BAB I
PENDAHULUAN
Darah memegang peranan inti dalam kehidupan manusia. Darah beredar dalam pembuluh darah membentuk suatu sistem sirkulasi, dengan jantung sebagai pompanya. Darah mengalir membawa oksigen untuk metabolisme sel dan berbagai zat lain yang dibutuhkan oleh tubuh. Gangguan pada darah atau sirkulasinya tentu membawa dampak yang sangat serius bagi tubuh. Salah satu jenis gangguan hematologi yang diturunkan secara genetik adalah talasemia.
Thalasemia adalah penyakit kelainan darah yaitu anemia hemolitik herediter yang diturunkan secara autosomal resesif dengan disebabkan oleh defek genetik pada pembentukan rantai globin. Penyakit ini baru muncul pada seseorang apabila ia memiliki dua gen talasemia yang berasal dari kedua orang tuanya yaitu satu dari ayah dan satu dari ibu.
Thalasemia tersebar diseluruh ras di mediterania, Timur tengah, India sampai Asia tenggara dan presentasi klinisnya bervariasi dari asimptomatik sampai berat hingga mengancam jiwa, tetapi tidak menutup kemungkinan penyakit ini dapat ditemukan dimana saja diseluruh dunia.
Saat ini, penyakit thalasemia merupakan penyakit genetika yang cukup banyak di Indonesia. Frekuensinya terus meningkat per tahun. Walupun begitu, masyarakat tidak menaruh perhatian yang cukup besar terhadap penyakit yang sudah menjadi salah satu penyakit genetika terbanyak ini. Hal ini disebabkan karena gejala awal dari penyakit sangat umum. Padahal gejala akhir yang ditimbulkan akan sangat fatal jika tidak ditangani secara akurat, cepat, dan tepat.
Melihat kenyataan ini, maka sebaiknya kita harus mewaspadai dengan cara mengetahui dengan benar informasi tentang penyakit ini, sehingga penyakit ini dapat diidentifikasi dan penanganannya pun dapat dilakukan secara dini dengan cara yang tepat.
BAB II
HEMOPOIESIS DAN HEMOGLOBIN
Proses pembentukkan sel darah yaitu hemopoiesis. Proses pembentukkan darah pertama kali terjadi pada fase prenatal yaitu di yolk sac (kantung kuning telur) pada janin usia 0-2 bulan, kemudian fase selanjutnya pada hepar dan lien pada janin usia 2-7 bulan, dan pada fase lanjut di sumsum tulang mulai janin usia 5-9 bulan. Pada post natal, pembentukan utama terjadi di sumsum tulang. Pada bayi dan anak, hematopoisis yang aktif terutama pada sumsum tulang termasuk bagian distal tulang panjang, hal ini berbeda dengan dewasa dimana hematopoisis terbatas pada vertebra, costae, sternum, pelvis, scapula, dan jarang berlokasi pada humerus dan femur. Pada keadaan patologis (sumsum tulang sudah tidak berfungsi atau adanya kebutuhan yang meningkat), pembentukan dapat terjadi di nodus limfatikus, lien, timus, hepar. Pembentukan darah di luar sumsum tulang ini disebut hemopoisis ekstra meduler.
Proses pembentukkan darah dimulai dari sel induk pluripoten yang berdiferensiasi menjadi sel induk limfoid dan sel progenitor myeloid campuran yang kemudian berdiferensiasi lagi.
Darah terdiri dari berbagai komponen yang penting, antara lain sel darah merah (eritosit), sel darah putih (leukosit), keping darah (trombosit) serta plasma. Fungsi leukosit adalah untuk melindungi tubuh terhadap infeksi. Fungsi dari trombosit adalah untuk mekanisme pembekuan darah sedangkan eritrosit membawa satu protein yaitu hemoglobin yang berfungsi dalam mengikat O2 di paru, membawanya ke peredaran darah dan melepaskannya ke sel dan jaringan tubuh.
Hemoglobin (Hb) tersusun atas heme yang merupakan cincin porfirin dalam ikatan dengan Fe dan globulin yang merupakan protein pendukung. Satu molekul hemoglobin mengandung 4 sub-unit. Masing-masing sub-unit tersusun atas satu molekul globin dan satu molekul heme.
Globulin terdiri atas 2 pasang rantai polipeptida, yaitu sepasang rantai α dan sepasang rantai non alpha (β,γ,δ). Kombinasi rantai polipeptida tersebut akan menentukan jenis hemoglobin. Hb A1(2α2β) merupakan lebih dari 96 % Hb total, Hb F (2α2γ) kurang dari 2% dan Hb A2 (2α2δ) kurang dari 3%.
Rantai polipeptida α tersusun atas 141 asam amino, sedangkan rantai non α tersusun atas 146 asam amino. Sintesis rantai α disandi oleh gen α1 dan gen α2 di kromosom 16, sedangkan gen yang mensintesis rantai β, rantai γ dan rantai δ terletak di kromosom 11.
Pada orang normal sintesis rantai α sama dengan rantai non alpha.
Sejak masa embrio, janin, anak hingga dewasa, sel darah merah memiliki 6 hemoglobin, antara lain :
• Hemoglobin embrional (Hb Gower1, Hb Gower2, Hb Portland) • Hemoglobin fetal (Hb-F)
• Hemoglobin dewasa (Hb-A1, Hb-A2) Hemoglobin embrional :
Selama masa gestasi 2 minggu pertama, eritoblas primitif dalam yolc sack membentuk rantai globin epsilon (ε) dan zeta (Z) yang membentuk Hb primitif yaitu Hb Gower1 (Z2ε2). Selanjutnya mulailah sintesis rantai α menggantikan rantai Z dan rantai γ menggantikan rantai ε sehingga membentuk Hb Gower2, Hb Portland. Pada masa gestasi 4-8 minggu yang ditemukan adalah Hb Gower 1 dan Hb Gower 2 dan menghilang pada masa gestasi 3 bulan.
Hemoglobin Fetal
Migrasi sel pruripoten stem sel dari yolc sack ke hati diikuti sintesi Hb fetal yang merupakan awal sintesis rantai Hb β. Setelah masa gestasi 8 minggu, muncul Hb-F yang paling dominan dan setelah janin berusia 6 bulan merupakan 90% Hb terdiri dari Hb-F dan kemudian menurun menjelang kelahiran, setelah bayi lahir dan setelah usia 6-12 bulan, HbF tetap ada tapi hanya ditemukan sedikit.
Hemoglobin Dewasa
Pada masa embrio, telah dideteksi HbA karena telah terjadi proses perubahan sintesis rantai γ menjadi rantai β dan selanjutnya globin β meningkat dan pada masa gestasi 6 bulan ditemukan HbA 5-10% dan waktu lahir 30%. Menginjak usia 6-12 bulan Hb sudah memperlihatkan gambaran Hb dewasa yaitu HbA1 dan HbA2 dan sedikit HbF
Lokus α β γ δ
Genotip α/α β/β γ/γ δ/δ
Polipetida
Hb yang
terbentuk α2β2 α2γ2 α2δ2 (HbA1) (HbF) (HbA2)
Struktur kimia hemoglobin memungkinkan molekul hemoglobin memiliki kemampuan untuk mengikat oksigen secara reversible. Zat besi dalam molekul heme secara langsung berfungsi sebagai pengikat oksigen. Hemoglobin memiilki struktur kuartener empat rantai polipeptida, masing-masing dengan satu tempat pegikatan oksigen. Sehingga satu molekul hemoglobin dapat mengikat 4 molekul oksigen.
BAB III
THALASEMIA
Thalasemia adalah salah satu dari penyakit genetik yang diwariskan dari orang tua kepada anaknya dimana terjadi kelainan sintesis hemoglobin yang heterogen akibat pengurangan produksi satu atau lebih rantai globin yang menyebabkan ketidakseimbangan produksi rantai globin.
II.A. SEJARAH
Sejarah thalasemia dimulai di eropa, dimana seorang peneliti bernama Riettedan Wintrobe mendeskripsikan mengenai adanya anemia mikrositik hipokrom yang tak terjelaskan pada anak-anak keturunan itali dan dilaporkan adanya anemia ringan pada kedua orangtua dari anak-anak-anak-anak yang mengidap anemia tersebut. Pada saat yang bersamaan, seorang dokter spesialis anak, Thomas Cooley juga mendeskripsikan suatu tipe anemia berat pada anak-anak yang berasal dari italia dimana beliau menemukan adanya nukleasi sel darah merah yang masif pada sapuan apus darah tepi yang semula diduga anemia eritroblastik. Namun tak lama, Cooley menyadari bahwa eritoblastik tidak spesifik pada temuan ini dan temuan ini sangat mirip dengan kelainan darah yang ditemukan oleh Riettedan. Sehingga kelainan darah ini dinyatakan sebagai bentuk homozigot dari anemia hipokrom mikrositik yang kemudian diberi labelisasi sebagai thalassemia mayor sedangkan bentuk ringannya dinamakan thalassemia minor. Kata thalassemia berasal dari bahasa yunani yaitu thalassa yang berarti “laut” dan emia yang berarti “berhubungan dengan darah”.
II.B. EPIDEMIOLOGI
WHO (2006) meneliti kira-kira 5% penduduk dunia adalah carrier dari 300-400 ribu bayi thalassemia yang baru lahir pertahunnya. Frekuensi gen thalassemia di Indonesia berkisar 3-10%. Berdasarkan angka ini, diperkirakan lebih 2000 penderita baru dilahirkan setiap tahunnya di Indonesia. Salah satu RS di Jakarta, sampai dengan akhir tahun 2003 terdapat 1060 pasien thalassemia mayor yang berobat jalan di Pusat Thalassemia Departemen Anak FKUI-RSCM yang terdiri dari 52,5 % pasien thalassemia β homozigot, 46,2 % pasien thalassemia HbE, serta
thalassemia α 1,3%. Sekitar 70-80 pasien baru, datang tiap tahunnya. Fakta ini mendukung thalasemia sebagai salah satu penyakit turunan yang terbanyak dan menyerang hampir semua golongan etnik dan terdapat di seluruh negara di dunia termasuk Indonesia.
II.C. PATOFISIOLOGI
Talasemia merupakan salah satu bentuk kelainan genetik hemoglobin yang ditandai dengan kurangnya atau tidak adanya sintesis satu rantai globin atau lebih, sehingga terjadi ketidak seimbangan jumlah rantai globin yang terbentuk. Mutasi gen pada globin alfa akan menyebabkan penyakit alfa- thalassemia dan jika itu terjadi pada globin beta maka akan menyebabkan penyakit beta-thalassemia
Secara genetik, gangguan pembentukan protein globin dapat disebabkan karena kerusakan gen yang terdapat pada kromosom 11 atau 16 yang ditempati lokus gen globin. Kerusakan pada salah satu kromosom homolog menimbulkan terjadinya keadaan heterozigot, sedangkan kerusakan pada kedua kromosom homolog menimbulkan keadaan homozigot (-/-).
Pada thalassemia homozigot, sintesis rantai menurun atau tidak ada sintesis sama sekali. Ketidakseimbangan sintesis rantai alpha atau rantai non alpha, khususnya kekurangan sintesis rantai β akan menyebabkan kurangnya pembentukan Hb.
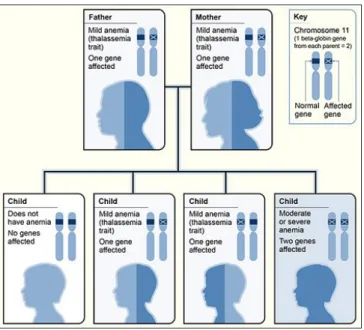
Ketidakseimbangan dalam rantai protein globin alfa dan beta disebabkan oleh sebuah gen cacat yang diturunkan. Untuk menderita penyakit ini, seseorang harus memiliki 2 gen dari kedua orang tuanya. Jika hanya 1 gen yang diturunkan, maka orang tersebut hanya menjadi pembawa/carier.
II.C.I. Thalasemia beta
Secara biokimia kelainan yang paling mendasar adalah menurunnya biosintesis dari unit β globin pada Hb A. Pada thalasemia β heterozigot, sintesis β globin kurang lebih separuh dari nilai normalnya. Pada thalasemia β homozigot, sintesis β globin dapat mencapai nol.
Karena adanya defisiensi yang berat pada rantai β, sintesis Hb A total menurun dengan sangat jelas atau bahkan tidak ada, sehingga pasien dengan thalasemia β homozigot mengalami anemia berat. Sebagai respon kompensasi, maka sintesis rantai γ menjadi teraktifasi sehingga
hemoglobin pasien mengandung proporsi Hb F yang meningkat. Namun sintesis rantai γ ini tidak efektif dan secara kuantitas tidak mencukupi.
Pada thalasemia β homozigot, sintesis rantai α tidak mengalami perubahan dan tidak mampu membentuk Hb tetramer. Ketidak-seimbangan sintesis dari rantai polipeptida ini mengakibatkan kelebihan adanya rantai α bebas di dalam sel darah merah yang berinti dan retikulosit. Rantai α bebas ini mudah teroksidasi. Mereka dapat beragregasi menjadi suatu inklusi protein (haeinz bodys), menyebabkan kerusakan membran pada sel darah merah dan destruksi dari sel darah merah imatur dalam sumsum tulang sehingga jumlah sel darah merah matur yang diproduksi menjadi berkurang sehingga sel darah merah yang beredar menjadi kecil, terdistorsi, dipenuhi oleh inklusi α globin, dan mengandung komplemen hemoglobin yang menurun dan memberikan gambaran dari Anemia Cooley/anemia mikrositik hipokrom yaitu hipokromik, mikrosisitk dan poikilositik.
Sel darah merah yang sudah rusak tersebut akan dihancurkan oleh limpa, hepar, dan sumsum tulang, menggambarkan komponen hemolitik dari penyakit ini. Sel darah merah yang mengandung jumlah Hb F yang lebih tinggi mempunyai umur yang lebih panjang.
Anemia yang berat terjadi akibat adanya penurunan oksigen carrying capacity dari setiap eritrosit dan tendensi dari sel darah merah matur (yang jumlahnya sedikit) mengalami hemolisa secara prematur.
Eritropoetin meningkat sebagai respon adanya anemia, sehingga sumsum-sumsum tulang dipacu untuk memproduksi eritroid prekusor yang lebih banyak. Namun mekanisme kompensasi ini tidak efektif karena adanya kematian yang prematur dari eritroblas. Hasilnya adalah suatu ekspansi sumsum tulang yang masif yang memproduksi sel darah merah baru.
Sumsum tulang mengalami ekspansi secara masif, menginvasi bagian kortikal dari tulang, menghabiskan sumber kalori yang sangat besar pada umur-umur yang kritis pada pertumbuhan dan perkembangan, mengalihkan sumber-sumber biokimia yang vital dari tempat-tempat yang membutuhkannya dan menempatkan suatu stress yang sangat besar pada jantung. Secara klinis terlihat sebagai kegalan dari pertumbuhan dan perkembangan, kegagalan jantung high output, kerentanan terhadap infeksi, deformitas dari tulang, fraktur patologis, dan kematian di usia muda tanpa adanya terapi transfusi.
Jika seseorang memiliki 1 gen beta globin normal, dan satu lagi gen yang sudah termutasi, maka orang itu disebut carier/trait.
Gambar diatas menunjukkan bahwa kedua orangtua merupakan carier/trait. Maka anaknya 25% normal, 50% carier/trait, 25% mewarisi 2 gen yang termutasi (thalasemia mayor).
II.C.II. Thalasemia alpha
Rantai globin yang berlebihan pada thalasemia α adalah rantai γ dan yang kurang atau hilang sintesisnya dalah rantai α. Rantai γ bersifat larut sehingga mampu membentuk hemotetramer yang meskipun relatif tidak stabil, mampu bertahan dan memproduksi molekul Hb yang lain seperti Hb Bart (γ4) dan Hb H (β4). Perbedaan dasar inilah yang mempengaruhi lebih ringannya manisfestasi klinis dan tingkat keparahan penyakitnya dibandingkan dengan thalasemia beta.
Patofisiologi thalasemia α sebanding dengan jumlah gen yang terkena. Pada thalasemia α homozigot (-/-) tidak ada rantai α yang diproduksi. Pasiennya hanya memiliki Hb Bart’s yang tinggi dengan Hb embrionik. Meskipun kadar Hb nya tinggi tapi hampir semuanya adalah Hb
Bart’s sehingga sangat hipoksik yang menyebabkan sebagian besar pasien lahir mati dengan tanda hipoksia intrauterin.
Bentuk thalasemia α heterozigot (α0 dan -α+) menghasilkan ketidakseimbangan jumlah rantainya tetapi pasiennya dapat mampu bertahan dengan HbH dimana kelainan ini ditandai dengan adanya anemia hemolitik karena HbH tidak bisa berfungsi sebagai pembawa oksigen.
Mutasi yang terjadi pada gen alpha globin disebut delesi.
II.D. KLASIFIKASI THALASEMIA DAN PRESENTASI KLINISNYA
Thalassemia α / minor
Penghapusan 4 gen- hydrops fetalis Penghapusan 3 gen- penyakit Hb H Penghapusan 2 gen ( trait thalasemia α° ) Penghapusan 1 gen ( trait thalasemia α+ )
Thalassemia β
Homozigot – thalassemia mayor Heterzigot- trait thalassemia
Thalassemia intermediate
Sindroma klinik yang disebabkan oleh sejenis lesi genetik
II.D.I. Thalasemia α II.
D.I.1. Thalasemia homozigot (α0)
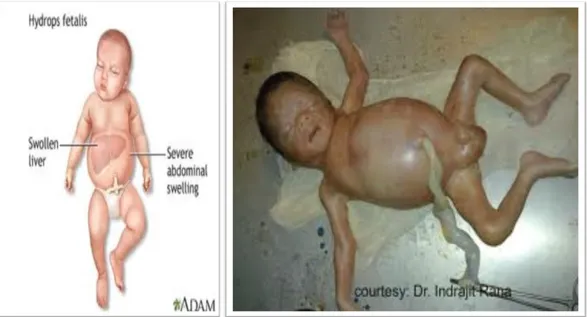
Sindrom hidrops Hb Bart’s biasanya terjadi dalam rahim. Bila hidup hanya dalam waktu pendek. Gambaran klinisnya adalah hidrops fetalis dengan edema permagna dan hepatosplenomegali. Kadar Hb 6-8 g/dl dengan eritrosit hipokromik dan beberapa berinti. Kadar Hb Bart’s 80% dan sisanya Hb portland. Biasanya keadaan ini disertai toksemia gravidarum, perdarahan post partum dan masalah karena hipertrofi plasenta. Pada pemeriksaan otopsi memperlihatkan adanya peningkatan kelainan bawaan. Beberapa bayi berhasil diselamatkan dengan transfusi tukar dan berulang serta pertumbuhannya bisa mencapai normal.
Gambar Hidrops fetalis :
II.
Ditandai anemia mikrositik hipokrom yang cukup berat (7-11 g/dL) dan splenomegali sedang dimana Hb H (β4) dapat dideteksi dalam sel darah merah dengan elektroforesis atau pada sediaan retikulosit. Pada kehidupan janin ditemukan Hb Bart (γ4). HbH bisa diketahui dengan bantuan brilian cresil blue yang akan menyebabkan pengendapan dan pembentukkan badan inklusi. Setelah splenektomi, umumnya bentukkan ini makin banyak di eritrosit. Pada beberapa kasus, penderita bisa tergantung transfusi sedangkan sebagian besar kasus umumnya penderita bisa tumbuh normal tanpa transfusi.
II.
D.I.3. Karier thalasemia α
Bisa berasal dari thalasemia α0 (-/αα) atau thalasemia (-α/-α). Biasanya asimptomatis, didapatkan anemia mikrositik hipokrom ringan dengan penurunan MCH dan MCV yang bermakna. Hb elektroforesisn normal dan pasien hanya bisa didiagnosis dengan analisa DNA. Pada masa neonatus, Hb Bart’s 5-10 % tapi tidak didapatkan HbH pada masa dewasa dan kadang bisa didapatkan inklusi pada eritrosit karier thalasemia α.
II.
D.I.4. Karier thalasemia α silent
Bentuk heterozigot karier thalasemia α+ (–α/αα). Memiliki gambaran darah yang abnormal tetapi dengan elektroforesis normal. Saat lahir 50% kasus memiliki Hb Bart’s 1-3% tapi tidak adanya Hb Bart’s tidak menyingkirkan diagnosa kasus ini.
II.D.II. Thalasemia β
Hampir semua anak dengan thalasemia β homozigot dan heterozigot memperlihatkan gejala klinis sejal lahir yaitu gagal tumbuh, infeksi berulang, kesulitan makan, kelemahan umum. Bayi tampak pucat dan terdapat splenomegali. Bila menerima transfusi berulang, pertumbuhannya bisa normal hingga pubertas.
Pada anak yang mendapat transfusi dan terapi chelasi (pengikat besi), anak bisa mencapai pubertas dan terus mencapai usia dewasa dengan normal. Bila terapi chelasi tidak adekuat, secara bertahap akan terjadi penumpukkan besi yang efeknya mulai nampak pada dekade pertama. Adolscent growth spurt tidak akan tercapai, komplikasi ke hati, endokrin, dan jantung.
• Facies cooley
Terjadi keaktifan sumsum tulang yang luar biasa pada tulang muka dan tulang tengkorak hingga nengakibatkan perubahan perkembangan tulang tersebut dan umumnya terjadi pada anak usia lebih dari 2 tahun
• Pucat yang berlangsung lama
Merupakan gejala umum pada penderita thalassemia, yang berkaitan dengan anemia berat. Penyebab anemia pada thalassemia bersifat primer dan sekunder. Primer adalah berkurangnya sintesis Hb A dan eritropoesis yang tidak efektif disertai penghancuran sel-sel eritrosit intramedular. Sedangkan yang sekunder mengakibatkan hemodilusi, dan destruksi eritrosit oleh sistem retikuloendotelial dalam limpa dan hati.
• Perut membuncit
Pada anak yang besar tampak perut yang membuncit akibat pembesaran hati dan limpa. Hati dan limpa membesar akibat dari hemopoisis ekstrameduler dan hemosiderosis. Dan akibat dari penghancuran eritrosit yang berlebihan itu dapat menyebabkan terjadinya peningkatan biliribin indirek, sehingga menimbulkan kuning pada penderita thalassemia dan kadang ditemui trombositopenia.
• Gagal tumbuh dan mudah terkena infeksi
• Karena pendeknya umur eritrosit menyebabkan hiperurikemi dan gout sekunder sering timbul
• Sering terjadi gangguan perdarahan akibat rombositopenia maupun kegagalan hati akibat penimbunan besi, infeksi dan hemapoiesis ekstramedular.
• Bila pasien ini mencapai pubertas, akan timbul komplikasi akibat penimbunan besi yaitu Keterlambatan menarke (pada anak perempuan) dan gangguan perkembangan sifat seks sekunder akibat dari hemosiderosis yang terjadi pada kelenjar endokrin. Selain pada kelenjar endokrin, hemosiderosis pada pankreas dapat menyebabkan diabetes mellitus. Siderosis miokardium menyebabkan komplikasi ke jantung.
Temuan Laboratorium
• Kelainan morfologi eritrosit pada penderita thalassemia-β° yang tidak ditransfusi adalah ekstrem. Disamping hipokromia dan mikrositosis berat, banyak ditemukan poikilositosit yang terfragmentasi, aneh (bizarre) dan sel target. Sejumlah besar eritrosit yang berinti ada di darah tepi, terutama setelah splenektomi. Inklusi intraeritrosit, yang merupakan presipitasi dari kelebihan rantai α, juga terlihat pasca splenectomi. Kadar Hb turun secara cepat menjadi kurang dari 5 g/dL kecuali jika transfusi diberikan. Kadar bilirubin serum tidak terkonjugasi meningkat. Kadar serum besi tinggi, dengan saturasi kapasitas pengikat besi. Gambaran biokimiawi yang nyata adalah adanya kadar Hb F yang sangat tinggi dalam
eritrosit. Senyawa dipiridol menyebabkan urin berwarna coklat gelap, terutama pasca splenektomi.
II.
D.II. Karier thalasemia β
Hampir tanpa gejala, umumnya dengan anemia ringan dan jarang didapatkan splenomegali. Adanya penurunan ringan kadar Hb dengan penurunan MCV dan MCH yang bermakna.
II.D.III. Intermedia thalasemia
Sindroma klinik yang disebabkan oleh sejenis lesi genetik. Anemia hipokrom mikrositik ( Hb 7-10 gr/dl ), hepatomegali dan splenomegali, deformitas menurun, kelebihan beban besi ( iron over load ).
II.E. PEMERIKSAAN PENUNJANG
Pemeriksaan laboratorium yang perlu untuk menegakkan diagnosis thalasemia ialah:
1. Darah
Pemeriksaan darah yang dilakukan pada pasien yang dicurigai menderita thalasemia adalah Darah rutin
Kadar hemoglobin menurun. Dapat ditemukan peningkatan jumlah lekosit, ditemukan pula peningkatan dari sel PMN. Bila terjadi hipersplenisme akan terjadi penurunan dari jumlah trombosit.
Hitung retikulosit
Hitung retikulosit meningkat antara 2-8 %. Gambaran darah tepi
Anemia pada thalassemia mayor mempunyai sifat mikrositik hipokrom. Pada gambaran sediaan darah tepi akan ditemukan retikulosit, poikilositosis, tear drops sel dan target sel.
Serum Iron & Total Iron Binding Capacity
Kedua pemeriksaan ini dilakukan untuk menyingkirkan kemungkinan anemia terjadi karena defisiensi besi. Pada anemia defisiensi besi SI akan menurun, sedangkan TIBC akan meningkat.
LFT
Kadar unconjugated bilirubin akan meningkat sampai 2-4 mg%. bila angka tersebut sudah terlampaui maka harus dipikir adanya kemungkinan hepatitis, obstruksi batu empedu dan cholangitis. Serum SGOT dan SGPT akan meningkat dan menandakan adanya kerusakan hepar. Akibat dari kerusakan ini akan berakibat juga terjadi kelainan dalam faktor pembekuan darah.
2. Elektroforesis Hb
Diagnosis definitif ditegakkan dengan pemeriksaan eleltroforesis hemoglobin. Pemeriksaan ini tidak hanya ditujukan pada penderita thalassemia saja, namun juga pada orang tua, dan saudara sekandung jika ada. Pemeriksaan ini untuk melihat jenis hemoglobin dan kadar Hb A2. petunjuk adanya thalassemia α adalah ditemukannya Hb Barts dan Hb H. Pada thalassemia β kadar Hb F bervariasi antara 10-90%, sedangkan dalam keadaan normal kadarnya tidak melebihi 1%.
3. Pemeriksaan sumsum tulang
Pada sumsum tulang akan tampak suatu proses eritropoesis yang sangat aktif sekali. Ratio rata-rata antara myeloid dan eritroid adalah 0,8. pada keadaan normal biasanya nilai perbandingannya 10 : 3.
4. Pemeriksaan roentgen
Ada hubungan erat antara metabolisme tulang dan eritropoesis. Bila tidak mendapat tranfusi dijumpai osteopeni, resorbsi tulang meningkat, mineralisasi berkurang, dan dapat diperbaiki dengan pemberian tranfusi darah secara berkala. Apabila tranfusi tidak optimal terjadi ekspansi rongga sumsum dan penipisan dari korteknya. Trabekulasi memberi gambaran mozaik pada tulang. Tulang terngkorak memberikan gambaran yang khas, disebut dengan “hair on end” yaitu menyerupai rambut berdiri potongan pendek pada anak besar.
Thalassemia sering kali didiagnosis salah sebagai anemia defisiensi Fe, hal ini disebabkan oleh karena kemiripan gejala yang ditimbulkan, dan gambaran eritrosit mikrositik hipokrom. Namun kedua penyakit ini dapat dibedakan, karena pada anemia defisiensi Fe didapatkan :
• Pucat tanpa organomegali
• Tidak tedapat besi dalam sumsum tulang
• Bereaksi baik dengan pengobatan dengan preparat besi
II.G. PENGOBATAN
Prinsip pengobatan pada pasien talasemia adalah :
terapi tranfusi darah untuk mencegah komplikasi dari anemia kronis pencegahan dari resiko kelebihan besi akibat terapi transfusi
penatalaksanaan splenomegali
Pada anak dengan thalassemia mayor beta membutuhkan pelayanan kesehatan yang terus menerus seumur hidupnya.
A. Tranfusi darah
Pemberian tranfusi darah ditujukan untuk mempertahankan dan memperpanjang umur atau masa hidup pasien dengan cara mengatasi komplikasi anemia, memberi kesempatan pada anak untuk proses tumbuh kembang, memperpanjang umur pasien. Terapi tranfusi darah dimulai pada usia dini ketika ia mulai menunjukkan gejala simtomatik. Transfusi darah dilakukan melalui pembuluh vena dan memberikan sel darah merah dengan hemoglobin normal. Untuk mempertahankan keadaan tersebut, transfusi darah harus dilakukan secara rutin karena dalam waktu 120 hari sel darah merah akan mati. Khusus untuk penderita beta thalassemia intermedia, transfuse darah hanya dilakukan sesekali saja, tidak secara rutin. Sedangkan untuk beta thalssemia mayor (Cooley’s Anemia) harus dilakukan secara teratur
Tranfusi darah diberikan bila Hb anak < 7 gr/dl dyang diperiksa 2x berturut dengan jarak 2 mingg dan bila kadar Hb > 7 gr/dl tetapi disertai gejala klinis seperti Facies Cooley, gangguan tumbuh kembang, fraktur tulang curiga adanya hemopoisis ekstrameduler. Pada penanganan
selanjutnya, transfusi darah diberikan Hb ≤8 gr/dl sampai kadar Hb 11-12 gr/dl. Darah diberikan dalam bentuk PRC, 3 ml/kgBB untuk setiap kenaikan Hb 1 g/dL.
B. Kelasi Besi
Pasien thalasemia dengan terapi tranfusi biasanya meninggal bukan karena penyakitnya tapi karena komplikasi dari tranfusi darah tersebut. Komplikasi tersebut adalah penumpukan besi diberbagai organ.
Desferoxamine diberikan setelah kadar feritin serum sudah mencapai 1000 mg/L atau saturasi transferin sudah mencapai 50 %, atau sekitar setelah 10 -20 kali transfusi. Pemberian dilakukan secara subkutan melalui pompa infus dalam waktu 8-12 jam dengan dosis 25-35 mg/kg BB/hari, minimal selama 5 hari berturut-turut setiap selesai transfusi darah. Dosis desferoxamine tidak boleh melebihi 50 mg/kg/hari. Evaluasi teratur terhadap toksisitas desferoxamin direkomendasikan pada semua pasien yang mendapat terapi ini.
Saat ini sudah tersedia kelasi besi oral, namun penggunaannya di Indonesia belum dilakukan.
C. Suplemen Asam Folat
Asam folat adalah vitamin B yang dapat membantu pembangunan sel-sel darah merah yang sehat. Suplemen ini harus tetap diminum di samping melakukan transfusi darah ataupun terapi khelasi besi.. Asam Folat 2x1 mg/hari untuk memenuhi kebutuhan yang meningkat.
D. Splenektomi
Indikasi :
limpa yang terlalu besar sehingga membatasi gerak pasien, menimbulkan peningkatan tekanan intra-abdominal dan bahaya terjadinya ruptur
D. Transplantasi sumsum tulang
Transplantasi sumsum tulang untuk talasemia pertama kali dilakukan tahun 1982. Transplantasi sumsum tulang merupakan satu-satunya terapi definitive untuk talasemia. Jarang dilakukan karena mahal dan sulit.
II.H. SKRINING DAN PENCEGAHAN II.H.1 SKRINING
Bila populasi tersebut hendak memiliki pasangan, dilakukan skrining premarital. Penting sekali menyediakan program konselin verbal dan tertulis mengenai hasil skring.
Alternatif lain, memeriksakan setiap wanita hamil muda berdasarkan ras. Skrining yang efektif adalah melalui eritrosit. Bila MCV dan MCH sesuai gambaran thalasemia, perkiraan kadar HbA harus diukur. Bila kadarnya normal, pasien dikirim ke pusat yang menganalisis gen. Penting
untuk memeriksa Hb elektroforesa pada kasus-kasus ini untuk mencari kemungkinan variasi struktural Hb.
II.H.2 PENCEGAHAN
Ada 2 pendekatan untuk menghindari thalasemia, yaitu :
• Karena karier thalasemia β bisa diketahui dengan mudah, skrining populasi dan konseling tentang pasangan bisa dilakukan. Bila heterozigot menikah, 1 dari 4 anak mereka bisa menjadi homozigot atau gabungan heterozigot
• Bila ibu heterozigot sudah diketahui sebelum lahir, pasangan bisa diperiksa dan bila termasuk karier, pasangan tersebut ditawari diagnosis prenatal dan terminasi kehamilan pada fetus dengan thalasemia β berat
BAB IV
PENUTUP
III. 1. KESIMPULAN
Thalassemia merupakan penyakit genetik yang disebabkan oleh ketidaknormalan pada protein globin yang terdapat di gen. Dapat menyerang siapa aja dengan berbagai etnik ras di seluruh dunia dan termasuk salah satu penyakit genetik kelainan darah yang terbanyak di Indonesia. Jika globin alfa yang rusak maka penyakit itu dinamakan alfa-thalassemia dan jika globin beta yang rusak maka penyakit itu dinamakan alfa thalassemia. Gejala yang terjadi dimulai dari anemia hingga gangguan tumbuh kembang. Pemeriksaan thalasemia bisa dilakukan melalui pemeriksaan darah, Hb elektroforesa, pemeriksaan sumsum tulang dan roentgen. Thalassemia harus sudah diobati sejak dini agar tidak berdampak fatal. Pengobatan yang dilakukan adalah dengan melakukan transfusi darah, meminum beberapa suplemen asam folat, terapi kelasi besi, splenektomi, hingga transplantasi sumsum tulang. Thalasemia bisa diketahui sedini mungkin dengan proses skrining.
DAFTAR PUSTAKA
1. Berhman, RE; Kliegman, RM ; Arvin: Nelson Ilmu Kesehatan Anak, volume 2, edisi 15. Penerbit Buku Kedokteran EGC, Jakarta : 2005, hal1708-1712
2. Berhman, RE; Kliegman, RM and Jensen, HB: Nelson Text Book of Pediatrics, 16th edition. WB Saunders company, Philadelphia: 2000, page 1630-1634
3. Permono, H. BAmbang; Sutaryo; Windiastuti, Endang; Abdulsalam, Maria; IDG Ugrasena: Buku Ajar Hematologi-Onkologi Anak, Cetakan ketiga. Penerbit Badan Penerbit IDAI, Jakarta : 2010, hlm 64-84
4. A.V. Hoffbrand and J.E. Pettit; alih bahasa oleh Iyan Darmawan : Kapita Selekta Haematologi, edisi ke 2. Penerbit Buku Kedokteran EGC, Jakarta : 1996, hal 66-85
5. Children's Hospital & Research Center Oakland. 2005. “What is Thalassemia and Treating Thalassemia”.
6. Markum : Buku Ajar Ilmu Kesehatan Anak jilid 1. FKUI, Jakarta : 1991, hal 331
7. Paediatrica Indonesiana, The Indonesian Journal of pediatrics and Perinatal Medicine, volume 46, No.5-6. Indonesian Pediatric Society, Jakarta: 2006, page 134-138