antioxidation bya-tocopherol and ubiquinol in homo-geneous solution and in aqueous dispersions of lipids:unrecognized consequences of lipid particle size as exem-plified by oxidation of human low density lipoprotein. Proc Natl Acad Sci USA 1993;90:45 – 9.
[43] Niki, E, Nakano M. Oxygen radicals in biological systems. Estrogens as antioxidants. In: Lester, P., Glazer, A.N., editors. Methods Enzymol 1990;186:330 – 333.
[44] Maiorino M, Zamburlini A, Roveri A, Ursini F. Prooxidant role of vitamin E in copper induced lipid peroxidation. FEBS 1993;330:1174 – 6.
[45] Neuzil J, Thomas SR, Stocker R. Requirement for, promotion, or inhibition by a-tocopherol of radical-induced initiation of plasma lipoprotein lipid peroxidation. Free Rad Biol Med 1997;22:57 – 71.
[46] Steinberg D, Parthasarathy S, Carew TE, Khoo JG, Witzum JL. Beyond cholesterol: modifications of low density lipoprotoein that increase its atherogenicity. N Engl J Med 1989;32:915 – 24. [47] Watson AD, Navab M, Hama SY, et al. Effect of platelet activating factor-acetylhydrolase on the formation and action of minimally oxidized low density lipoprotein. J Clin Invest 1995;95:774 – 82.
[48] Huber LA, Scheffler E, Poll T, Ziegler R, Dresel HA. 17 beta-estradiol inhibits LDL oxidation and cholesterol ester for-mation in cultured macrophages. Free Radic Res Commun 1990;8:167 – 73.
[49] Negre-Salvayre A, Pieraggi MT, Mabile L, Salvayre R. Protec-tive effect of 17b-estradiol against the cytotoxicity of minimally oxidized LDL to cultured bovine aortic endothelial cells. Atherosclerosis 1993;99:207 – 17.
[50] Maziere C, Auclair M, Ronveaux MF, Salmon S, Santus R, Maziere JC. Estrogens inhibit copper and cell-mediated modifi-cation of low density lipoprotein. Atherosclerosis 1991;89:175 – 82.
[51] Tang M, Abplanalp W, Ayres S, Subbiah R. Superior and distinct antioxidant effects of selected estrogen metabolites on lipid peroxidation. Metabolism 1996;45:411 – 4.
.