MODIFIKASI PEMBENTUKAN SPECTRUM PADA METODE
SPECTRAL ALIGNMENT UNTUK PENGOREKSIAN DNA
SEQUENCING ERROR
GERRY INDRAMADES ALMI
DEPARTEMEN ILMU KOMPUTER
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM INSTITUT PERTANIAN BOGOR
BOGOR 2014
PERNYATAAN MENGENAI SKRIPSI DAN
SUMBER INFORMASI SERTA PELIMPAHAN HAK CIPTA
Dengan ini saya menyatakan bahwa skripsi berjudul Modifikasi Pembentukan Spectrum Pada Metode Spectral Alignment untuk Pengoreksian DNA Sequencing Error adalah benar karya saya dengan arahan dari komisi pembimbing dan belum diajukan dalam bentuk apa pun kepada perguruan tinggi mana pun. Sumber informasi yang berasal atau dikutip dari karya yang diterbitkan maupun tidak diterbitkan dari penulis lain telah disebutkan dalam teks dan dicantumkan dalam Daftar Pustaka di bagian akhir skripsi ini.Dengan ini saya melimpahkan hak cipta dari karya tulis saya kepada Institut Pertanian Bogor.
Bogor, Agustus 2014 Gerry Indramades Almi NIM G64100122
ABSTRAK
GERRY INDRAMADES ALMI. Modifikasi Pembentukan Spectrum pada Metode Spectral Alignment untuk Pengoreksian DNA Sequencing Error. Dibimbing oleh WISNU ANANTA KUSUMA
DNA sequence assembly berbasis graf biasanya digunakan untuk menghasilkan contigs dengan cara merangkai reads yang dihasilkan oleh DNA sequencer generasi kedua. Namun demikian, DNA sequence assembly berbasis graf ini sensitif terhadap keberadaan sequencing error. Adanya sequencing error akan meningkatkan kompleksitas graf. Sementara itu, setiap proses sequencing selalu menghasilkan sequencing error. Penelitian ini bertujuan untuk memperbaiki kinerja dari pengoreksian DNA sequencing error berbasis spectral alignment dengan mengimplementasikan pendekatan statistika ketika membentuk spectrum. Pendekatan ini menghasilkan spektrum dari solid tuple dengan memilih tuple yang termasuk ke dalam 75% tertinggi dari distribusi kemunculan tuple. Reads yang mengandung sequencing error dikoreksi dengan sekuens referensi yang termasuk di dalam spektrum solid tuple ini. Evaluasi dilakukan dengan menggunakan Velvet, sebuah perangkat lunak DNA assembly. Hasil evaluasi menunjukkan bahwa pendekatan ini dapat mereduksi kompleksitas graf hingga 45% dibandingkan hasil yang diperoleh pendekatan sebelumnya.
Kata kunci: DNA sequencing error, spectral alignment, DNA sequence assembly, pendekatan statistika
ABSTRACT
GERRY INDRAMADES ALMI. Modification of Spectrum Generation on Spectral Alignment Method for DNA Sequencing Error Correction. Supervised by WISNU ANANTA KUSUMA
Graph based DNA sequence assembly is actually used to generate contigs form reads produced by second generation sequencer. However, graph based DNA sequence assembly is very sensitive against sequencing error. The existence of sequencing errors will increase the complexity of graph. Meanwhile, every process of sequencing always produce sequencing errors. This research aims to improve the performance of DNA sequencing error correction based on the spectral alignment by implementing a statistical approach. This approach generate the spectrum of solid tuple by choosing tuples that belong to the highest 75% of the tuple occurencies distribution. Reads containing sequencing errors are corrected using tuple references that belong to the solid tuple spectrum. Evaluation is conducted using Velvet, a DNA assembly software. The results show that our approach can reduce the complexity of graph produced by the previous approach up to 45%.
Keywords: DNA sequencing error, spectral alignment, DNA sequence assembly, statistical approach
Skripsi
sebagai salah satu syarat untuk memperoleh gelar Sarjana Ilmu Komputer
pada
Departemen Ilmu Komputer
MODIFIKASI PEMBENTUKAN SPECTRUM PADA METODE
SPECTRAL ALIGNMENT UNTUK PENGOREKSIAN DNA
SEQUENCING ERROR
GERRY INDRAMADES ALMI
DEPARTEMEN ILMU KOMPUTER
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM INSTITUT PERTANIAN BOGOR
BOGOR 2014
Penguji: Toto Haryanto, S.Kom, M.Si
Judul Skripsi : Modifikasi Pembentukan Spectrum pada Metode Spectral Alignment untuk Pengoreksian DNA Sequencing Error Nama : Gerry Indramades Almi
NIM : G64100122
Disetujui oleh
Dr Eng Wisnu Ananta Kusuma, ST MT Pembimbing I
Diketahui oleh
Dr Ir Agus Buono, MSi MKom Ketua Departemen
PRAKATA
Puji dan syukur penulis panjatkan kepada Allah subhanahu wa ta’ala atas segala karunia, rahmat dan ridho-Nya sehingga karya ilmiah ini berhasil diselesaikan. Penelitian ini berfokus pada bidang bioinformatika dengan menitikberatkan pada pengoreksian DNA sequence sebagai salah satu praproses untuk merangkai sequence hasil proses sequencing menjadi whole genome. Hal yang menjadi motivasi penulis dalam memilih topik ini yaitu bioinformatika sebagai salah satu bidang ilmu yang selalu menawarkan metode-metode baru untuk menyelesaikan masalah-masalah terutama pada bidang biologi molekuler melalui bantuan teknologi informasi.
Terima kasih penulis ucapkan kepada Bapak Dr Eng Wisnu Ananta Kusuma, ST MT selaku pembimbing yang senantiasa selalu membimbing, mengawasi dan mengingatkan penulis pada penelitian ini. Tak lupa pula penulis menyampaikan terima kasih kepada ayah, ibu dan adik yang selalu mendukung dan mendoakan selama penelitian ini berlangsung. Ungkapan terima kasih juga disampaikan kepada teman-teman satu bimbingan yaitu Huda, Yuda, Alfat, dan Delly serta rekan-rekan satu angkatan Ilmu Komputer angkatan 47 yang secara langsung dan tidak langsung membantu penulis pada penelitian ini.
Semoga karya ilmiah ini bermanfaat bagi perkembangan ilmu pengetahuan secara umum dan ilmu komputer pada khususnya.
Bogor, Agustus 2014 Gerry Indramades Almi
DAFTAR ISI
DAFTAR TABEL vi DAFTAR GAMBAR vi PENDAHULUAN 1 Latar Belakang 1 Perumusan Masalah 1 Tujuan Penelitian 2 Manfaat Penelitian 2Ruang Lingkup Penelitian 2
METODE PENELITIAN 2
Akuisisi data DNA sequence 2
Implementasi Metode Spectral Alignment 3
Koreksi Data DNA sequence 4
Evaluasi 5
HASIL DAN PEMBAHASAN 6
Simulasi Data DNA sequence dengan MetaSim 6
Perangkat Lunak Pendeteksi dan Pengkoreksi Error 6
Perbandingan hasil koreksi perangkat lunak 9
Evaluasi hasil koreksi perangkat lunak 10
SIMPULAN DAN SARAN 12
Simpulan 12
Saran 12
DAFTAR PUSTAKA 12
DAFTAR TABEL
1 Spesifikasi organisme yang digunakan pada penelitian 3 2 Konfigurasi parameter untuk membangkitkan data DNA sequence 6 3 Perbandingan hasil eksekusi perangkat lunak 9 4 Perbandingan waktu eksekusi perangkat lunak 10 5 Hasil DNA sequence assembly dengan Velvet 11
DAFTAR GAMBAR
1 Ilustrasi penentuan solid tuple 4
2 Persamaan matematika jarak levenshtein 5
3 Diagram alir tahapan kerja perangkat lunak yang dihasilkan 6
4 Pseudocode pembentukan spectrum 7
5 Ilustrasi penentuan anggota himpunan T-string 7 6 Contoh inisialisasi matriks pemberian skor penilaian 8 7 Contoh matriks pemberian skor penilaian yang telah terisi 9 8 Hasil konstruksi graf dengan fragmen tanpa error 10 9 Hasil konstruksi graf dengan fragmen mengandung error 11
PENDAHULUAN
Latar Belakang
Teknologi DNA sequencing saat ini telah mengalami banyak perkembangan, salah satunya teknologi DNA sequencing generasi kedua. Teknologi DNA sequencing generasi kedua dapat menghasilkan reads yang lebih banyak dan dengan biaya yang lebih murah jika dibandingkan dengan teknologi Sanger sequencing, meskipun panjang reads yang dihasilkan jauh lebih pendek. Salah satu contohnya adalah Illumina Genome Analyzer yang dapat menghasillkan 1.5 miliar base-pairs (bp) dengan panjang read 36 dalam satu kali eksekusi selama 60 jam (Shi et al. 2009).
Mayoritas teknologi DNA assembly yang sudah mapan saat ini dirancang agar dapat bekerja optimal ketika merakit reads hasil Sanger sequencing. Teknologi ini didesain untuk merangkai reads berukuran panjang (Shi et al. 2009). Oleh karena itu diperkenalkanlah beberapa teknologi DNA assembly yang baru, salah satunya yang menggunakan pendekatan k-mer. Akan tetapi pendekatan ini tidak dapat merangkai bagian yang berulang (repeat) dengan akurat (Shi et al. 2009). Untuk mengatasi masalah ini, dikembangkanlah metode DNA sequence assembly dengan pendekatan Eulerian path pada graf De-Bruijn (Pevzner et al. 2001).
DNA sequence assembly dengan pendekatan graf De-Bruijn ternyata mampu mengatasi data reads yang pendek dan banyak, serta dapat mengatasi masalah repeat. Masalahnya adalah DNA sequence assembly dengan pendekatan graf sangat sensitif terhadap error. Padahal setiap proses sequencing pasti menghasilkan error. Jika data yang dimasukan mengandung error, graf yang dihasilkan dapat menjadi lebih kompleks dari yang seharusnya. Error tersebut harus diperbaiki sebelum reads dirangkai dengan DNA assembler berbasis graf. Oleh karena itu, penelitian ini dilakukan untuk membangun sistem yang dapat mendeteksi dan mengoreksi error pada DNA sequence.
Salah satu penelitian yang terkait pengkoreksian DNA sequencing dilakukan oleh Caesar et al (2013) dengan menerapkan metode spectral alignment. Pada penelitian tersebut, Caesar et al (2013) menggunakan nilai threshold untuk menentukan solid tuple yang menjadi bagian dari spectrum. Akan tetapi, hasil evaluasi yang didapat oleh Caesar et al (2013) belum terlalu baik. Jumlah node yang dihasilkan mencapai 8-10 kali lipat dibandingkan jumlah node yang dihasilkan oleh data tanpa error. Pada penelitian ini, penentuan solid tuple dilakukan dengan memperhatikan peringkat frekuensi kemunculan tuple. Diharapkan perangkat lunak yang dihasilkan dapat mendeteksi dan mengoreksi error dengan lebih baik dibandingkan perangkat lunak yang dihasilkan pada penelitian sebelumnya.
Perumusan Masalah
Untuk meningkatkan hasil koreksi yang didapat, ada dua pendekatan yang dapat dilakukan. Pendekatan pertama adalah dengan mengganti keseluruhan metode dengan metode yang dianggap lebih baik. Pendekatan kedua adalah
2
dengan melakukan perubahan terhadap metode yang sedang digunakan, sehingga didapat hasil koreksi yang lebih baik. Pendekatan yang digunakan pada penelitian ini adalah pendekatan kedua.
Rumusan masalah pada penelitian ini adalah:
1 Perubahan seperti apa yang harus dilakukan di tahap pembentukan spectrum pada metode spectral alignment sehingga didapat hasil koreksi DNA sequencing error yang lebih baik?
2 Apakah hasil yang didapatkan lebih baik dibandingkan perangkat lunak yang dihasilkan oleh Caesar et al (2013)?
Tujuan Penelitian
Penelitian ini bertujuan untuk membangun perangkat lunak pengoreksi error pada DNA sequence dengan mengubah cara pembentukan spectrum pada metode spetral alignment, dan membandingkan hasil koreksinya dengan data yang belum dikoreksi dan data hasil koreksi penelitian sebelumnya (Caesar et al 2013).
Manfaat Penelitian
Penelitian ini diharapkan dapat mempercepat proses DNA sequence assembly. Reads hasil proses sequencing yang dikoreksi dengan perangkat lunak ini dapat mengurangi kompleksitas graf yang dihasilkan. Dengan demikian contigs yang dihasilkan akan lebih panjang dan proses DNA sequence assembly dapat dilakukan dengan lebih cepat.
Ruang Lingkup Penelitian Ruang lingkup penelitian ini antara lain :
1 Data DNA sequence yang digunakan adalah data hasil simulasi dengan MetaSim menggunakan error model Empirical (Solexa).
2 Proses perakitan DNA sequence pada tahap evaluasi menggunakan perangkat lunak Velvet (Zerbino dan Birney 2008).
METODE PENELITIAN
Secara garis besar, penelitian ini dibagi menjadi empat tahap, yaitu akuisisi data DNA sequence, implementasi metode spectral alignment, koreksi data DNA sequence, dan evaluasi hasil koreksi.
Akuisisi data DNA sequence
Data DNA sequence yang digunakan pada penelitian ini diunduh dari
website NCBI1. Data tersebut merupakan data hasil proses sekuensing bermacam-macam makhluk hidup. Dari data tersebut, dipilihlah tiga organisme seperti yang
1 ftp://ftp.ncbi.nlm.nih.gov/genomes/Bacteria/all.fna.tar.gz
3 tercantum dalam Tabel 1. Data ke-3 organisme itu selanjutnya akan diproses dengan menggunakan perangkat lunak MetaSim (Richter et al. 2008), untuk disimulasikan dan menghasilkan fragmen-fragmen yang mengandung error. Fragmen-fragmen yang mengandung error itulah yang menjadi masukan perangkat lunak yang dikembangkan pada penelitian ini.
Tabel 1 Spesifikasi organisme yang digunakan pada penelitian
Organisme Panjang sekuens
Lengkap (bp) Gi
Acetobacter pasteurianus IFO
3283-01 plasmid pAPA01-060 1815 258513334
Lactobacillus plantarun
WCFS1 plasmid pWCFS101 1917 54307228
Staphylococcus aureus subsp.
Aureus ED98 plasmid pAVY 1442 262225764
Implementasi Metode Spectral Alignment
Pada tahap ini, dibuat sebuah perangkat lunak yang mampu mengkoreksi error pada data DNA sequence dengan menerapkan metode spectral alignment. Penelitian ini menggunakan library SeqAn (Döring et al. 2008). SeqAn dibangun menggunakan bahasa C++ berisi pustaka-pustaka yang mengimplementasikan algoritme dan struktur data yang efisien untuk mengelola dan memanipulasi data DNA sequence. Metode spectral alignment terdiri atas empat bagian, yaitu penentuan solid dan weak tuple, pembentukan spectrum dari solid tuple, pembentukan T-string, dan pencarian anggota T-string yang paling mirip dengan reads masukan (Shi et al. 2009).
Solid Tuple dan Weak Tuple
Setelah data masukan dibaca, ditentukan tuple yang termasuk solid tuple atau weak tuple. Tuple yang tidak termasuk solid tuple disebut weak tuple. Untuk menentukan jenis tuple, didefinisikan hal berikut. Diberikan himpunan data sequence hasil pembacaan yang disebut R = {r1, r2, r3, ...,rk}, dan | ri | = L, dengan
ri∈ {A,C,G,T}L untuk seluruh i pada selang l ≤ i ≤ k. Simbol A, C, G dan T
merupakan kode nuklotida dengan A untuk adenin, C untuk sitosin, G untuk guanin dan T untuk timin.
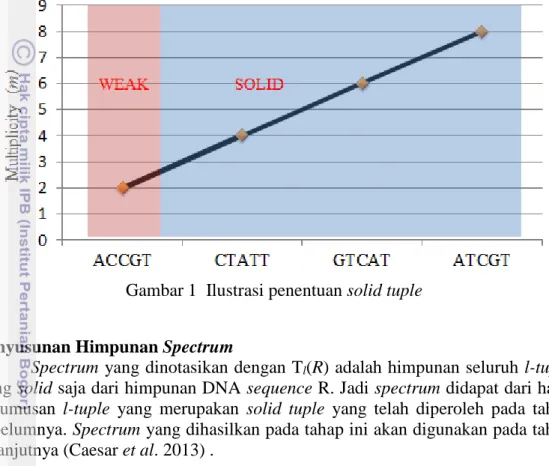
Pada penelitian yang dilakukan oleh Caesar et al (2013), parameter yang digunakan untuk menentukan solid tuple adalah, panjang dari tuple (l) dan multiplicity (m). Multiplicity adalah jumlah kemunculan sebuah tuple pada himpunan R. Pada penelitian ini, parameter yang digunakan tidak hanya panjang (l) dan multiplicity (m) saja, pendekatan statistika juga akan digunakan dengan mengambil 75% tertinggi dari distribusi kemunculan tuple, seperti yang diilustrasikan pada Gambar 1. Jadi, yang termasuk solid tuple adalah tuple yang memiliki panjang (l) dan memiliki nilai (m) yang termasuk 75% tertinggi dari keseluruhan tuple. Sebagai contoh, R = {AAGC, AAAG, AACT, AGGT, AACC}. Sebuah himpunan l-tuple dengan panjang 2, disebut juga 2-tuple berisi
4
{AA, AG, AC, GT}, jika menerapkan parameter di atas, maka yang merupakan solid tuple adalah {AA, AG, AC} karena AA, AG, dan AC memiliki nilai (m) masing-masing empat, tiga, dan dua, sehingga termasuk ke dalam himpunan tuple dengan nilai multiplicity 75% tertinggi dari seluruh tuple. Tuple yang bukan solid tuple dianggap sebagai weak tuple.
Gambar 1 Ilustrasi penentuan solid tuple
Penyusunan Himpunan Spectrum
Spectrum yang dinotasikan dengan Tl(R) adalah himpunan seluruh l-tuple yang solid saja dari himpunan DNA sequence R. Jadi spectrum didapat dari hasil perumusan l-tuple yang merupakan solid tuple yang telah diperoleh pada tahap sebelumnya. Spectrum yang dihasilkan pada tahap ini akan digunakan pada tahap selanjutnya (Caesar et al. 2013) .
Koreksi Data DNA sequence
Pendeteksian Error
Setelah spectrum Tl(R) selesai dibentuk, langkah selanjutnya adalah menentukan reads mana saja yang merupakan anggota T-string dan bukan anggota T-string. T-string merupakan himpunan reads yang seluruh substring-nya ada didalam Tl(R). Reads yang bukan merupakan anggota T-string akan masuk ke dalam himpunan F, yaitu himpunan yang seluruh anggotanya merupakan DNA sequence yang mengandung error.
Pengoreksian Error
Setelah T-string dan himpunan F terbentuk, setiap anggota F akan dikoreksi satu demi satu, dengan membandingkannya dengan setiap anggota T-string dan menggantinya dengan anggota T-T-string yang paling mirip. Metode yang digunakan untuk mengukur kemiripan dua sekuens tersebut adalah dengan menggunakan jarak Levenshtein atau sering juga disebut sebagai edit distance (Levensthein 1966).
5
Jarak Levenshtein
Jarak Levensthein adalah langkah minimum yang dibutuhkan untuk mengubah satu string menjadi string lain dengan single-character edit (insertion, deletion, subtitution). Misalkan terdapat dua buah string, yakni a dan b, jarak levensthein dapat dirumuskan seperti pada Gambar 2.
Gambar 2 Persamaan matematika jarak levenshtein
Untuk setiap satu aksi yang diterapkan dihitung sebagai 1 skor. Sebagai contoh:
o kitten → bitten, menghasilkan skor 1 (substitution “k” menjadi “b”) o kitten → kitty, menghasilkan skor 2 (substitution karakter “e” menjadi “y”,
deletion karakter “n” di akhir string)
Semakin kecil nilai jarak levenshtein yang dihasilkan, semakin mirip kedua string. Tetapi pada penelitian ini, skor yang diberikan setiap single-character edit adalah -1, sehingga semakin besar nilai jarak levenshtein, semakin mirip kedua string.
Evaluasi
Evaluasi dilakukan dengan memasukan empat dataset ke dalam perangkat lunak DNA sequence assembler. Empat dataset tersebut meliputi, dataset hasil koreksi dengan perangkat lunak penelitian ini, dataset hasil koreksi dengan perangkat lunak penelitian sebelumnya, dataset tanpa error dan dataset dengan error yang tidak dikoreksi. Pada penelitian ini, sequence assembler yang digunakan adalah Velvet. Velvet adalah perangkat lunak yang terdiri atas algoritme-algoritme untuk memanipulasi graf De Bruijn dalam rangka melakukan DNA sequence assembly (Zerbino dan Birney 2008). Setelah diperoleh graf dari ke empat dataset tersebut, jumlah node yang dihasilkan oleh masing-masing dataset akan dibandingkan satu sama lain. Jumlah node merepresentasikan kompleksitas graf yang terbentuk. Semakin banyak error pada data, semakin banyak node yang terbentuk, dengan demikian semakin kompleks graf tersebut. Dataset tanpa error akan memiliki jumlah node paling sedikit dibandingkan dataset yang memiliki error dan dataset hasil koreksi.
6
HASIL DAN PEMBAHASAN
Simulasi Data DNA sequence dengan MetaSim
Untuk dataset yang menjadi input perangkat lunak penelitian ini, simulasi dilakukan pada setiap organisme sebanyak satu kali. Simulasi dilakukan dengan perangkat lunak MetaSim. Simulasi dilakukan untuk menghasilkan data yang mengandung error. Error model yang digunakan adalah Empirical (Solexa) dengan panjang per fragmen adalah 36. Pada error model yang digunakan, jenis error yang ditimbulkan dibatasi hanya pada error subtitusi saja dengan distribusi seragam. Konfigurasi parameter secara lengkap dapat dilihat pada Tabel 2.
Tabel 2 Konfigurasi parameter untuk membangkitkan data DNA sequence
Organisme Number of reads or mate pairs Error model DNA clone size distribution type Mean Second parameter Acetobacter pasteurianus 2000 Empirical - Solexa Normal 36 0 Lactobacillus plantarun 2000 Empirical - Solexa Normal 36 0 Staphylococcus aureus 1500 Empirical - Solexa Normal 36 0
Untuk dataset yang akan menjadi input perangkat lunak penelitian sebelumnya (Caesar et al. 2013), seluruh parameternya sama dengan parameter yang ada pada Tabel 2. Untuk dataset tanpa error, parameter yang digunakan juga sama seperti parameter pada Tabel 2, namun Error model yang digunakan adalah Exact, yang berarti tanpa error.
Perangkat Lunak Pendeteksi dan Pengkoreksi Error
Membaca fail masukan berformat FASTA Membangkitkan permutasi dari A, C, G, T dengan panjang l Menyusun spectrum Menyusun himpunan T-String dan himpunan F Koreksi seluruh anggota himpunan F Tulis fail keluaran berformat FASTA
7 Secara garis besar, tahapan kerja perangkat lunak pendeteksi dan pengoreksi error DNA sequence dapat dilihat pada Gambar 3. Perangkat lunak menerima input berupa fail FASTA (.fna) yang berisi fragmen-fragmen yang mengandung error hasil simulasi dengan MetaSim. Setelah isi fail dibaca dan disimpan ke dalam memori, sistem akan membangkitkan permutasi tanpa perulangan dari nukleotida A, C, G dan T. Panjang permutasi (l) yang digunakan adalah lima, sama seperti penelitian yang dilakukan Caesar et al (2013). Selanjutnya, kumpulan permutasi yang dihasilkan akan disebut sebagai pool.
Gambar 4 Pseudocode pembentukan spectrum
Setelah pool terbentuk, selanjutnya akan dibentuk himpunan spectrum. Himpunan spectrum berisikan tuple-tuple yang termasuk ke dalam solid tuple. Solid tuple sendiri merupakan tuple yang memiliki panjang l dan termasuk 75% tertinggi dalam hal nilai multiplicity-nya. Pseudocode pembentukan spectrum dapat dilihat pada Gambar 4.
8
Selanjutnya akan dibentuk himpunan T-string dengan merujuk pada himpunan spectrum. Himpunan T-string adalah himpunan reads yang seluruh substring-nya terdapat pada himpunan spektrum, sehingga dapat juga dikatakan sebagai himpunan reads yang tidak mengandung error. Reads-reads yang tidak termasuk ke dalam himpunan T-string akan masuk ke dalam himpunan F, yakni himpunan reads yang mengandung error. Contoh penentuan anggota himpunan T-string dapat dilihat pada Gambar 5.
Setelah himpunan T-string dan F terbentuk, anggota himpunan F dikoreksi satu demi satu dengan membandingkan setiap anggotanya dengan setiap anggota himpunan T-string. Setiap anggota F akan dihitung nilai kedekatannya dengan setiap anggota T-string. Nilai kedekatan dihitung dengan jarak Levensthein (Levensthein 1966). Penghitungan jarak Levensthein dilakukan dengan penjajaran global Needleman-Wunsch yang terdapat pada library SeqAn (Rmaeker 2012). Algoritme Needleman-Wunsch adalah algoritma yang digunakan untuk mencari penjajaran global yang memiliki nilai optimal dari dua buah sekuens (Utomo 2013). Setelah seluruh nilai jarak levenshtein seluruh anggota himpunan F terhadap seluruh anggota T-string diketahui, dipilih anggota T-string dengan nilai tertinggi untuk menggantikan anggota himpunan F yang mengandung error.
Hasil keluaran perangkat lunak ini adalah sebuah fail FASTA (.fna) berisi fragmen-fragmen DNA hasil koreksi yang akan digunakan pada tahap evaluaasi. Penjajaran global dengan algoritme Needleman-Wunsch
Algoritme Needleman-Wunsch menghitung semua informasi yang terdapat pada dua buah sequence sehingga jika kedua sequence berukuran n, maka kompleksitas waktunya adalah O(n2) (Utomo 2013). Scoring scheme atau nilai penskoran yang digunakan pada penelitian ini adalah nilai penskoran jarak levenshtein, yakni 0 untuk match, -1 untuk mismatch dan gap (Rmaeker 2012). Algoritme ini dibagi menjadi 2 tahap, yaitu:
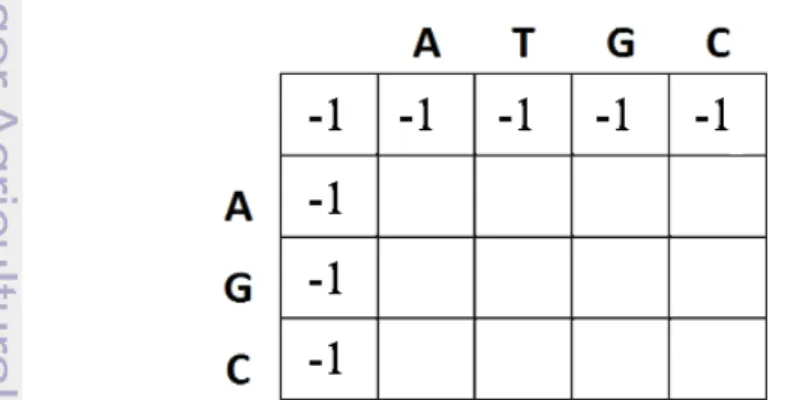
1. Inisialisasi
Pada tahap ini, dibentuk matriks dengan ukuran (m+1)×(n+1) dengan m dan n adalah panjang masing-masing kedua string. Selanjutnya kolom dan baris pertama diisi dengan nilai gap, yakni -1. Nilai gap adalah nilai yang diberikan ketika membandingkan karakter dengan karakter kosong. Contoh inisialisasi matriks dapat dilihat pada Gambar 6.
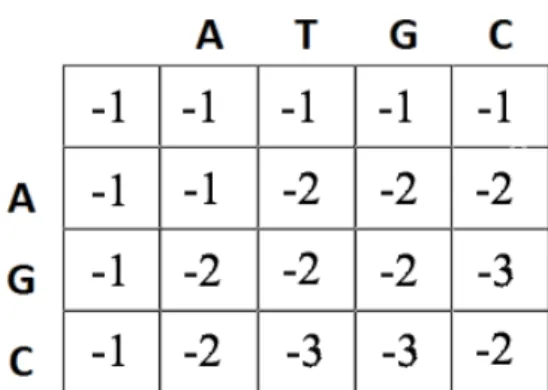
9 2. Pengisian Matriks
Pengisian matriks dilakukan dengan persamaan sebagai berikut:
M[i,j] = Max �
𝑀𝑀[𝑖𝑖 − 1, 𝑗𝑗] + 𝑤𝑤 𝑀𝑀[𝑖𝑖, 𝑗𝑗 − 1] + 𝑤𝑤 𝑀𝑀[𝑖𝑖 − 1, 𝑗𝑗 − 1] + 𝑆𝑆[𝑖𝑖, 𝑗𝑗]
dengan w adalah nilai gap dan S[i,j] adalah nilai match/mismatch. Contoh pengisian matriks dapat dilihat pada Gambar 7.
Gambar 7 Contoh matriks pemberian skor penilaian yang telah terisi
Pada matriks di atas, nilai akhir yang diperoleh adalah nilai yang berada pada posisi pojok kanan bawah, yaitu -2. Nilai akhir tersebut adalah jarak levenshtein antara string ATGC dan AGC. Dengan cara yang sama, jika string ATGC dan AGCC dibandingkan, nilai akhir yang diperoleh adalah -3. Sehingga dapat disimpulan string ATGC lebih mirip dengan AGC dibandingkan dengan AGCC.
Perbandingan hasil koreksi perangkat lunak
Jumlah anggota spectrum yang digunakan pada penelitian ini lebih sedikit dibandingkan yang digunakan oleh Caesar et al (2013), seperti yang ditunjukan pada Tabel 3. Karena jumlah anggota spectrum yang digunakan lebih sedikit, maka jumlah himpunan T-string yang dihasilkan menjadi lebih sedikit dan membuat jumlah reads error yang terdeteksi semakin banyak.
Tabel 3 Perbandingan hasil eksekusi perangkat lunak
Organisme Metode Jumlah reads Jumlah anggota spectrum Jumlah anggota T-string Jumlah reads yang terdeteksi sebagai error Acetobacter pasteurianus Penelitian ini 2000 768 563 1437 Caesar et al (2013) 2000 895 1606 394
10
Tabel 3 Perbandingan hasil eksekusi perangkat lunak (lanjutan)
Organisme Metode Jumlah reads Jumlah anggota spectrum Jumlah anggota T-string Jumlah reads yang terdeteksi sebagai error Lactobacillus plantarum Penelitian ini 2000 768 531 1469 Caesar et al (2013) 2000 909 1573 427 Staphylococcus aureus Penelitian ini 1500 768 709 791 Caesar et al (2013) 1500 811 1015 485
Waktu eksekusi yang dibutuhkan perangkat lunak penelitian ini lebih lama dibandingkan perangkat lunak Caesar et al (2013), seperti yang ditunjukan pada Tabel 4. Hal ini dikarenakan jumlah error yang harus dikoreksi semakin banyak, sehingga waktu eksekusi menjadi lebih lama.
Tabel 4 Perbandingan waktu eksekusi perangkat lunak
Organisme Metode Waktu eksekusi (ms)
Acetobacter pasteurianus Penelitian ini 524301 Caesar et al (2013) 461769
Lactobacillus plantarum Penelitian ini 512051
Caesar et al (2013) 483340
Staphylococcus aureus Penelitian ini 370102
Caesar et al (2013) 344651 Evaluasi hasil koreksi perangkat lunak
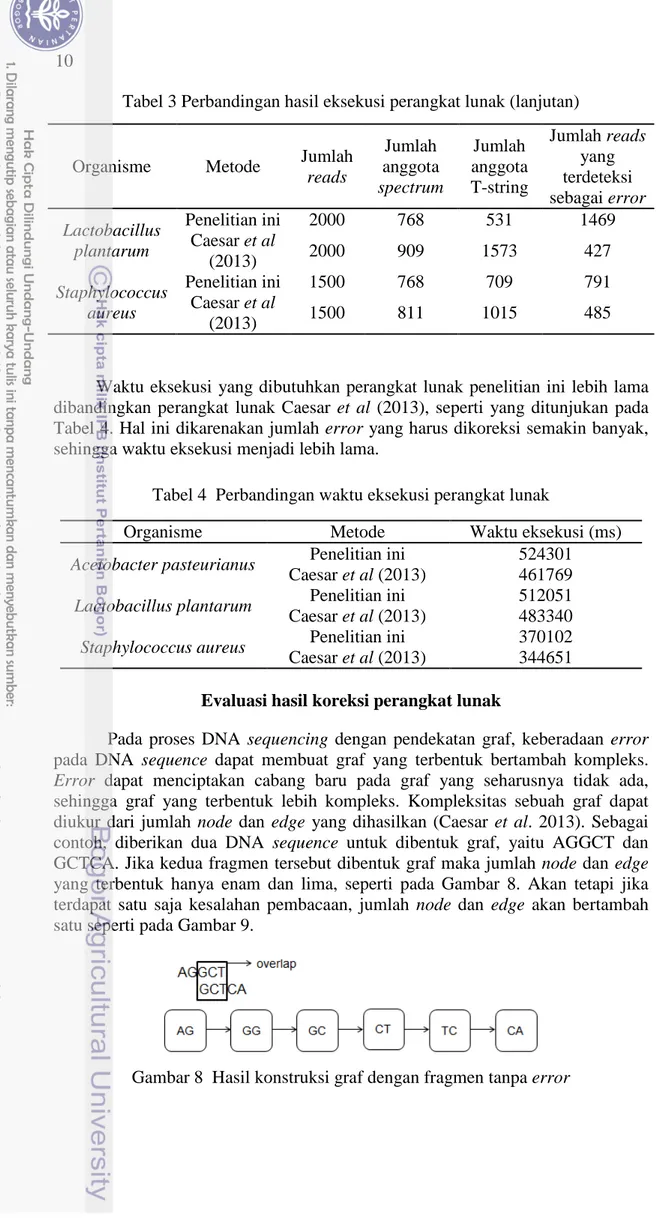
Pada proses DNA sequencing dengan pendekatan graf, keberadaan error pada DNA sequence dapat membuat graf yang terbentuk bertambah kompleks. Error dapat menciptakan cabang baru pada graf yang seharusnya tidak ada, sehingga graf yang terbentuk lebih kompleks. Kompleksitas sebuah graf dapat diukur dari jumlah node dan edge yang dihasilkan (Caesar et al. 2013). Sebagai contoh, diberikan dua DNA sequence untuk dibentuk graf, yaitu AGGCT dan GCTCA. Jika kedua fragmen tersebut dibentuk graf maka jumlah node dan edge yang terbentuk hanya enam dan lima, seperti pada Gambar 8. Akan tetapi jika terdapat satu saja kesalahan pembacaan, jumlah node dan edge akan bertambah satu seperti pada Gambar 9.
11
Gambar 9 Hasil konstruksi graf dengan fragmen mengandung error Evaluasi hasil koreksi dengan Velvet
Evaluasi dilakukan dengan menggunakan empat dataset. Setiap dataset terdiri atas tiga organisme, sehingga jumlah keseluruhan data yang digunakan ada dua belas. Empat dataset terdiri atas, data yang mengandung error, data yang tidak mengandung error, data yang mengandung error dan telah dikoreksi dengan perangkat lunak penelitian kali ini dan data yang mengandung error dan telah dikoreksi dengan perangkat lunak penelitian sebelumnya. Data tersebut dievaluasi dengan perangkat lunak DNA sequence assembler yaitu Velvet. Keluaran dari Velvet adalah graf De Bruijn yang merupakan hasil assembly dari data masukan.
Hasil keluaran Velvet disimpan ke dalam dua fail, yaitu “PreGraph” dan “LastGraph”. Fail “PreGraph” berisi daftar node dari graf yang dihasilkan dari data masukan. Adapun fail “LastGraph” berisi daftar node dari graf yang dihasilkan dari data masukan dan dilakukan koreksi data oleh Velvet itu sendiri. Sehingga fail yang perlu diperhatikan adalah “PreGraph”.
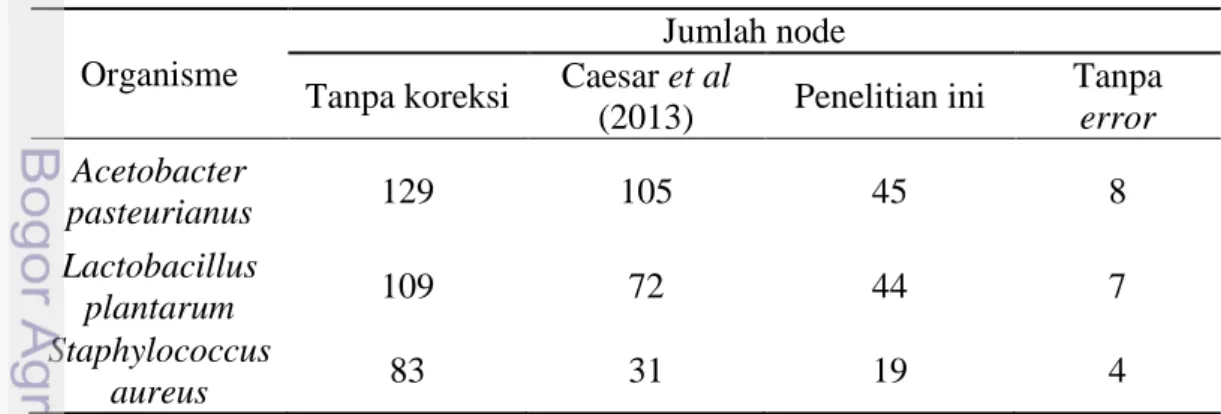
Pada setiap menjalankan Velvet, diperlukan masukan berupa nilai hash length atau k. Parameter hash length atau k adalah panjang k-mers yang dimasukkan ke dalam hash table. Nilai k harus ganjil, lebih kecil dari maxkmerhash yaitu 31 bp, dan lebih kecil dari panjang per fragmen data. Nilai k yang digunakan pada penelitian kali ini adalah 17. Hasil proses DNA sequence assembly menggunakan Velvet dapat dilihat pada Tabel 5.
Tabel 5 Hasil DNA sequence assembly dengan Velvet
Organisme
Jumlah node Tanpa koreksi Caesar et al
(2013) Penelitian ini Tanpa error Acetobacter pasteurianus 129 105 45 8 Lactobacillus plantarum 109 72 44 7 Staphylococcus aureus 83 31 19 4
Pada Tabel 5 di atas, dataset dengan label “Tanpa koreksi” berisi data sequence dengan error, dataset dengan label “Caesar et al (2013)” berisi data sequence yang dikoreksi dengan perangkat lunak dari penelitian sebelumnya, dataset “Hasil penelitian ini” berisi data sekuens yang dikoreksi dengan perangkat lunak penelitian kali ini dan dataset “Tanpa error” berisi data sekuens tanpa error.
12
Dataset yang dikoreksi dengan perangkat lunak penelitan kali ini menghasilkan graf yang lebih sederhana jika dibandingkan dengan penelitian sebelumnya, dilihat dari jumlah node yang lebih sedikit.
Hal ini dapat dicapai karena parameter penentuan solid tuple yang digunakan pada penelitian ini, lebih ketat dibandingkan dengan penelitian sebelumnya. Pada penelitian ini, jumlah solid tuple digunakan hanya 75% dari keseluruhan tuple. Adapun pada penelitian sebelumnya, jumlah solid tuple yang digunakan berkisar antara 82-91% dari keseluruhan tuple. Karena solid tuple yang digunakan lebih sedikit, maka jumlah anggota spectrum juga akan berkurang, yang akan berakibat berkurangnya jumlah anggota himpunan T-string. Jika jumlah anggota T-string lebih sedikit, maka jumlah fragmen yang dideteksi sebagai error akan semakin banyak, dan akhirnya hasil koreksi menjadi lebih maksimal.
SIMPULAN DAN SARAN
Simpulan
Penelitian ini telah mampu menghasilkan perangkat lunak yang dapat mendeteksi dan mengoreksi error yang ada pada DNA sequence. Kinerja keseluruhan yang dicapai lebih baik dibandingkan kinerja perangkat lunak penelitian sebelumnya (Caesar et al. 2013), terlihat dengan lebih sederhananya graf yang dihasilkan dan lebih banyaknya jumlah error yang terdeteksi. Perangkat lunak ini dapat mereduksi rata-rata 45% dari jumlah node dan mampu mengoreksi error 1.6 - 3.6 kali lebih banyak dibanding perangkat lunak yang dihasilkan Caesar et al (2013). Namun demikian, waktu eksekusi yang diperlukan oleh perangkat lunak ini lebih lama jika dibandingkan perangkat lunak yang dihasilkan oleh Caesar et al (2013).
Saran
Untuk penelitian selanjutnya, pengoreksian error dapat dilakukan dengan mengganti setiap substring dari reads masukan dengan anggota spectrum yang paling mirip. Penggantian nilai presentase untuk penentuan solid tuple dapat dicoba untuk mendapatkan hasil koreksi yang lebih baik. Hamming distance dapat digunakan sebagai pengukur kemiripan string. GPU paralel processing juga dapat diterapkan untuk mempersingkat waktu eksekusi yang dibutuhkan.
DAFTAR PUSTAKA
Caesar N, Kusuma WA, Wijaya SH. 2013. DNA sequencing error correction using spectral alignment Di dalam: International Conference on Advanced Computer Science and Information Systems (ICACSIS). hlm 279-284.
Döring A, Weese D, Rausch T, Reinert K. 2008. SeqAn an efficient, generic C++ library for sequence analysis. BMC Bioinformatics 9(1):11.
13 Levenshtein VI. 1966. Binary codes capable of correcting deletions, insertions and
reversals. Soviet Physics Doklady 10:707.
Pevzner PA, Tang H, Waterman MS. 2001. An Euler path approach to DNA fragment assembly. Proc. Natl. Acad. Sci. 98:9478-9753.
Richter DC, Ott F, Auch AF, Schmid R, Huson DH. 2008. MetaSim A sequencing simulator for genomics and metagenomics. PLoS ONE 3(10).
Rmaeker. 2012. Pairwise Sequence Alignment [Internet]. [diunduh 2014 Jun 10]; Tersedia pada: http://trac.seqan.de/wiki/Tutorial/PairwiseSequenceAlignment. Shi H, Schmidt B, Liu W, Wittig WM. 2009. Accelerating error correction in
high-throughput short-read DNA sequencing data with CUDA Di dalam: IEEE International Symposium on Parallel and Distributed Processing. hlm 1-8. Utomo AW. 2013. Penjajaran global sekuen DNA menggunakan algoritme
Needleman-Wunsch [skripsi]. Bogor (ID): Institut Pertanian Bogor.
Zerbino DR, Birney W. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18(5):821-829.
14
RIWAYAT HIDUP
Penulis lahir di Jakarta pada tanggal 17 Desember 1992. Penulis merupakan sulung dari 2 bersaudara dari pasangan Bapak H.Ir.Amul Yanuar dan Ibu Ratna Heni. Penulis menghabiskan masa pendidikan dasar, menengah dan atas di Kota Bekasi. Tahun 2009, penulis lulus dari SMA Negeri 2 Bekasi. Di tahun yang sama pula penulis diterima sebagai mahasiswa Institut Pertanian bogor di departemen Ilmu Komputer lewat jalur SNMPTN.
Selama menjalani kuliah di IPB penulis berpartisipasi dalam kepanitiaan Programming Competition Pesta Sains Nasional 2012. Penulis juga sempat melaksanakan praktek kerja lapang selama 2 bulan di Pusat Data Informasi dan Standardisasi Badan Pengkajian dan Penerapan Teknologi, Serpong pada tahun 2013.