The preparation of copper-oxidized LDL for the measurement of
oxidized LDL antibodies by EIA
Maria F. Lopes-Virella *, Sinikka Koskinen, Marina Mironova, David Horne,
Richard Klein, Charlyne Chassereau, Candace Enockson, Gabriel Virella
Di6ision of Endocrinology,Diabetes and Medical Genetics,Department of Medicine,Ralph H. Johnson VA Medical Center,
Department of Microbiology and Immunology,Medical Uni6ersity of South Carolina,Charleston,SC, USA
Received 9 June 1999; received in revised form 25 August 1999; accepted 3 November 1999
Abstract
In the present study we try to define the optimal conditions for preparation of copper-oxidized low-density lipoprotein (oxLDL) to be used for the assay of oxLDL antibodies by enzyme immunoassay (EIA). Oxidation of LDL was monitored by measuring the formation of conjugated dienes at 234 nm and the generation of fluorescent products with emission at 430 nm when excitation is performed at 360 nm. The generation of immunogenic epitopes was evaluated by testing the reactivity of aliquots collected at different times during the oxidation process with human sera with high oxLDL antibody levels and with a purified human oxLDL antibody. The values of fluorescence emission at 430 nm correlated best with reactivity with oxLDL antibodies; strong reactivity was usually associated with values greater than 1.1 U. The time needed for fluorescence emission to reach maximum levels varied between 6 and 14 h for most LDL, but it was considerably longer in a few LDL preparations. The maximal reactivity of oxLDL with oxLDL antibodies was observed when the LDL oxidation reaction was stopped 4 or more hours after the fluorescence readings reached their peak. At this stage of the oxidation reaction, apolipoprotein B fragmentation and aggregation were observed as shown by Western blot analysis. The CV for 13 EIA runs of two reference oxLDL antibodies reacting with four different pools of standardized oxLDL prepared according to the stated guidelines was 14.5 and 3.9%, confirming the reproducibility of our oxidation conditions. © 2000 Elsevier Science Ireland Ltd. All rights reserved.
Keywords: Arteriosclerosis; Autoimmunity; Autoantibodies; Oxidized low-density lipoprotein antibodies; Oxidized low-density lipoprotein antibody enzyme immunoassay; Oxidized low-density lipoprotein
www.elsevier.com/locate/atherosclerosis
1. Introduction
Oxidized low-density lipoprotein (oxLDL) has been found to have an important role in the development of atherosclerosis. The pathogenic mechanisms by which oxLDL leads to arteriosclerosis have been the object of intense investigation, and remain to be fully defined. It has been clearly established that oxLDL is immuno-genic, leading to the formation of autoantibodies [1 – 8] and immune complexes [9,10] which may have a patho-genic role. Antibodies reactive with oxLDL (anti-oxLDL) have been detected [1 – 10] and isolated [11]
from the serum of patients with vascular disease and from the serum of apparently healthy subjects. Studies showing a correlation between increased levels of oxLDL antibodies and the presence or progression of vascular lesions [1,3 – 6,8] support the pathogenic role of these antibodies, as do the observations that oxLDL antibodies recognize epitopes in atherosclerotic lesions of rabbits and humans but not in normal arteries [12 – 14]. However, other groups have failed to demon-strate any significant correlation between the detection of oxLDL antibodies and clinical or radiological find-ings of atherosclerosis [2,7].
One possible explanation for these contradictory ob-servations may be the lack of standardization of the assays used for the measurements of oxLDL antibodies by different groups. Enzyme immunoassay (EIA) is the most commonly used method for the determinations of
* Corresponding author. Present address: Ralph H. Johnson VAMC, Strom Thurmond Building, Room 530, 114 Doughty Street, PO Box 250766, Charleston, SC 29403, USA. Tel.:+1-843-577-5011, ext. 6854; fax: +1-843-953-6480.
E-mail address:[email protected] (M.F. Lopes-Virella).
oxLDL antibodies [1 – 11]. We and others [1] have encountered problems with batch-to-batch variability in reactivity of oxLDL antibodies when copper oxidized LDL has been used as antigen in EIA. This seems to reflect variations in the degree of oxidation of the different LDL preparations used to perform the assay. One of the parameters which is usually not carefully controlled is the duration of the oxidation reaction, which is stopped after an arbitrarily defined time pe-riod. This may result in the use of insufficiently oxi-dized LDL preparations. However, even when the reaction is allowed to proceed to its maximum, the extent of modification varies in different LDL prepara-tions. The most widely used method to determine the extent of LDL modification is the measurement of thiobarbituric acid reactive substances (TBARS) [15]. This method is known to have low specificity [16] and, in our experience, fails to reflect the batch-to-batch variability of oxLDL for the products (epitopes) recog-nized by oxLDL antibodies. Alternative methods to determine the extent of LDL modification include the measurement of lipid peroxides, conjugated dienes and aldehyde formation, all indicative of lipid peroxidation during the early phase of the oxidation process [16,17] as well as the measurement of fluorescent products with emission at 430 nm when excitation is performed at 360 nm, indicative of apolipoprotein B modification [16,18]. Thus, we decided to determine whether continuous monitoring of the oxidation of LDL by sensitive tech-niques would give a more precise indication about the extent of LDL modification needed to react well with serum ox-LDL antibodies and therefore allow a more rational approach to consistently prepare oxLDL for the assay. The techniques chosen for our study were the measurement of conjugated dienes and of fluorescent compounds formed during LDL oxidation. The results presented in this article show that monitoring the fluorescent compounds allows us to determine the stage and extent of LDL oxidation associated with a better recognition by oxLDL antibodies. This criterion is far superior to reaction time or to the TBARS assay to determine if and when the optimal degree of LDL oxidation has been reached, thus allowing us to prepare batches of oxLDL which exhibit consistent reactivity with oxLDL antibodies.
2. Materials and methods
2.1. Lipoprotein isolation, modification and characterization
Blood for lipoprotein isolation was collected from normal healthy volunteers in 0.4 mmol/l EDTA after 12 h of fasting. The donors used for this purpose were normolipemic healthy volunteers, not receiving
pre-scription medication for any acute or chronic condition and without family history of coronary artery disease, peripheral vascular disease, or stroke. None of the volunteers was receiving anti-oxidant therapy. LDL was isolated from individual or pooled plasma, after density adjustment (1.019Bdensity B1.063 g/ml) with potassium bromide (KBr), by preparative ultracentrifu-gation at 50 000 rpm for 17 h on a Beckman L-80 ultracentrifuge, using a type 70 Ti rotor [19]. LDL, isolated as described, was washed by
ultracentrifuga-tion, dialyzed against a 0.15-mol/l sodium chloride
solution containing 300 mmol/l EDTA, pH 8.0, passed
through an Acrodisc filter (0.22-mm pore size) in order
to sterilize and remove aggregates, and stored under nitrogen in the dark at 4°C.
For oxidation, we used two individual LDL prepara-tions and eight different LDL pools, each one of the pools containing LDL isolated from three different individuals. All pools and individual LDL preparations were oxidized shortly after blood collection, usually within 2 – 3 weeks and never exceeding 4 weeks after blood collection (except when otherwise noted). Of the LDL pools four were split into two aliquots. Of these aliquots four were oxidized during the first 2 weeks after blood collection; of the remaining four aliquots, two were oxidized 5 – 5.5 weeks after blood collection, and the remaining two were oxidized 9 weeks after blood collection.
Oxidation was performed according to the protocol described by Steinbrecher et al. [20]. To remove residual KBr and EDTA prior to starting the oxidation reac-tion, the LDL was passed through a PD-10 column (Pharmacia Biotech, Uppsala, Sweden). Phosphate buffered saline (PBS), pH 7.4, was oxygenated at 2
l/min for 10 min and LDL was diluted by PBS to a
final concentration of 1500 mg/l of ApoB. A stock
solution of copper chloride (CuCl2, 10 mmol/l in
dis-tilled water) was added to the LDL preparation to a
final concentration of 40 mmol/l. After addition of
CuCl2, the LDL preparation was filtered through a
0.22-mm filter and incubated at 37°C. In order to track
the degree of oxidation, two aliquots of every LDL preparation, adjusted to an ApoB concentration of 50 mg/l, were used for continuous monitoring of fluores-cence and formation of conjugated dienes. Preliminary comparisons of data generated by continuous monitor-ing as described above and by analysis of timed aliquots collected from the batch undergoing oxidation
after dilution to 50 mg of ApoB/l showed identical
results. Fluorescence was monitored at 37°C on a
lu-minescence spectrophotometer (SLM-AMINCO®Series
The monitored samples were not stirred, to more closely reproduce the conditions of oxidation of the experimental batches. The values were expressed as fluorescence units, using a 0.1-mg/ml quinine solution
to adjust the sensitivity of the fluorometer (the gain was adjusted to obtain a reading of 2.6 U with the quinine solution). Conjugated dienes were measured on a Beckman spectrophotometer (model DU 640 with temperature control) at 234 nm [16,17]. The tem-perature was set at 37°C and the cells were not stirred.
Small aliquots of the main LDL preparation were harvested under sterile conditions at different stages of the oxidation reaction. The oxidation in each col-lected sample was stopped by the addition of EDTA and butyl-hydroxytoluene (BHT) to a final
concentra-tion of 0.3 and 0.2 mmol/l, respectively. Copper was
then removed from the oxLDL samples by overnight dialysis against 0.15 mol/l NaCl containing 0.3 mmol/
l EDTA, pH 8.0. After dialysis the oxLDL samples were filtered through a 0.22-mm filter to remove large
aggregates. The final protein concentration was deter-mined after filtration by a Lowry assay [21]. Recovery
data was consistently at 80% of the initial protein
concentration for samples oxidized for periods not ex-ceeding 24 h. Protein recovery in samples oxidized for longer periods of time was slightly lower, usually not below 70% of the initial protein concentration.
The degree of LDL denaturation associated with copper oxidation was assessed by two separate crite-ria: Western blot analysis and agarose gel
elec-trophoresis. For both assays we used samples
collected at different oxidation times. For Western blot, the samples were dissolved in a buffer contain-ing 63 mmol/l Tris – HCl, 2% SDS, 10% glycerol, 0.01
mmol/l BHT, and 0.001% bromophenol blue (pH
6.8), heated for 3 min in a boiling water bath, and then run for 2.5 h, at 100 V in a 4-20% PAGE gel in
25 mmol/l Tris 190 mmol/l glycine buffer, pH 8.3,
containing 0.1% SDS (Novex, San Diego, CA). After electrophoresis, the proteins were electrotransfered to
PVDF membranes in 15 mmol/l Tris-glycine buffer,
pH 8.3, with 20% (v/v) methanol. Membranes were
blocked with 5% fat-free dry milk for 1 h at room temperature, and then incubated for another hour with a 1:30 000 dilution of rabbit antiserum to hu-man apolipoprotein B (Behring, Somerville, NJ) in TBS-T-5% milk solution. After incubation with this antiserum the membranes were washed five times with TBS-T and then incubated for 1 h with a 1:20 000 dilution of sheep anti-rabbit IgG HRP conjugated an-tibody in the same buffer. Afterwards the membranes were washed five times with TBS-T and the im-munoreactive proteins were visualized by treating the membranes with ECL detection reagents (Amersham,
Arlington Heights, IL) followed by exposure of Ko-dak X-Omat film.
Agarose gel electrophoresis of native and modified
LDL was performed in 0.5% (w/v) gel, prepared in
0.05 M sodium barbital buffer, pH 8.6 and subjected to 25-mA constant current for 2.5 h at 4°C. The gels were fixed for 1 h in 5% glacial acetic acid, 75%
ethanol (v/v) and air dried. LDL bands were
visual-ized after staining the gel in a saturated solution of oil red O (0.04% in 60% ethanol, stored at 4°C).
2.2. Human oxLDL antibodies
To test the reactivity of the various preparations of copper-oxidized LDL prepared as described above, six human sera with concentrations of oxLDL antibody
ranging from 20 to 204 mg/l determined by
enzy-moimmunoassay were used. A purified human
oxLDL antibody with a total antibody concentration
of 88 mg/l was also used to test the reactivity of
some oxLDL batches. This oxLDL antibody was iso-lated from human serum by affinity chromatography as described previously [11,22], had a dissociation constant (Kd) of 6.6×10−
9
mol/l, and was consti-tuted predominantly by IgG (68.5%). None of the reference antibodies was obtained from the same indi-viduals who donated plasma for LDL isolation.
Pre-vious studies have shown that isolated oxLDL
antibodies have marked cross-reactivity with MDA-LDL, but react very poorly with glycated MDA-LDL,
na-tive LDL, oxidized human serum albumin, or
cardiolipin [11].
2.3. Enzyme immunoassay
The reactivity of different oxLDL preparations with oxLDL antibodies was tested by a competitive en-zyme immunoassay (EIA) we have previously
de-scribed [2,22]. The assay was conducted on
flat-bottom Immulon type 1 EIA plates (Dynatech, Chantilly, VA). The wells were coated by overnight
incubation at 4°C with 100 ml of oxLDL (7.5 mg/l
LDL in 1 mol/l carbonate-bicarbonate buffer, pH
9.6). Serum samples prediluted 1/10 in PBS
contain-ing 1% bovine serum albumin (BSA, Cohn Fraction
V- cat. cA-7906, St Louis, MO, USA) (PBS-BSA)
and isolated antibody were separated into two 200-ml
aliquots. One of the aliquots was absorbed with an
equal volume of oxLDL (400 mg/ml in PBS-BSA) and
the other (unabsorbed) was mixed with an equal vol-ume of PBS-BSA. The final dilution of all serum
samples was 1/20. All samples (absorbed and
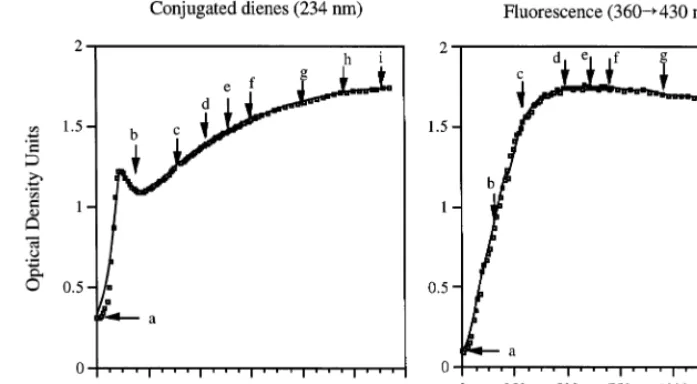
Fig. 1. Results of a 24-h continuous monitoring during copper oxidation of a LDL pool of conjugate diene formation, measured by spectrophotometry at 234 nm (left panel) and of ApoB-emitted fluorescence, determined by exciting the LDL solution at 360 nm and measuring emitted fluorescence at 430 nm (right panel). The arrows and letters indicate the aliquots harvested for testing their reactivity with reference sera with known concentrations of oxLDL antibodies (Fig. 2) and LDL denaturation by Western blot and agarose gel electrophoresis (Fig. 3).
washed four times with 0.05% Tween 20 (Sigma) in PBS, pH 7.8. The absorbed and unabsorbed aliquots
were spun at 9000×g in an Eppendorf centrifuge
(model 5413) for 30 min. The supernatants of the centrifuged samples, unabsorbed and absorbed, were tested at two final dilutions: 1:2 and 1:4 for purified
antibodies and 1:20 and 1:40 for sera. Then 100 ml of
both dilutions were transferred to the wells of the oxLDL-coated plates and after overnight incubation at
4°C and washing, 150ml of peroxidase-conjugated
rab-bit anti-human IgG (IgG fraction) (Cappel Organon Teknika, Durham, NC), diluted 1:5000 in PBS-BSA, was added to each well. After incubation for 1 h at 4°C, the unbound conjugate antibody was washed off, and 0.5 mmol/l of 2,2%
-azino-di-(3-ethylbenzthiazoline-6-sul-fonate) (ABTS, Sigma, St Louis, MO) and 3% (v/v)
hydrogen peroxide in a 45-mmol/l concentration of
citric acid buffer, pH 4.0, was added as substrate. Color was allowed to develop for 10 min at room tempera-ture, in the dark. The reaction was stopped with 0.1
mol/l citric acid, pH 2.1, and the absorbance was
measured at 414 nm in a VMax enzyme-linked im-munosorbent assay reader (Molecular Devices, Sunny-vale, CA). The optical density values were calculated by subtracting the OD values measured on oxLDL-ab-sorbed aliquots from those measured on unaboxLDL-ab-sorbed aliquots at identical dilutions. A human oxLDL anti-body standard was used to calculate the concentration of oxLDL antibody in serum and isolated antibody preparations as described in an earlier publication [22]. In all EIA studies testing different oxLDL prepara-tions the same oxLDL preparation was used to coat the wells of the Immulon plates and to absorb free anti-body on antianti-body-containing samples.
3. Results
The longitudinal changes in the formation of fluores-cent compounds and of conjugated dienes during oxi-dation of a LDL pool (pool 1) for 24 h are presented in Fig. 1. The nine aliquots of this LDL pool (labeled ‘a’ to ‘i’ in the figure) were obtained at different stages of oxidation and tested for their reactivity with five differ-ent reference sera (Fig. 2). In the example presdiffer-ented in Fig. 1, fluorescence emission at 430 nm reached its
maximum level 6 – 7 h after starting oxidation. We
observed maximal reactivity with oxLDL antibodies 4 h
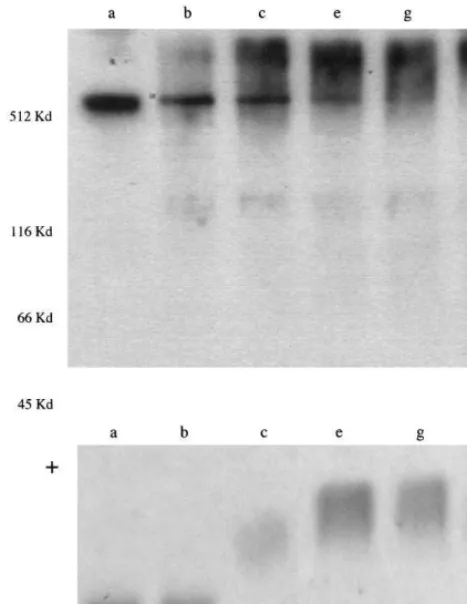
Fig. 3. Top panel: Reproduction of a Western blot analysis of the degree of LDL fragmentation and aggregation associated with differ-ent times of oxidation. Native LDL (a) and five samples collected at the times shown in Fig. 1 were separated by SDS – PAGE and exposed to a polyclonal rabbit human ApoB antibody. Samples ‘g’ and ‘i’ had maximal reactivity with reference oxLDL antibodies (Fig. 2). The positions of molecular weight standards included in the run are indicated on the left margin of the figure. Bottom panel: Repro-duction of an agarose gel electrophoretogram of the samples sepa-rated by Western blot. The samples were applied at origin (O).
conjugated diene peak nor the OD reading of that peak correlated with the maximal fluorescence readings. Af-ter the early 234-nm peak is reached, the OD values continued to slowly increase. This secondary increase in OD is not due to continuing formation of lipid perox-ides but rather to the decomposition of lipid peroxperox-ides to aldehydes and other products, which also absorb light at 234 nm. The decomposition of lipid hydroper-oxides is associated with fragmentation of apolipo-protein B and aggregation of the fragments. A Western blot study of LDL aliquots collected at different times of LDL oxidation showed that aliquots with maximal antibody reactivity (aliquots ‘e’ and ‘g’, Figs. 1 and 2) showed virtually complete disappearance of the 512-kDa ApoB fraction, replaced by small molecular weight fractions and larger molecular weight aggregates (Fig. 3). On agarose gel electrophoresis, aliquots ‘e’ and ‘g’ showed a marked increase in electronegativity, relative to native LDL and aliquots collected at earlier times. Based on these results and the results obtained with one other LDL pool and two LDL preparations iso-lated from different individuals, we decided to monitor the reaction with fluorescence emission data (due to the fact that the peak of fluorescence was closer in time than the early conjugated diene peak to optimal oxida-tion) and to stop the reaction 4 – 6 h after the peak fluorescence value was reached. Spectrophotometric and fluorescence data obtained with 13 LDL pools oxidized and monitored under the outlined conditions showed that the average time needed to reach maximal
fluorescence values was 1094 h, with a maximum of
19 h.
The wide range of time required to reach maximal modification was even more dramatically illustrated with an LDL pool that showed increasing fluorescence values for over 24 h of oxidation (Table 1). In that particular study monitoring was done at various time periods and the highest values of fluorescence were recorded in aliquots harvested after 48 h of oxidation, which also showed maximum reactivity with reference oxLDL antibodies. Both parameters remained stable when oxidation of LDL was continued for an addi-tional 24 h after peak fluorescence was reached.
The experiments described above suggest that once the maximum levels of fluorescence and antibody reac-later. The antibody reactivity of oxLDL aliquots
har-vested at later times (up to 24 h of oxidation) remained unchanged (Fig. 2). Monitoring optical density values at 234 nm showed a very early peak, approximately 2 h after starting the reaction, preceding the stabilization of
fluorescence values by 5 h (Fig. 1). The time needed
to reach the conjugated diene early peak correlates well with the time needed to reach maximal fluorescence emission, but neither the time needed to reach the
Table 1
Longitudinal follow-up of fluorescence and reactivity with reference oxLDL antibodies of LDL harvested at 2, 10, 12, 24 and 48 h of oxidationa
2 10
Time of oxidation (h) 12 24 48 72
1.800 1.680
0.440
Fluorescence (360–430 nm) 2.600 3.801 3.930
0.106 0.253 0.242
Reactivity with purified antibody 0.350 0.696 0.695
0.024 0.065 0.067 0.158 0.342 0.361
Reactivity with reference serum
Table 2
Reactivity of oxLDL aliquots of an LDL pool harvested at 3, 17, 36, and 47 h, after the fluorescence reached the maximum level with purified oxLDL antibody, and two reference sera containing 47 mg/l of oxLDL antibody (serum 1) and 204 mg/l of oxLDL antibody (serum 5)a
17 h
Antibody 3 h 36 h 47 h
0.576
Purified Ab 0.774 0.755 0.789
0.424 0.460
0.208 0.466
Serum 1
0.608
Serum 5 0.478 0.543 0.551
aReactivity is expressed as optical density values calculated by subtracting the OD values obtained on oxLDL-absorbed aliquots from those measured on unabsorbed aliquots. Final dilutions were 1/20 for serum and 1/2 for isolate.
aliquots. All aliquots were dialyzed against PBS-EDTA, filtered, and stored at 4°C, protected from light and under a nitrogen-rich atmosphere. One aliquot of each of the four pools was oxidized shortly after blood collection, as indicated. Stored aliquots from two of the pools were oxidized 5 and 5.5 weeks after blood collec-tion and the other two aliquots were oxidized 9 weeks after blood collection. There was an apparent inverse correlation between pre-oxidation storage time and the time elapsed before maximal LDL oxidation was reached (Table 3). On the other hand, prolonged pre-oxidation storage appeared to result in a lower degree of oxidation, as reflected by lower fluorescence and conjugated dienes values (Table 3). While all the freshly oxidized LDL preparations reached fluorescence values in excess of 1.1, only one of the stored LDL pools (pool 6) reached a level slightly above 1.0 (1.02).
We also tested the reactivity of the eight different oxLDL aliquots with reference antibodies (Table 4). The OD values (expressed as the difference in the readings obtained with unabsorbed and absorbed aliquots) varied between 0.46 and 0.52 when tested with reference serum 1 and between 0.76 and 0.85 when tested with reference serum 5. For all aliquots oxidized after more than 4 weeks of storage time the OD values were lower than those measured with aliquots oxidized after a shorter storage time.
Finally, we investigated the reproducibility of anti-body assays when samples were tested against different oxLDL preparations prepared according to what ap-peared to be optimal conditions, i.e. the oxidation reaction was stopped 4 – 6 h after maximal fluorescence was reached, and the fluorescence value exceeded 1.1 OD U. We used two quality control sera, one
contain-ing a low antibody concentration (47 mg/l) and the
other containing a high antibody concentration (204 mg/l), in a series of 13 EIA runs in which four different tivity are reached, those parameters may remain stable
for a considerable period of time. To better assess this point we carried out an additional experiment in which oxidation of a fourth LDL pool was continued for as long as 47 h after the maximum level of fluorescence was reached. Values of fluorescence and conjugated dienes were monitored continuously until 20 h of oxida-tion. After that, fluorescence and conjugated dienes values were determined simultaneously in aliquots col-lected at specific times. The maximum level of fluores-cence (F430=1.9) was reached after 9 h of oxidation
and it remained stable for a period of 47 h. The reactivity with oxLDL antibodies showed an increase between the aliquots collected at 3 and 17 h after the peak of fluorescence was reached (the 3-h aliquot was collected too early to reflect maximal reactivity), and remained stable at the maximum level in the LDL samples harvested 36 and 47 h after the maximum level of fluorescence was reached (Table 2).
To estimate the effect of storage of native LDL on the kinetics of oxidation we prepared four LDL pools (pools 5 – 8) and divided each one of them into two
Table 3
Study of the effect of LDL storage prior to oxidation on the levels of fluorescence and conjugated dienes measured after oxidationa
Conjugated dienes Fluorescence
LDL pool no.
Peak fluorescence value (360–430
Time to reach the maximal level of OD (234 nm) after 12 h of oxidation
fluorescence (h) nm)
Fresh LDLb Stored LDLc Fresh LDL Stored LDL Fresh LDL Stored LDL
1.89
aOne half of four LDL pools was oxidized within 4 weeks of isolation of the different LDL fractions included in the pool (fresh LDL) and the other half was oxidized after 5–9 weeks of storage (stored LDL). The time of oxidation needed to reach the maximum level of fluorescence and the corresponding values for fluorescence, as well as the levels of conjugated dienes after 12 h of oxidation are presented in the table.
bOxidized within 2 weeks of blood collection.
Table 4
Study of the effect of LDL storage prior to oxidation on the reactivity with reference oxLDL antibodiesa
LDL pool no. Reactivity with reference serum 1 Reactivity with reference serum 5
Stored LDLc
Fresh LDLb Fresh LDL Stored LDL
5 0.47 0.36 0.76 0.63
0.42 0.85
0.46 0.67
6
0.52
7 0.35 0.84 0.62
0.54
8 0.36 0.84 0.69
aThe processing of LDL pools was as detailed in Table 3. Reactivity is expressed as optical density values calculated by subtracting the OD 414 values corresponding to the reactivity of a given oxLDL pool with an unabsorbed aliquot of a reference oxLDL-containing sera and the OD414 corresponding to the reactivity of the same oxLDL pool with a pre-absorbed aliquot of the same reference serum.
bOxidized within 2 weeks of blood collection
cPool 5 was oxidized 5 weeks after blood collection; pool 6 was oxidized 5.5 weeks after blood collection; pools 7 and 8 were oxidized 9 weeks after blood collection.
pools of oxLDL were used as antigens. Fig. 4
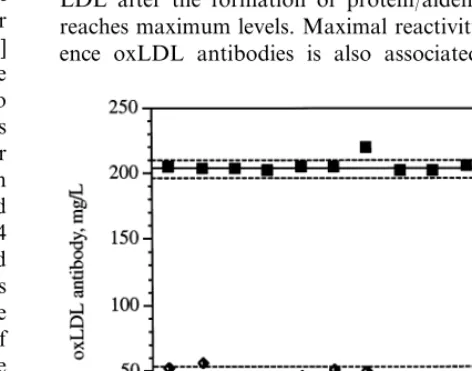
repro-duces graphically the values (mean91 S.D.) obtained
with these two quality control samples; the coefficients of variation calculated from these values were of 14.4% for the low control and 3.9% for the high control. These CV compared favorably with between-run CV calculated with the same samples using a single LDL pool (7.4 and 3.0%, respectively; n=5).
4. Discussion
The lack of consistency in the results of anti-oxLDL surveys published by different groups [1 – 11] has strongly suggested that at least some of the contradic-tions between different studies may result from the lack of standardization of the assays currently used for measurement of oxLDL antibodies. We and others [1] have used copper oxidized LDL as antigen in the antibody assay, because of its similar characteristics to the LDL isolated from human atheromatous plaques [5 – 9]. The protocols used by different groups for preparation of copper oxidized LDL have in common the fact that the oxidation reaction is usually stopped after a given period of time, usually between 18 and 24 h [3,5 – 8,11]. The degree of oxidation has been assessed most often by measurements of TBARS [15] and, less frequently, of conjugated dienes [3,16,23,24]. Since the lack of sensitivity and accuracy of the measurements of TBARS is well known [16], we decided to measure conjugated dienes to monitor the formation of lipid hydroperoxides during LDL oxidation, and to measure fluorescence to monitor the modification of ApoB and
the formation of protein/aldehyde adducts. We then
studied the correlation between these parameters and the reactivity with reference sera with known oxLDL antibody contents and with an oxLDL antibody iso-lated by affinity chromatography.
The formation of conjugated dienes can be measured spectrophotometrically by monitoring absorbance at a
wavelength of 234 nm. To monitor the degree of apo-protein oxidation we relied on the measurement of fluorescent products generated as a consequence of conjugation of aldehyde fragments released by oxidized fatty acids to amino groups [18]. These fluorescent compounds show a maximum emission at 430 nm when excitation is performed at 360 nm.
Since the immunoreactivity of oxLDL is dependent on the modification of ApoB, it was hoped that the fluorescence values would closely reflect the structural changes of ApoB that are associated with immuno-genicity. This postulate was supported by our findings, showing that fluorescence monitoring gives reliable in-formation on the degree of modification of LDL and that purified oxLDL antibody reacts most avidly with
LDL after the formation of protein/aldehyde adducts
reaches maximum levels. Maximal reactivity with refer-ence oxLDL antibodies is also associated with
trophoretic evidence of denaturation, evident in West-ern blot analysis (aggregation, fragmentation) and in agarose gel electrophoresis (increased negative charge). Peak fluorescence values also give valuable indications about antibody reactivity. Our LDL preparations and pools with good reactivity reached fluorescence values above 1.1 U and we have found that when this level of modification is not reached, the corresponding batches of oxLDL do not react adequately with our reference oxLDL antibodies.
Stopping LDL oxidation 4 – 6 h after the fluorescence values reach their peak and ensuring that the peak fluorescence level is above 1.1 has allowed us to obtain oxLDL preparations that react optimally and consis-tently with defined oxLDL antibodies. In contrast, the conjugated diene data could not be reliably linked to the reactivity with reference oxLDL antibodies. The distinct and early peak of conjugated dienes observed at 234 nm during the oxidation reaction may permit esti-mation of the optimal oxidation time, since it usually precedes the fluorescence peak by 4 – 6 h. However, there is a poor correlation between the levels of fluores-cence and the levels of conjugated dienes in the same LDL preparations, and just monitoring the conjugate diene peak would not allow determination of the extent of fluorescence.
Our observations underscored the need to stop the oxidation reaction when it is complete, rather than at a fixed time. On the other hand, the reactivity of oxLDL with defined antibodies seems to become stable 4 – 6 h after the fluorescence values reached their peak. In one experiment in which an LDL pool was oxidized for a total of 56 h, antibody reactivity remained stable even when LDL was collected 47 h after the maximum level of fluorescence was reached (Table 1). However, to avoid possible interference of excessive LDL fragmen-tation, which is known to be more pronounced after long periods of oxidation, we prefer to stop oxidation within 4 – 6 h after fluorescence has reached its peak. The stability of the immunodominant epitopes of oxLDL during the oxidative process was greater than expected. However, when we tried to oxidize LDL aliquots that had been stored for more than 4 weeks prior to oxidation we observed inconsistent results. Monitoring of fluorescence and conjugated dienes showed that peak values were reached in shorter times, and the peak values were lower for LDL aliquots oxidized more than 4 weeks after blood collection than for those oxidized shortly after LDL isolation. Also, the reactivity of LDL aliquots oxidized more than 4 weeks after blood collection with reference sera was lower than the reactivity of LDL aliquots oxidized soon after LDL isolation. Thus, it appears probable that prolonged LDL storage induces either a mild degree of LDL delipidation or a change in the degree of satura-tion of the LDL fatty acids. Either change could cause
not only a reduction in the generation of conjugated dienes during copper oxidation but also in the forma-tion of fluorescent compounds and, consequently, a relative paucity of the epitopes recognized by oxLDL antibodies. This has led us to establish that for prepara-tion of oxLDL to be used in antibody assays we should use LDL pools stored for as short a time as possible, never exceeding 4 weeks after blood collection.
A crucial test for the validity of our protocol for oxLDL preparation was to verify that the reactivity of reference oxLDL antibodies would remain constant when different oxLDL preparations were used. This was confirmed using data from a total of 13 separate runs in which four different oxLDL pools were used as antigens and the same two quality control samples were included in the runs (Fig. 4). The calculated concentra-tions for the quality control samples remained very stable, and the coefficients of variation calculated with the data generated in those 13 runs compared favorably with between run CV determined for the same quality controls using a single batch of oxLDL. Thus, when oxLDL is prepared from a pool of freshly isolated LDL and the oxidation reaction is stopped 4 – 6 h after a maximum fluorescence value is reached and the fluores-cence value is greater than 1.1 U, the resulting oxLDL preparations are remarkably homogeneous in their re-activity with oxLDL antibodies. The use of oxLDL prepared according to these principles has resulted in increased consistency of the data generated in our oxLDL antibody assay [2], as reflected by closer agree-ment of the values for the quality control samples included in each run.
In conclusion, by carefully defining the conditions for preparation of copper-oxidized LDL and by carefully monitoring the oxidation reaction we have been able to obtain oxLDL preparations that exhibit consistent be-havior in our oxLDL antibody assay. Monitoring fluorescence as the oxidation reaction progresses allows us to determine when the reaction should be stopped for individual LDL pools and, more importantly, al-lows us to discard insufficiently modified LDL prepara-tions. Such preparations do not behave consistently in the oxLDL antibody assay and would otherwise not be identified until the results of the oxLDL antibody enzy-moimmunoassay became available.
Acknowledgements
Foun-dation (Dr S. Koskinen). Dr Sinikka Koskinen is a post-doctoral fellow of the Juvenile Diabetes Founda-tion InternaFounda-tional. The authors wish to thank Gregor Krings for technical assistance and Rebecca Rollins for editorial assistance.
References
[1] Salonen JT, Yla¨-Herttuala S, Yamamoto R, Butler S, Korpela H, Salonen R, et al. Autoantibody against oxidised LDL and progression of carotid atherosclerosis. Lancet 1992;339:883 – 7. [2] Virella G, Virella I, Leman RB, Pryor MB, Lopes-Virella MF.
Anti-oxidized low-density lipoprotein antibodies in patients with coronary heart disease and normal healthy volunteers. Int J Clin Lab Res 1993;23:95 – 101.
[3] Maggi E, Chiesa R, Melissano G, Castellano R, Astore D, Grossi A, et al. LDL oxidation in patients with severe carotid atherosclerosis. A study of in vitro and in vivo oxidation mark-ers. Arterioscler Thromb 1994;14:1892 – 9.
[4] Puurunen M, Ma¨ntta¨ri M, Manninen V, Tenkanen L, Alfthan G, Ehnholm C, et al. Antibody against oxidized low-density lipoprotein predicting myocardial infarction. Arch Intern Med 1994;154:2605 – 9.
[5] Bellamo G, Maggi E, Poli M, Agosta FG, Bollati P, Finardi G. Autoantibodies against oxidatively modified low-density lipo-proteins in NIDDM. Diabetes 1995;44:60 – 6.
[6] Bergman C, Wu R, de Faire U, Lefvert AK, Swedenborg J. Patients with early-onset peripheral vascular disease have in-creased levels of autoantibodies against oxidized LDL. Arte-rioscler Thromb Vasc Biol 1995;15:441 – 5.
[7] Uusitupa MIJ, Niskanen L, Luoma J, Vilja P, Mercuri M, Rauramaa R, et al. Autoantibodies against oxidized LDL do not predict atherosclerotic vascular disease in non-insulin-dependent diabetes mellitus. Arterioscler Thromb Vasc Biol 1996;16:1236 – 42.
[8] Raitakari OT, Pitka¨nen O-P, Lehtima¨ki T, Lahdenpera¨ S, Hide-hiro I, Yla¨-Herttuala S, et al. In vivo low density lipoprotein oxidation relates to coronary reactivity in young men. J Am Coll Cardiol 1997;30:97 – 102.
[9] Maggi E, Bellazzi R, Gazo A, Seccia M, Bellomo G. Autoanti-bodies against oxidatively-modified LDL in uremic patients un-dergoing dialysis. Kidney Int 1994;46:869 – 76.
[10] Mironova M, Virella G, Virella-Lowell I, Lopes-Virella MF. Anti-modified LDL antibodies and LDL-containing immune complexes in IDDM patients and healthy controls. Clin Im-munol Immunopathol 1997;85:73 – 82.
[11] Mironova M, Virella G, Lopes-Virella MF. Isolation and char-acterization of human antioxidized LDL antibodies. Arterioscler Thromb Vasc Biol 1996;16:222 – 9.
[12] Haberland ME, Fong D, Cheng L. Malondialdehyde-altered protein occurs in atheroma of Watanabe heritable hyperlipi-demic rabbits. Science 1988;241:215 – 8.
[13] Boyd HC, Gown AM, Wolfbauer G, Chait A. Direct evidence for a protein recognized by a monoclonal antibody against oxidatively modified LDL in atherosclerotic lesions from a Watanabe heritable hyperlipidemic rabbit. Am J Pathol 1989;135:815 – 25.
[14] Palinski W, Rosenfeld ME, Yla¨-Herttuala S, Gurtner GC, Socher SS, Butler SW, et al. Low density lipoprotein undergoes oxidative modification in vivo. Proc Natl Acad Sci USA 1989;86:1372 – 6.
[15] Warren L. The thiobarbituric acid assay of sialic acids. J Biol Chem 1959;234:1971 – 5.
[16] Esterbauer H, Gebicki J, Puhl H, Ju¨rgens G. The role of lipid peroxidation and antioxidants in oxidative modification of LDL. Free Radic Biol Med 1992;13:341 – 90.
[17] Kim RS, LaBella FS. Comparison of analytical methods for monitoring autooxidation profiles of authentic lipids. J Lipid Res 1987;28:1110 – 7.
[18] Cominacini L, Garbin U, Davoli R, Miccioli R, Bosello O, Gaviraghi G, et al. A simple test for predisposition to LDL oxidation based on the fluorescence development during copper-catalyzed oxidative modification. J Lipid Res 1991;32:349 – 58. [19] Lopes-Virella MF, Sherer GK, Lees AM, Wohltmann H,
Mayfield R, Sagel J, et al. Surface binding, internalization, and degradation by cultured human fibroblasts of LDL isolated from type 1 (insulin-dependent) diabetic patients: changes with metabolic control. Diabetologia 1982;22:430 – 6.
[20] Steinbrecher UP. Oxidation of human low density lipoprotein results in derivatization of lysine residues of apolipoprotein B by lipid peroxide decomposition products. J Biol Chem 1987;262:3603 – 8.
[21] Lowry OH, Rosebrough NJ, Farr AL, Randall AJ. Protein measurement with the Folin phenol reagent. J Biol Chem 1951;193:265 – 75.
[22] Koskinen S, Enockson C, Lopes-Virella MF, Virella G. Prepara-tion of a human standard for determinaPrepara-tion of the levels of antibodies to oxidatively modified low-density lipoproteins. Clin Diagn Lab Immunol 1998;5:817 – 22.
[23] Drake TA, Hannani K, Fei H, Lavi S, Berliner JA. Minimally oxidized low-density lipoprotein induces tissue factor expression in cultured human endothelial cells. Am J Pathol 1991;138:601 – 7.
[24] Liao F, Berliner JA, Mehrabian M, Navab M, Demer LL, Lusis AJ, et al. Minimally modified low density lipoprotein is biologi-cally active in vivo in mice. J Clin Invest 1991;87:2253 – 7.