SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S.Farm)
Program Studi Ilmu Farmasi
Oleh:
Maria Yolanda
NIM : 068114001
FAKULTAS FARMASI UNIVERSITAS SANATA DHARMA
i
SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S.Farm)
Program Studi Ilmu Farmasi
Oleh:
Maria Yolanda
NIM : 068114001
FAKULTAS FARMASI UNIVERSITAS SANATA DHARMA
ii
SECARA KROMATOGRAFI CAIR KINERJA TINGGI FASE TERBALIK
Yang diajukan oleh:
Maria Yolanda
NIM : 068114001
telah disetujui oleh:
Pembimbing
iii
SECARA KROMATOGRAFI CAIR KINERJA TINGGI FASE TERBALIK
Oleh: Maria Yolanda NIM : 068114001
Dipertahankan di hadapan Panitia Penguji Skripsi Fakultas Farmasi
Universitas Sanata Dharma pada tanggal : 29 Mei 2010
Mengetahui Fakultas Farmasi Universitas Sanata Dharma
Dekan
Rita Suhadi, M.Si., Apt.
Pembimbing
Prof. Dr. Sudibyo Martono, M.S., Apt.
Panitia Penguji :
1. Prof. Dr. Sudibyo Martono, M.S., Apt. ……….
2. Christine Patramurti, M.Si., Apt. ……….
Lewat
at tebing dan jurang yang menak
wat jalan yang berputar-putar yang
arah tujuannya
an yang membuatmu kehilangan y
sayangi
Itu bukan karena Dia tak pedul
mendidik dan membentukmu jadi
ang membawa berkat bagi sesam
membuat segala sesuatu in
waktunya,
Let go and Let God
Kupersembahkan ka
Bapak dan ibuku yan
Hugo dan Ndayu
Para sahabat dan almamat
v
Yang bertanda tangan di bawah ini, saya mahasiswa Universitas Sanata Dharma:
Nama : Maria Yolanda
Nomor Mahasiswa : 068114001
Demi pengembangan ilmu pengetahuan, saya memberikan kepada Perpustakaan Universitas Sanata Dharma karya ilmiah saya yang berjudul:
OPTIMASI DAN VALIDASI METODE PENETAPAN KADAR ASPARTAM DALAM MINUMAN SERBUK BERAROMA SECARA KROMATOGRAFI CAIR KINERJA TINGGI FASE TERBALIK
Beserta perangkat yang diperlukan (bila ada). Dengan demikian saya memberikan kepada Perpustakaan Universitas Sanata Dharma hak untuk menyimpan, mengalihkan dalam bentuk media lain, mengelolanya dalam bentuk pangkalan data, mendistribusikan secara terbatas, dan mempublikasikannya di Internet atau media lain untuk kepentingan akademis tanpa perlu meminta ijin dari saya maupun memberikan royalti kepada saya selama tetap mencantumkan nama saya sebagai penulis.
Demikian pernyataan ini yang saya buat dengan sebenarnya.
Dibuat di Yogyakarta
Pada tanggal: 15 April 2010
Yang menyatakan
vi
Puji dan syukur penulis panjatkan kepada Allah Bapa Yang Maha Kuasa atas
segala limpahan berkat dan kasih-Nya sehingga penelitian dan penyusunan skripsi
yang berjudul “Optimasi Dan Validasi Metode Penetapan Kadar Aspartam Dalam
Minuman Serbuk Beraroma Secara Kromatografi Cair Kinerja Tinggi Fase Terbalik”
dapat diselesaikan dengan baik. Skripsi ini disusun sebagai salah satu syarat untuk
meraih gelar Sarjana Farmasi (S.Farm) di Fakultas Farmasi, Universitas Sanata
Dharma, Yogyakarta.
Dalam pelaksanaan penelitian hingga selesainya penyusunan skripsi ini,
penulis mendapat banyak dukungan dan bantuan dari berbagai pihak. Oleh karena itu,
penulis mengucapkan terima kasih kepada:
1. Rita Suhadi, M.Si., Apt. selaku Dekan Fakultas Farmasi Universitas Sanata
Dharma Yogyakarta.
2. Prof. Dr. Sudibyo Martono, M.S., Apt. selaku dosen pembimbing yang
dengan sabar memberikan pengarahan, masukan, kritik dan saran baik selama
penelitian maupun penyusunan skripsi ini.
3. Christine Patramurti, M.Si., Apt. selaku dosen pembimbing akademik dan
dosen penguji, atas bimbingan, saran, kritik, nasehat dan semangat yang telah
vii
Mas Bimo, Pak Parlan, Mas Kunto, Mas Otok, dan Pak Timbul yang telah
banyak membantu selama penelitian di laboratorium.
6. Ibu Ani Fatima atas bantuan dalam pengadaan baku aspartam sehingga
penelitian dapat berjalan lancar.
7. Keluarga keduaku Kak Ivonne, Kak Nora, Tere, Cici, Esti. Terima kasih
sudah menemani dan mendukung di masa-masa penuh perjuangan ini. Kalian
begitu berarti.
8. Adhitya Eka Prasetya, teman seperjuangan dan tempat berbagi keluh kesah
selama penelitian dan penyusunan skripsi. Terima kasih partner.
9. Gregorius Adhi dan Yudhi Pradana atas suka, duka, dan semangat yang kita
bagi bersama.
10. Sahabat mungilku: Uut, Dewi, Sinta, atas kebersamaan, dukungan dan
persahabatan kita.
11. Robertus V. Mahartantyo, atas kasih, senyum, air mata dan pengalaman
berharga yang membuatku belajar sangat banyak.
12. Keluarga besar Ceria: mami, Kak Ivonne, Mikha, Mery, Oktin, Okvi, Putri,
Tyas, Sherly, Tere, Esti, Cici, Lia, atas bantuan dan dukungan selama di
viii
14. Teman bermain dan belajar, Dani, Nika, Adit, Uut, Phita dan Aya. Mari kita
bermain (dan belajar).
15. Teman-teman FST angkatan 2006, atas tawa, canda, kebersamaan dan
kekompakan yang begitu indah dan tak terlupakan.One for the FST.
16. Keluarga besar Paduan Suara Mahasiswa Cantus Firmus. Suatu kebanggaan
bisa eksis bersama kalian. Nyayian kita akan selalu tersimpan di hati.
17. Semua pihak yang tidak dapat disebutkan satu per satu yang telah membantu
penulis dalam mewujudkan skripsi ini. Tidak tertulis di sini bukan berarti
tidak tertulis di hati.
Penulis menyadari bahwa masih banyak kekurangan dalam penyusunan skripsi
ini, sehingga segala kritik dan saran yang membangun sangat penulis harapkan.
Semoga skripsi ini membantu dan bermanfaat bagi pembaca pada khususnya dan
ilmu pengetahuan pada umumnya.
Yogyakarta, 15 April 2010
ix
tidak memuat karya atau bagian karya orang lain, kecuali yang telah disebutkan
dalam kutipan dan daftar pustaka sebagaimana layaknya karya ilmiah.
Yogyakarta, 15 April 2010
Penulis,
x
penggunaan aspartam dapat memicu timbulnya berbagai macam penyakit sehingga penggunaannya pada berbagai jenis makanan dan minuman perlu dimonitor. Penelitian ini bertujuan untuk mengetahui validitas metode Kromatografi Cair Kinerja Tinggi (KCKT) fase terbalik yang digunakan dalam penetapan kadar aspartam.
Penelitian ini bersifat eksperimental deskriptif, menggunakan metode KCKT fase terbalik dengan kolom Kromasil-100 C18 250 x 4,6 mm, 5μm, fase gerak bufer fosfat pH 4 : asetonitril (80 : 20),flow rate1,4 mL/menit, dan detektor UV 214 nm.
Parameter validasi yang diteliti meliputi presisi, akurasi, linearitas, Limit of Detection (LOD) dan Limit of Quantitation (LOQ). Hasil penelitian menunjukkan metode memiliki linearitas yang baik pada konsentrasi 2,0 – 6,0 mg/100 mL (r = 0,999). Nilai recovery dan CV berturut-turut untuk level kadar rendah, sedang dan tinggi adalah 99,0% dan 0,46%; 101,1% dan 1,13%; 100,1% dan 1,12%. Nilai LOD untuk aspartam adalah 0,11 mg/100 mL sedangkan nilai LOQ 0,38 mg/100 mL. Berdasarkan hasil tersebut maka metode ini valid untuk penetapan kadar aspartam.
xi
instead of sugar. Several researches conclude that the usage of aspartame can touch off the emergence of some diseases and that is why its users need monitoring. This research is aimed to discover validity of High Performance Liquid Chromatography (HPLC) method of reversed phase which is employed in aspartame determination.
This is a non experimental descriptive research using HPLC method with reversed phase by using Kromasil column-100 C18 250x4.6 mm, 5 μm, phosphate buffer mobile phase pH 4 : acetonitrile (80 : 20), flow rate 1.4 mL/minute, and detector UV 214 nm.
Validity parameter is observed included precision, accuracy, linearity,Limit of Detection(LOD), andLimit of Quantitation(LOQ). Result of the research shows that the method holds a decent linearity on concentration 2.0 – 6.0 mg/100 mL (r = 0.999). Recovery point and successive CV to low level, medium, and high are 99.0% and 0.46%; 101.1% and 1.13%; 100.1% and 1.12%. LOD point to aspartame is 0.11 mg/100 mL while LOQ is 0.38 mg/100mL. Based on the result, it can be summed up that this particular method is valid to aspartame determination.
xii
HALAMAN JUDUL ...i
HALAMAN PERSETUJUAN PEMBIMBING ... ii
HALAMAN PENGESAHAN SKRIPSI...iii
HALAMAN PERSEMBAHAN ...iv
HALAMAN PERSETUJUAN PUBLIKASI... v
KATA PENGANTAR ...vi
PERNYATAAN KEASLIAN KARYA ...ix
INTISARI ... x
ABSTRACT...xi
DAFTAR ISI... xii
DAFTAR TABEL...xvi
DAFTAR GAMBAR ... xvii
DAFTAR LAMPIRAN...xix
BAB I PENGANTAR ... 1
A. Latar Belakang ... 1
1. Permasalahan... 3
2. Keaslian Penelitian... 4
3. Manfaat Penelitian ... 4
xiii
C. Aspartam ... 7
1. Deskripsi Aspartam ... 7
2. Penggunaan Aspartam ... 8
3. Kajian Keamanan ... 9
4. Metode Analisis Aspartam ... 10
D. Kromatografi Cair Kinerja Tinggi... 11
1. Kromatografi Partisi Fase Terbalik... 12
2. Fase Diam ... 13
3. Fase Gerak ... 13
4. Kolom dan Kinerjanya... 15
5. Pengekoran Puncak Kromatogram ... 19
6. Analisis Kualitatif dan Kuantitatif ... 21
E. Parameter Validasi Metode Analisis ... 22
1. Akurasi... 22
2. Presisi... 23
3. Selektivitas... 24
4. Linearitas dan Rentang ... 24
5.Limit Of Detection (LOD) dan Limit Of Quantitation (LOQ)... 24
xiv
B. Variabel Penelitian ... 27
1. Variabel bebas ... 27
2. Variabel tergantung ... 27
3. Variabel terkendali ... 27
C. Bahan-bahan Penelitian ... 28
D. Alat-alat Penelitian ... 28
E. Tata Cara Penelitian... 29
1. Optimasi Metode KCKT ... 29
2. Verifikasi Sistem ... 31
3. Validasi Metode... 32
F. Analisis Hasil ... 33
BAB IV. HASIL DAN PEMBAHASAN ... 36
A. Optimasi Metode ... 36
1. Penentuan Panjang Gelombang Pengamatan ... 36
2. Optimasi Fase Gerak ... 37
3. Pengamatan Waktu Retensi Aspartam ... 46
B. Verifikasi Sistem KCKT ... 50
1. Verifikasi akurasi pompa... 50
xv
2. Linearitas ... 55
3. Akurasi... 56
4. Presisi... 57
5.LODdanLOQ ...57
BAB V. KESIMPULAN DAN SARAN... 60
A. Kesimpulan ... 60
B. Saran ... 61
DAFTAR PUSTAKA ... 62
xvi
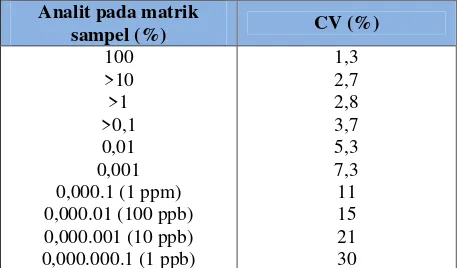
eluen strength
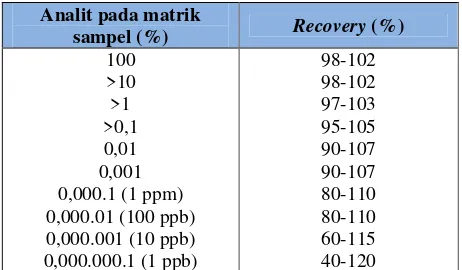
Tabel II. Kriteria %recoveryyang diijinkan ... 22
Tabel III. Kriteria presisi yang diijinkan untuk konsentrasi analit yang berbeda ... 23
Tabel IV. Hasil optimasiflow ratefase gerak bufer fosfat pH 4 : asetonitril (80 : 20) ... 44
Tabel V. Data penyimpanganflow ratepada uji akurasi pompa... 51
Tabel VI. Hasil uji presisi sistem injeksi ... 52
Tabel VII. Data kurva baku aspartam... 53
Tabel VIII. Hasil penetapanrecoveryaspartam ... 56
xvii
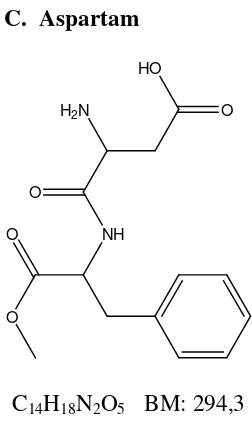
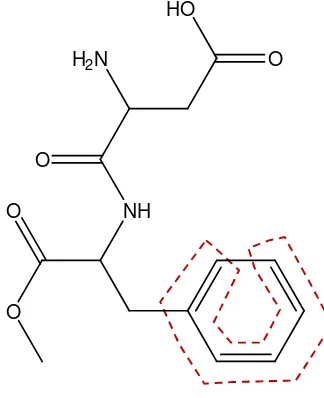
Gambar 1. Struktur kimia aspartam ... 7
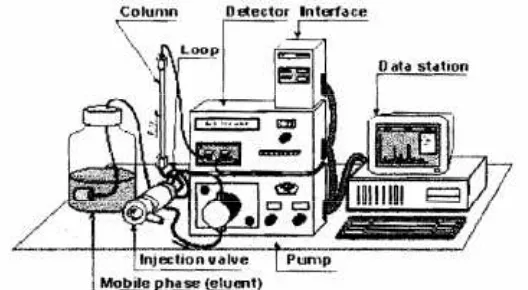
Gambar 2. Peralatan KCKT ... 12
Gambar 3. Difusi Eddy ... 17
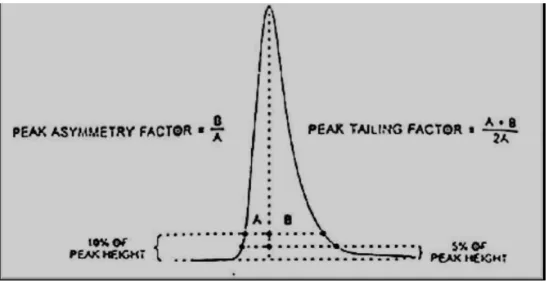
Gambar 4. PenentuanPeak AsymmetrydanPeak Tailing Factor... 20
Gambar 5. Distribusi dalam fase diam dan fase gerak... 21
Gambar 6. Gugus kromofor aspartam ... 36
Gambar 7. Spektrum panjang gelombang aspartam ... 37
Gambar 8. Reaksi ionisasi aspartam ... 39
Gambar 9. Degradasi aspartam ... 40
Gambar 10. Kromatogram optimasi fase gerak bufer fosfat pH 4 : asetonitril (95 : 5) ... 41
Gambar 11. Kromatogram optimasi fase gerak bufer fosfat pH 4 : asetonitril (90 : 10) ... 41
Gambar 12. Kromatogram optimasi fase gerak bufer fosfat pH 4 : asetonitril (85 : 15) ... 42
Gambar 13. Kromatogram optimasi fase gerak bufer fosfat pH 4 : asetonitril (80 : 20) ... 42
xviii
Gambar 17. Kromatogram waktu retensi sampel... 47
Gambar 18. Kromatogram waktu retensi aspartam + baku aspartam ... 48
Gambar 19. Ikatan aspartam dengan fase gerak... 49
Gambar 20. Ikatan aspartam dengan fase diam ... 50
xix
Lampiran 2. Contoh perhitungan N dan HETP ... 67
Lampiran 3. Kromatogram hasil optimasiflow ratefase gerak bufer fosfat pH 4 : asetonitril (80 : 20) ... 68
Lampiran 4. Tabel hasil penetapan presisi injeksi ... 73
Lampiran 5. Kromatogram penetapan presisi injeksi... 74
Lampiran 6. Kromatogram kurva baku aspartam replikasi I... 81
Lampiran 7. Kromatogram kurva baku aspartam replikasi II ... 86
Lampiran 8. Kromatogram kurva baku aspartam replikasi III ... 91
Lampiran 9. Penimbangan baku dan contoh perhitungan kadar baku ... 96
Lampiran 10. Kromatogram hasil validasi metode replikasi I ... 97
Lampiran 11. Kromatogram hasil validasi metode replikasi II... 100
Lampiran 12. Kromatogram hasil validasi metode replikasi III ... 103
Lampiran 13. Data penimbangan dan perhitunganrecovery... 106
1
A. Latar Belakang Penelitian
Zat-zat pemanis makanan dan minuman pengganti gula saat ini banyak
digunakan dalam aneka jenis makanan dan minuman. Zat tersebut ditambahkan untuk
memberikan persepsi rasa manis namun tidak memiliki nilai gizi.
Salah satu jenis makanan atau minuman yang menggunakan pemanis
pengganti gula adalah minuman serbuk beraroma. Minuman minuman serbuk
beraroma adalah minuman ringan yang dikonsumsi dengan cara melarutkan serbuk
tersebut kedalam air sesuai dengan saran penyajian yang biasanya dicantukan pada
kemasan. Untuk meningkatkan minat konsumen akan produknya, produsen biasanya
menambahkan bahan tambahan seperti pemanis buatan untuk meningkatkan cita rasa
pada produk minuman serbuk beraroma tersebut.
Aspartam adalah salah satu pemanis buatan yang paling laris digunakan saat
ini. Aspartam merupakan pemanis buatan dari golongan gula non-sakarida yang
banyak dipakai untuk produk-produk diet atau produk rendah kalori. Dibuat dengan
menggabungkan 2 buah asam amino, yaitu fenilalanin dan asam aspartat dengan
derajat kemanisan sekitar 160 sampai 200 kali gula pasir dan hampir tidak
mengandung kalori.
Beberapa penelitian menunjukkan bahwa penggunaan aspartam sebagai
seperti penyakit Alzheimer, epilepsi, tumor otak, stroke dan penyakit jantung.
Walaupun demikian, pemakaian aspartam sebagai pemanis buatan masih diijinkan
oleh FDA (Food and Drug Administration) namun dengan batas-batas pemakaian yang dianjurkan agar penggunaannya aman (Maisons and Alfort, 2002).
Di Indonesia, Berdasarkan Peraturan Kepala POM No. HK
00.05.5.1.4547/2004 pasal 6 ayat 3, produk pangan yang mengandung aspartam wajib
mencantumkan peringatan untuk pasien fenilketonuria yang tertulis jelas pada
kemasan produk. Berdasarkan Peraturan Kepala POM No. HK 00.05.5.1.4547/ 2004
batas maksimum penggunaan aspartam adalah 50 mg/kg berat badan per hari.
Penetapan kadar aspartam dan beberapa pemanis buatan lainnya dalam
minuman ringan pernah dilakukan Hayun, dkk. (2004) dengan metode Kromatografi
Cair Kinerja Tinggi dengan detektor UV. Fase diam yang digunakan adalah C-18
Latek (15 cm x 4,0 mm) dan fase gerak asetonitril : bufer asetat (5 : 95).
Perbedaan antara penelitian ini dengan penelitian Hayun dkk. terletak pada
komposisi fase gerak, fase diam dan sampel yang digunakan. Pada penelitian ini
digunakan fase diam C-18 merekKromasil (25 cm x 4,6 mm) dan fase gerak buffer fosfat : asetonitril (80 : 20), sedangkan sampel yang digunakan adalah minuman
serbuk beraroma. Dengan adanya perbedaan tersebut maka perlu dilakukan kembali
optimasi dan validasi untuk penetapan kadar aspartam dalam minuman serbuk
beraroma karena sistem KCKT dalam penelitian Hayun dkk. belum tentu
Aspartam bersifat polar dengan gugus yang mudah terionisasi mudah
terionisasi yaitu COOH dan NH2 karena adanya pasangan elektron bebas (Anonim,
2005). Oleh karena itu dipilih sistem KCKT fase terbalik dimana fase diamnya
bersifat non polar dan fase geraknya polar sehingga aspartam memiliki afinitas yang
lebih besar pada fase gerak dan pemisahan menjadi lebih efisien.
Metode Kromatografi Kinerja Tinggi Fase Terbalik adalah metode terpilih
yang digunakan dalam analisis aspartam dalam minuman serbuk beraroma.
Berdasarkan hasil penelitian ini diharapkan dapat diperoleh metode penetapan kadar
aspartam menggunakan metode KCKT yang memiliki akurasi, presisi, linearitas, dan
spesifisitas yang baik. Penelitian ini merupakan penelitian pendahulu dari penelitian
lain mengenai penetapan kadar aspartam dalam minuman serbuk beraroma secara
KCKT fase terbalik.
1. Permasalahan
Berdasarkan latar belakang dapat dirumuskan permasalahan sebagai berikut:
Apakah metode Kromatografi Cair Kinerja Tinggi fase terbalik dengan fase diam
C-18 dan fase gerak asetonitril : bufer fosfat pH 4 (20 : 80 v/v) yang digunakan dalam
penetapan kadar aspartam dalam minuman serbuk beraroma memiliki validitas yang
2. Keaslian Penelitian
Berdasarkan sumber informasi yang diperoleh penulis, validasi penetapan
kadar aspartam dalam minuman serbuk beraroma secara KCKT fase terbalik belum
pernah dilakukan. Namun penelitian mengenai kadar aspartam dan beberapa jenis
pemanis buatan lainnya pernah dilakukan oleh Hayun, dkk. (2004) pada sampel
minuman ringan bersoda secara KCKT menggunakan kolom C-18 Latex (15 cm x 4,0
mm) dan fase gerak campuran asetonitril : buffer asetat pH 5 (5 : 95) dengan detektor
UV pada panjang gelombang 254 nm.
3. Manfaat Penelitian
Manfaat teroritis penelitian ini adalah memberikan informasi tentang validasi
metode Kromatografi Cair Kinerja Tinggi untuk penetapan kadar aspartam.
Manfaat metodologis penelitian ini adalah memberikan informasi tentang
pengembangan metode analisis untuk penetapan kadar aspartam agar nantinya dapat
digunakan sebagai metode analisis alternatif untuk penetapan kadar aspartam.
B. Tujuan Penelitian
Untuk mengetahui apakah metode Kromatografi Cair Kinerja Tinggi fase
terbalik dengan fase diam C-18 dan fase gerak asetonitril : bufer fosfat pH 4 (20 : 80
v/v) yang digunakan pada penetapan kadar aspartam dalam minuman serbuk
5
A. Bahan Tambahan Makanan
Berdasarkan Peraturan Menteri Kesehatan RI No. 722/Menkes/Per/IX/1988
Bahan Tambahan Makanan (BTM) adalah bahan yang ditambahkan ke dalam
makanan untuk mempengaruhi sifat atau bentuk pangan baik yang mempunyai atau
tidak mempunyai nilai gizi, antara lain pemanis buatan, bahan pengawet, pewarna,
penyedap rasa dan aroma, anti gumpal dan pengental. Ketentuan lain yang mengatur
penggunaan bahan tambahan makanan adalah larangan bagi setiap orang yang
memproduksi pangan untuk diedarkan menggunakan bahan tambahan makanan yang
dinyatakan terlarang atau melampaui ambang batas maksimal yang telah ditetapkan.
Penambahan BTM secara umum bertujuan untuk meningkatkan nilai gizi
makanan, memperbaiki nilai sensori makanan, dan memperpanjang umur simpan
makanan. Selain tujuan-tujuan di atas, BTM sering digunakan untuk meproduksi
makanan untuk kelompok khusus seperti penderita diabetes, pasien yang baru
mengalami operasi, orang-orang yang menjalankan diet rendah kalori atau rendah
lemak, dan sebagainya. Berbagai BTM yang digunakan untuk maksud tersebut di
B. Pemanis Buatan
Pemanis merupakan senyawa kimia yang sering ditambahkan dan digunakan
untuk keperluan produk olahan pangan, industri, dan minuman serta makanan
kesehatan. Pemanis berfungsi untuk meningkatkan cita rasa dan aroma, memperbaiki
sifat fisik, sebagai pengawet, memperbaiki sifat-sifat kimia, mengembangkan jenis
minuman dan makanan dengan jumlah kalori terkontrol, dan sebagai bahan substitusi
pemanis utama (Sakidja, 1989).
Berdasarkan Keputusan Kepala Badan Pengawas Obat Dan Makanan
Republik Indonesia Nomor : HK.00.05.5.1.4547, pemanis buatan adalah bahan
tambahan pangan yang dapat menyebabkan rasa manis pada produk pangan yang
tidak atau sedikit mempunyai nilai gizi atau kalori, hanya boleh ditambahkan ke
dalam produk pangan dalam jumlah tertentu.
Pemanis buatan adalah bahan tambahan yang dapat menyebabkan rasa manis
pada pangan, tetapi tidak memiliki nilai gizi. Beberapa pemanis buatan yang telah
dikenal dan banyak digunakan adalah sakarin, siklamat, aspartam, dulsin, sorbitol
sintetis, nitro-propoksi-anilin (Sakidja, 1989).
Pemanis buatan diperoleh secara sintetis melalui reaksi-reaksi kimia di
laboratorium ataupun skala industri. Karena diperoleh melalui proses sintetis dapat
dipastikan bahan tersebut mengandung senyawa-senyawa sintetis. Penggunaan
pemanis buatan perlu diwaspadai karena dalam takaran yang berlebih dapat
menimbulkan efek samping yang merugikan kesehatan manusia. Hasil penelitian
dan bersifat karsinogenik. Oleh karena itu Organisasi Kesehatan Dunia (World Health Organization/WHO) telah menetapkan batas-batas yang disebut Acceptable Daily Intake(ADI) atau kebutuhan per orang per hari, yaitu jumlah yang dapat dikonsumsi tanpa menimbulkan risiko. Sejalan dengan itu, di negara-negara Eropa, Amerika dan
juga di Indonesia telah ditetapkan standar penggunaan pemanis buatan pada produk
makanan (Ambarsari dkk., 2009).
Menurut peraturan Menteri Kesehatan RI No. 722/Menkes/Per/IX/88, produk
pangan yang menggunakan pemanis buatan harus mencantumkan jenis dan jumlah
pemanis buatan dalam komposisi bahan atau daftar bahan pada tabel.
C. Aspartam
Gambar 1. Struktur kimia aspartam
1. Deskripsi Aspartam
Aspartam atau Aspartil fenilalanin metil ester (APM) dengan rumus kimia
-aspartyl-L-phenylalanine-1-methyl ester dengan 220 kali tingkat kemanisan sukrosa dengan nilai kalori sebesar 0,4 kkal/ merupakan senyawa yang tidak berbau,
berbentuk tepung kristal berwarna putih, sedikit larut dalam air, dan berasa manis.
Aspartam memiliki tingkat kemanisan relatif sebesar 60 sampai g atau setara dengan
1,67 kJ/g (Gelardi, 1986).
Aspartam mengandung tak kurang dari 98,0% dan tidak lebih dari 102,0%
C14H18N2O5.Kelarutan aspartam dalam air tergantung pada pH dan suhu. Kelarutan
maksimum tercapai pada pH 2,2 (20 mg/mL pada 250C) (Anonim, 1995).
2. Penggunaan Aspartam
Mengacu pada asam amino pembentuk aspartam, maka aspartam bukanlah
termasuk suatu bahan pemanis nonkalori karena seperti protein, aspartam
dimetabolisme menjadi asam amino penyusunnya dan memiliki nilai energi 4 kkal/g.
Tetapi karena dalam penggunaannya, 100 gram sukrosa dapat diganti dengan 1 gram
aspartam maka dapat dikatakan bahwa aspartam merupakan bahan pemanis nonkalori
(Sakidja, 1989).
Pemanis buatan aspartam merupakan pemanis buatan yang diijinkan
digunakan dalam pembuatan produk makanan sesuai Peraturan Menteri Kesehatan
N0.722 tahun 1988. Aspartam banyak digunakan oleh orang yang membatasi jumlah
kalori misalnya penderita diabetes, orang yang obesitas atau yang menjaga berat
badan. Aspartam juga berfungsi sebagai penegas rasa terutama cita rasa buah
Berdasarkan Keputusan Kepala Badan Pengawas Obat Dan Makanan
Republik Indonesia Nomor : HK.00.05.5.1.4547 aspartam tidak diizinkan
penggunaannya pada produk pangan olahan tertentu untuk dikonsumsi oleh
kelompok tertentu meliputi bayi, balita, ibu hamil, ibu menyusui dalam upaya
memelihara dan meningkatkan kualitas kesehatannya.
3. Kajian Keamanan
Aspartam tersusun atas asam amino sehingga dalam tubuh akan mengalami
metabolisme seperti halnya asam amino pada umumnya. Aspartam terurai menjadi
asam amino L-asam aspartat dan L-fenilalanin yang merupakan asam amino
penyusun protein dalam makanan sehari-hari sehingga tidak akan menimbulkan efek
berbahaya (Sakidja, 1989).
Aspartam merupakan salah satu bahan tambahan makanan yang telah
mengalami uji dan percobaan yang mendalam serta menyeluruh dan telah disetujui
oleh US-FDA. Pada tahun 1974, US-FDA telah mengabulkan usulan mengenai
penggunaan aspartam sebagai pemanis pada makanan kering. Tetapi karena
banyaknya laporan mengenai keamanan aspartam bagi kesehatan, pemasaran
aspartam baru diizinkan pada tahun 1981 (Sakidja, 1989). Selain itu tahun 2006 AFC
Panel dari Badan Keamanan Pangan Eropa (EFSA) mengevaluasi carcinogenicity
aspartam dan berkesimpulan bahwa dari segi keamanan tidak ada alasan untuk
Penggunaan aspartam bagi orang yang menderita penyakit turunan yang
dikenal sebagai fenilketonuria perlu mendapat perhatian khusus. Orang yang
menderita penyakit tersebut tidak mampu memetabolisme fenilalanin. Berlebihnya
jumlah fenilalanin dalam tubuh penderita dapat menyebabkan kerusakan otak yang
dapat mengakibatkan cacat mental, karena adanya penumpukan fenilalanin di otak
(Sakidja, 1989).
Di Indonesia, Berdasarkan Peraturan Kepala POM No. HK
00.05.5.1.4547/2004 pasal 6 ayat 3, produk pangan yang mengandung aspartam wajib
mencantumkan peringatan yang tertulis jelas pada kemasan produk bagi pasien
fenilketonuria. Berdasarkan Peraturan Kepala POM No. HK 00.05.5.1.4547/ 2004
batas maksimum penggunaan aspartam adalah 50 mg/kg berat badan per hari.
4. Metode Analisis Aspartam
Aspartam dapat ditentukan secara kualitatif dengan kromatografi lapis tipis
(KLT) didasarkan pada pemisahan dengan KLT karena perbedaan afinitas aspartam
terhadap fase diam dan fase geraknya. Fase diam untuk penetapan kadar aspartam ini
adalah silika gel 60 GF 254, sedangkan fase geraknya adalah sistem pengembang
n-butanol : asam asetat glasial : air (2 : 1 : 1). Untuk menampakkan noda digunakan
larutan ninhidrin 0,2% dalam air dan larutan brom 1% dalam karbon tetra klorida.
Noda dilihat di bawah lampu UV pada panjang gelombang 254 nm (Sakidja, 1989).
Penentuan aspartam secara kuantitatif dapat dilakukan secara titrasi
biru (Anonim, 2000). Penetapan kadar aspartam dalam minuman dan makanan yang
paling umum digunakan saat ini adalah dengan metode KCKT fase terbalik
menggunakan fase diam C18 dan dua jenis fase gerak yang dapat dipilih yaitu
metanol : bufer asetat atau asetonitril : bufer fosfat (Nollet, 2000). Penetapan kadar
aspartam dengan metode KCKT pernah dilakukan Hayun dkk. (2004) dengan sampel
minuman ringan bersoda menggunakan fase diam C-18 Latek (15 cm x 4,0 mm) dan
fase gerak asetonitril : buffer asetat pH 5 (5 : 95). Metode ini memiliki akurasi dan
presisi yang cukup baik namun waktu analisisnya sangat lama yaitu lebih dari 30
menit.
D. Kromatografi Cair Kinerja Tinggi
Kromatografi Cair Kinerja Tinggi adalah suatu sistem kromatografi yang fase
geraknya dialirkan dengan cepat dengan bantuan pompa dan hasilnya dideteksi
dengan detektor (Gritter et al., 1985). Tujuan analisis dengan KCKT yaitu
didapatkannya pemisahan yang baik dalam waktu yang relatif singkat (Mulja dan
Gambar 2. Peralatan KCKT
1. Kromatografi Partisi Fase Terbalik
Prinsip kromatografi partisi didasarkan pada partisi linarut antara dua pelarut
yang tidak bercampur yang ada pada fase diam dan fase gerak. Fase diam (polar atau
nonpolar) disalutkan pada penyangga dan dikemas ke dalam kolom. Jika linarut
ditambahkan ke dalam sistem yang terdiri atas dua pelarut yang tidak bercampur dan
keseluruhan sistem dibiarkan setimbang, linarut akan tersebar antara dua fase
menurut persamaan:
m s
C C K
Dengan K adalah koefisien distribusi dan Cs dan Cm adalah konsentrasi linarut
berturut-turut dalam fase diam dan fase gerak (Johnson dan Stevenson, 1991).
Pada kromatografi fase terbalik, fase diamnya bersifat non polar, biasanya
digunakan hidrokarbon dan fase geraknya relatif bersifat polar seperti air, metanol
Kolom yang biasa digunakan pada kromatografi partisi fase terbalik adalah
kolom dengan kemasan fase terikat, yang bersifat stabil karena fase diamnya terikat
secara kimia pada penyangga, sehingga tidak mudah terbawa oleh fase gerak.
Penyangga pada kemasan fase terikat biasanya terbuat dari silika yang sudah
diseragamkan, berpori dan umumnya partikel mempunyai diameter 3,5 atau 10 µm
(Skoog et al., 1994).
2. Fase Diam
KCKT fase terbalik menggunakan fase diam yang berupa senyawa organik,
dimana senyawa organik ini terikat secara kimia dengan gugus silanol pada
permukaan silika. Hal ini yang menyebabkan permukaan silika menjadi bersifat non
polar. Dalam kromatografi jenis ini, senyawa lebih polar terelusi lebih dahulu,
kemudian diikuti senyawa non polar (Munson, 1991).
Fase diam yang biasa digunakan pada kromatografi partisi fase terbalik adalah
oktadesilsilan (ODS). Selain ODS, dikenal pula silika dengan substitusi oktil (C8).
Panjang pendeknya rantai karbon mempengaruhi tertambatnya senyawa pada fase
diam. Kolom dengan rantai panjang bersifat retensif, sehingga senyawa yang
mempunyai sifat mirip dengan kolom akan tertambat lebih lama (Munson, 1991).
3. Fase Gerak
Fase gerak merupakan salah satu faktor yang mempengaruhi pemisahan.
beberapa sifat yang perlu diperhatikan yaitu, fase gerak yang digunakan harus murni
tanpa cemaran, sesuai dengan detektor, dapat melarutkan cuplikan, memiliki
viskositas yang rendah dan memungkinkan memperoleh kembali cuplikan dengan
mudah, jika diperlukan (Johnson and Stevenson, 1991).
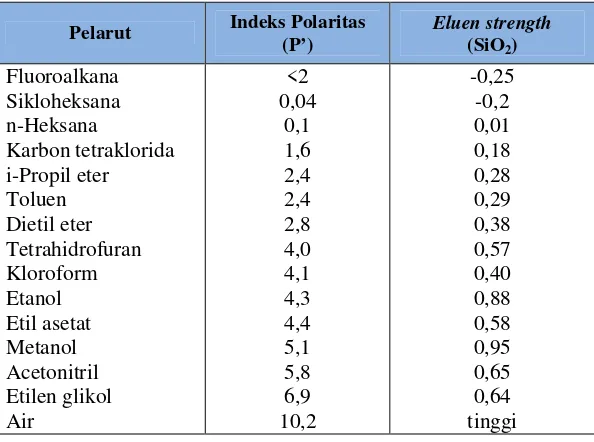
Kepolaran pelarut merupakan ukuran kekuatan pelarut atau kemampuan
pelarut untuk mengelusi suatu senyawa. Kandungan utama fase gerak pada
kromatografi fase terbalik adalah air. Pelarut yang dapat campur dengan air seperti
metanol, etanol, asetonitril, dioksan, tetrahidrofuran dan dimetilformamida
ditambahkan untuk mengatur kepolaran fase gerak (Munson, 1991).
Polaritas fase gerak dalam KCKT sangat mempengaruhi kromatogram yang
dihasilkan, sehingga perlu diperhitungkan komposisi campuran pelarut yang akan
digunakan. Berdasarkan nilai P’(indeks polaritas), maka besarnya polaritas campuran
pelarut dapat dihitung dengan persamaan berikut :
Pcampuran=∑ '= 1P1’ + 2P2’ + ...+ nPn’
Nilai Pcampuran adalah polaritas campuran, P’ menyatakan indeks polaritas,
merupakan fraksi pelarut dalam campuran dan n adalah jenis pelarut yang digunakan
(Skoog, 1985).
Berikut ini adalah daftar pelarut berikut nilai indeks polaritas dan eluen
Tabel I. Nilai indeks polaritas daneluen strengthpelarut
Eluen strength merupakan ukuran kemampuan fase gerak menarik analit dari fase diam. Waktu retensi analit akan turun jika digunakan fase gerak dengan eluen strength rendah, sebaliknya waktu retensi analit akan meningkat jika digunakan fase gerak denganeluen strengthtinggi (Snyder et al.,1997).
4. Kolom dan Kinerjanya
Keberhasilan atau kegagalan analisis tergantung pada pemilihan kolom dan
kondisi kerja yang tepat. Ukuran kinerja kolom dapat dilihat dari kemampuan kolom
dalam memisahkan senyawa (Johnson dan Stevenson, 1978). Batasan yang paling
sering digunakan yaitu bilangan lempeng teoritik dan faktor resolusi (Munson, 1991).
N L HETP
H
a) Teori Lempeng (Plate Theory)
Salah satu ukuran kinerja kolom adalah jumlah lempeng teoritik yang dihitung
dengan persamaan:
= 16 = 5,54
Nilai w adalah lebar alas, w1/2adalah lebar alas puncak pada setengah tinggi puncak,
dan tRadalah waktu retensi (Sastrohamidjojo, 2001).
Jumlah pelat teori berbanding lurus dengan panjang kolom. Karena panjang
kolom bermacam-macam, maka diperlukan ukuran efisiensi kolom yang tidak
bergantung pada panjang kolom. Tinggi atau jarak yang setara dengan pelat, H atau
Height Equivalent to a Theoritical Plate(HETP), merupakan ukuran efisiensi kolom yang lebih disukai karena memungkinkan perbandingan antara kolom yang
panjangnya berlainan. H berkaitan dengan jumlah pelat teori menurut persamaan
berikut:
L menunjukkan panjang kolom biasanya dalam mm, dan N menunjukkan jumlah
pelat teoritis (Johnson dan Stevenson, 1978).
b) Teori Laju (Rate Theory)
Pada waktu migrasi, analit mengalami transfer antara fase diam dan fase gerak
berkali-kali. Waktu tinggal pada fase diam maupun fase gerak tidak teratur dan
tergantung pada tersedianya energy termal dari lingkungannya yang memungkinkan
migrasi di dalam kolom tidak teratur. Hal ini mengakibatkan laju rata-rata analit
relatif terhadap fase gerak bervariasi sehingga terjadi pelebaran puncak analit.
Faktor-faktor utama penyebab terjadinya pelebaran puncak yaitu:
1) Difusi Eddy merupakan aliran tidak teratur yang menyebabkan
terjadinya pencampuran konvektif. Difusi Eddy disebabkan oleh banyak
kemungkinan pada kemasan kolom yang kurang baik.
Gambar 3. Difusi Eddy
Gambar 3 menunjukkan proses terjadinya difusi Eddy. Nomor 1 menunjukkan
analit yang keluar lebih dahulu karena melewati kolom dengan partikel
berukuran besar dan kurang kompak. Nomor 2 menunjukkan analit keluar
lebih lambat dari nomor 1 karena ukuran partikel yang lebih kecil dan lebih
kompak daripada nomor 1. Nomor 3, analit keluar paling akhir, hal ini terjadi
karena melewati bagian kolom dengan ukuran partikel halus dan kompak.
2) Difusi Longitudinal merupakan efek dari gerakan random molekul
analit dalam fase gerak karena adanya perbedaan konsentrasi.
3) Transfer massa non-ekuillibrium merupakan efek laju ekuilibrasi
pada umumnya aliran fase gerak terlalu cepat untuk mendapatkan ekuilibrium
antara kedua fase (Noegrohati, 1994).
Hubungan antara ketiga faktor dapat digambarkan dengan persamaan Van Deemter:
= + + . + .
A = difusi Eddy
B = difusi longitudinal
C = transfer massa non-ekuilibrium
= rata-rataflow ratelinear fase gerak
H = HETP (Noegrohati,1994)
Faktor resolusi adalah ukuran pemisahan dari dua puncak. Daya pisah (Rs)
dapat diukur dengan persamaan:
2
Nilai tR2dan tR1 adalah waktu retensi senyawa, diukur pada titik maksimum puncak
dan t adalah selisih antara tR2 dan tR1. Nilai w2 dan w1 adalah lebar alas puncak
dinyatakan dalam satuan waktu.
Untuk memperbaiki resolusi dapat dinyatakan dengan parameter-parameter
Berdasarkan rumus tersebut terlihat bahwa resolusi merupakan fungsi dari 3 faktor,
yaitu selektivitas kolom yang tergantung pada α, faktor kapasitas yang tergantung
pada nilai k’ dan faktor efisiensi yang tergantung pada nilai N (Noegrohati, 1994).
5. Pengekoran Puncak Kromatogram
Peak yang tidak simetris sering dijumpai bila konsentrasi sampel dala fase gerak terlalu besar. Apabila kapasitas kolom lebih besar pada konsentrasi yang lebih
rendah, bagian eluen dengan konsentrasi yang lebih rendah akan bergerak lebih
lambat dari pada bagian dengan konsentrasi solut lebih tinggi, sehingga peak yang terjadi tidak simetris dengan bidang bagian depan naik dengan tajam sedangkan
bidang di bagian belakang turun dengan landai (tailing). Keadaan sebaliknya dikenal sebagai leading atau fronting. Penyebab tailing antara lain karena ketidaksesuaian antara analit dengan kolom, pengemasan kolom yang tidak seragam dan faktor yang
terjadi di luar kolom seperti pada injektor (Noegrohati, 1994).
Parameter yang digunakan untuk menilai bentuk puncak adalah peak asymmetry factor (As). Nilai As diukur pada 10% dari tinggi peak (Snyder et al.,
1997). Kolom yang baik akan menghasilkan nilai Assebesar 0,95-1,1. Cara lain untuk
5% dari tinggi peak. Peak asymmetry factor dan peak tailing factor dapat dihitung seperti pada gambar 4 berikut:
Gambar 4. PenentuanPeak AsymmetrydanPeak Tailing Factor (Snyder et al., 1997)
Residu silanol yang tidak bereaksi dengan fase diam pada saat pembuatan
karena adanya halangan sterik dapat memberikan kepolaran yang tidak dikehendaki
dan dapat menyebabkan pengekoran puncak kromatogram. Hal ini terjadi karena
analit lebih terdistribusi dalam fase diam. Untuk mengurangi gugus silanol yang
masih bebas, perlu dilakukan pengikatan dengan trimetilklorosilan yang dapat
mencapai gugus silanol karena ukurannya lebih kecil dibandingkan organoklorosilan
lain. Penambahan trimetilklorosilan dapat menutupi gugus silanol yang bebas dalam
jumlah yang cukup banyak, namun tidak semua gugus dapat tertutupi (Gritter et al.,
1985; Skoog et al., 1998).
Distribusi analit dalam fase gerak dan fase diam pada saat terjadinya tailing
Gambar 5. Distribusi dalam fase diam dan fase gerak (Kuwana, 1980)
6. Analisis Kualitatif dan Kuantitatif
Selain sebagai metode utama untuk memisahkan senyawa dalam sampel,
KCKT juga digunakan untuk analisis kualitatif dan kuantitatif senyawa yang telah
dipisahkan. Berdasarkan kromatogram diperoleh informasi mengenai waktu retensi
suatu senyawa. Waktu retensi ini spesifik untuk tiap senyawa sehingga dapat
digunakan sebagai salah satu dasar uji kualitatif (Noegrohati, 1994). Analisis
kualitatif bertujuan untuk membuktikan ada tidaknya senyawa tertentu dalam sampel
dengan cara membandingkan waktu retensi senyawa murni dengan waktu retensi
senyawa yang dimaksud dalam sampel (Gritter et al., 1985).
luas area peak lebih disukai dibanding pengukuran berdasarkan tinggi peak karena tidak terpengaruh oleh pelebaranpeak(Noegrohati, 1994).
E. Parameter Validasi Metode Analisis
Validasi metode analisis adalah suatu tindakan penilaian terhadap parameter
tertentu, berdasarkan percobaan laboratorium, untuk membuktikan bahwa parameter
tersebut memenuhi persyaratan untuk penggunaannya (Harmita, 2004).
Beberapa parameter analisis yang harus dipertimbangkan dalam validasi
metode analisis antara lain akurasi, presisi, selektivitas, linearitas dan rentang,limit of detectiondanlimit of quantitation.
1. Akurasi
Akurasi atau kecermatan adalah ukuran yang menunjukkan derajat kedekatan
hasil analisis dengan kadar analit yang sebenarnya. Akurasi dinyatakan dengan persen
perolehan kembali (recovery) analit yang ditambahkan (Harmita, 2004).
Kriteria % perolehan kembali yang diijinkan pada setiap konsentrasi analit
pada matriks dapat dilihat pada table II di bawah ini:
Tabel II. Kriteria %recoveryyang diijinkan (Harmita, 2004)
Analit pada matrik
2. Presisi
Presisi adalah ukuran yang menunjukkan derajat kesesuaian antara hasil uji
individual, diukur melalui penyebaran hasil individual dari rata-rata jika prosedur
diterapkan secara berulang-ulang pada sampel-sampel yang diambil dari campuran
yang homogen. Presisi seringkali diukur sebagai persen Relative Standard Deviation
(RSD) atauCoefficient of Variation(CV) (Harmita, 2004).
Kriteria presisi diberikan jika metode memberikan nilai CV 2% atau kurang.
Akan tetapi nilai ini fleksibel tergantung pada kadar analit yang terdapat dalam
sampel yang dianalisis (Harmita, 2004). Kriteria presisi berdasarkan nilai CV yang
diijinkan pada setiap konsentrasi analit pada matriks dapat dilihat pada table III di
bawah ini:
Tabel III. Kriteria presisi yang diijinkan untuk konsentrasi analit yang berbeda (Yuwono dan Indrayanto, 2005)
3. Selektivitas
Selektivitas merupakan kemampuan suatu metode untuk mengukur dengan
akurat respon analit diantara seluruh komponen sampel yang mungkin ada dalam
matriks sampel (Mulja dan Hanwar, 2003).
4. Linearitas dan rentang
Linearitas merupakan kemampuan suatu metode (pada rentang tertentu)
untuk mendapatkan hasil uji yang secara langsung proporsional dengan konsentrasi
analit di dalam sampel (Anonim, 2007). Suatu metode dikatakan memiliki linearitas
yang baik jika nilai koefisien korelasi (r) > 0,99 atau R2> 0,997 (Chan et al., 2004).
Rentang adalah jarak antara level terbawah dan teratas suatu metode analisis
yang telah dipakai untuk mendapatkan presisi, akurasi dan linearitas yang bisa
diterima (Anonim, 2007).
5. Limit Of Detection(LOD) danLimit Of Quantitation(LOQ)
LOD adalah jumlah terkecil analit dalam sampel yang dapat dideteksi yang
masih memberikan respon signifikan dibandingkan dengan blangko. LOD merupakan
parameter uji batas (Harmita, 2004). LOQ adalah kadar terkecil dari suatu analit yang
masih dapat dianalisis dengan hasil yang tetap memenuhi syarat akurasi dan presisi
LOD dan LOQ dapat dihitung dengan rumus:
a. Limit Of Detection
=3 /
b. Limit Of Quantitation
=10 /
Dengan Sy/x adalah simpangan baku, dan Sl adalahslopeatau nilai b pada persamaan
garis y = bx + a (Harmita, 2004).
F. Landasan Teori
Metode KCKT digunakan dalam penetapan kadar aspartam dalam sampel
minuman serbuk beraroma berdasarkan perbedaan interaksi aspartam dan senyawa
lain dalam sampel terhadap fase diam dan fase gerak yang digunakan. Metode
KCKT yang digunakan dipilih fase terbalik karena aspartam bersifat polar sehingga
memiliki afinitas yang lebih besar pada fase gerak. Sistem KCKT fase terbalik yang
digunakan perlu diketahui kondisi optimalnya sehingga dapat memberikan hasil yang
G. Hipotesis
Dari rumusan permasalahan, dapat disusun hipotesis sebagai berikut:
Metode KCKT fase terbalik dengan fase diam C-18 dan fase gerak asetonitril : bufer
fosfat pH 4 (20 : 80 v/v) yang digunakan dalam penetapan kadar aspartam dalam
minuman serbuk beraroma memiliki validitas yang baik dilihat dari dari akurasi,
27
A. Jenis Penelitian
Penelitian yang dilakukan ini termasuk penelitian eksperimental deskriptif.
B. Variabel Penelitian
1. Variabel bebas
Variabel bebas yaitu variabel yang direncanakan untuk diberi pengaruhnya
terhadap variabel tergantung. Variabel bebas dalam penelitian ini adalah
perbandingan fase gerak yang digunakan yaitu asetonitril : bufer fosfat pH 4 danflow rateyang digunakan.
2. Variabel tergantung
Variabel tergantung yaitu titik permasalahan pada penelitian. Variabel
tergantung pada penelitian ini adalah nilai akurasi, presisi, linearitas dan rentang,
selektivitas/spesifikasi,Limit Of Detection,Limit Of Quatitation.
3. Variabel terkendali
Variabel terkendali yaitu variabel yang secara teoritis mempunyai pengaruh
penelitian ini adalah kemurnian pelarut yang digunakan. Untuk mengatasinya
digunakan pelarut yang memiliki kemurnian tinggi yaitu pelarutpro analysis.
C. Bahan-bahan Penelitian
Bahan yang digunakan dalam penelitian ini adalah aspartam kualitasworking standard(The NutraSweet Company) denganCertificate of Analysis(CoA) terlampir pada lampiran 1, KH2PO4, asam fosfat dan, metanol (p.a. Merck), akuabides (PT.
Ikapharmindo Putramas).
D. Alat-alat Penelitian
Alat-alat yang digunakan dalam penelitian ini adalah seperangkat KCKT yang
terdiri dari pompa merek Shimadzu LC-10 AD, detektor UV Vis merek Shimadzu
SPD 10 AV, CBM 101 merekShimadzu, seperangkat komputer merekACER, printer merek Hewlett Packard Deskjet 670 C, Injektor jenis katup suntik model 77251, kolom C-18 merekKromasil 4,6 mm x 25 cm , syringe merek Hamilton Part, alat
E. Tata Cara Penelitian 1. Optimasi Metode KCKT
a. Penentuan panjang gelombang pengamatan. Timbang seksama lebih
kurang 4 mg baku aspartam kemudian dilarutkan dalam akuabides hingga volume 10,0
mL. Kemudian diambil sebanyak 0,5 mL kemudian dimasukkan ke dalam labu ukur
10,0 mL dan diencerkan dengan akuabides hingga tanda. Larutan ini dibaca
absorbansinya pada panjang gelombang 190-260 nm dengan spektrofotometri UV.
Kemudian diperoleh kurva hubungan panjang gelombang dan absorbansi aspartam.
b. Pembuatan fase gerak. Fase gerak yang digunakan dalam penelitian ini
adalah campuran bufer fosfat pH 4 dan asetonitril dengan perbandingan 95 : 5; 90 : 10;
85 : 15 dan 80 : 20 (v/v). Bufer fosfat dibuat dengan melarutkan 0,85 g KH2PO4dalam
akuabides hingga volume 500,0 mL kemudian ditambahkan beberapa tetes asam fosfat
sampai pH 4. Masing-masing perbandingan fase gerak dibuat sesuai volume yang
dibutuhkan kemudian digojog dan disaring dengan penyaring Whatmann organik dengan bantuan pompa vakum lalu didegassingselama 15 menit.
c. Pembuatan larutan stok aspartam. Larutan stok dibuat dengan cara
menimbang seksama lebih kurang 10 mg baku aspartam kemudian dilarutkan dalam
fase gerak asetonitril : bufer fosfat pH 4 hingga volume 50,0 mL.
d. Pembuatan larutan baku aspartam 4 mg/100 mL. Larutan stok aspartam
dipipet sebanyak 2,0 mL kemudian dimasukkan ke dalam labu ukur 10,0 mL dan
e. Pembuatan larutan sampel. Timbang seksama kurang lebih 0,686 g
sampel, diencerkan dengan fase gerak hingga 10,0 mL. Pipet 2,0 mL masukkan ke
dalam labu ukur 10,0 mL dan encerkan dengan fase gerak hingga tanda. Larutan
tersebut disaring denganMilliporedan didegassingselama 15 menit.
f. Pengamatan waktu retensi aspartam. Aspartam dengan konsentrasi 4
mg/100 mL dipipet sejumlah 1,0 mL disaring dengan millipore dan didegassing
menggunakanultrasonic bathselama 15 menit. Larutan diinjeksikan sejumlah 50 µ L ke KCKT dengan fase gerak dan flow rate tertentu serta detektor di set pada panjang gelombang pengamatan. Berdasarkan kromatogram diamati waktu retensi aspartam.
g. Optimasi fase gerak untuk KCKT. Fase gerak yang digunakan adalah
campuran asetonitril : bufer fosfat pH 4 dengan perbandingan 20 : 80 ; 15 : 85 ; 10 : 90
; dan 5 : 95 (v/v). Masing-masing perbandingan fase gerak dibuat sesuai dengan
volume yang dibutuhkan kemudian digojog dan disaring dengan penyaring Whatmann
anorganik dengan bantuan pompa vakum. Fase gerak kemudian didegassingselama 15 menit menggunakanultrasonicator.Dipilih perbandingan fase gerak yang memberikan pemisahan terbaik.
h. Optimasi flow rate untuk KCKT. Optimasi flow rate menggunakan fase gerak asetonitril : bufer fosfat pH 4 (20 : 80) yang memberikan pemisahan terbaik.
yang memberikan kromatogram paling baik berdasarkan waktu tambat (tR), HETP, dan
jumlah pelat teori (N).
2. Verifikasi Sistem
a. Verifikasi akurasi pompa. Alirkan fase gerak asetonitril : bufer fosfat pH 4
(20 : 80) pada fllow rate 1 mL/menit kemudian ditampung pada labu ukur 10,0 mL dengan merek dan ketelitian yang sama. Lakukan sebanyak 7 kali. Catat waktu yang
diperlukan untuk menampung 10,0 mL fase gerak dalam labu. Hitung nilai CV dari waktu yang diperlukan untuk menampung 10,0 mL fase gerak dan hitung % perbedaan
nilaiflow ratehasil pengukuran dengan nilaiflow rateyang diatur pada alat. Perbedaan nilaiflow rate yang diijinkan adalah1% (Lam, 2009).
b. Verifikasi presisi sistem injeksi. Larutan baku 4 mg/100 mL aspartam
diinjeksikan sebanyak 7 kali ke dalam sistem KCKT dengan fase gerak asetonitril :
bufer fosfat pH 4 dengan perbandingan 20:80 danflow rate1,4 mL/menit. Hitung nilai
3. Validasi Metode
a. Pembuatan seri larutan baku aspartam. Seri larutan baku aspartam dibuat
dengan memipet 1,0; 1,5; 2,0; 2,5; dan 3,0 mL larutan stok aspartam kemudian
dimasukkan dalam labu ukur 10,0 mL dan diencerkan hingga tanda sehingga diperoleh
seri konsentrasi larutan baku aspartam 2,0; 3,0; 4,0; 5,0; dan 6,0 mg/100 mL.
b. Pembuatan kurva baku. Seri larutan baku 2,0; 3,0; 4,0; 5,0 dan 6,0 mg/mL
disaring dengan millipore dan didegassing menggunakan ultrasonic bath selama 15 menit. Larutan diinjeksikan sejumlah 50 µL ke KCKT dengan flow rate fase gerak yang optimum dan detektor di set pada panjang gelombang 214 nm.Berdasarkan
kromatogram dilihat AUC pada waktu retensi yang sesuai dengan waktu retensi
aspartam. Dibuat kurva regresi linear yang menyatakan hubungan antara kadar
aspartam lawan nilai AUC, kemudian ditentukan persamaan garis regresi linear serta
nilai koefisien korelasinya, dan nilai tersebut akan diterima apabila lebih dari 0,99
(Chan et al., 2004).
c. Recovery kurva baku. Timbang seksama kurang lebih 10 mg baku aspartam kemudian dilarutkan dengan fase gerak hingga volume 50,0 mL. Dari larutan
tersebut diambil sebanyak 1,0; 2,0; dan 3,0 mL kemudian masing-masing dimasukkan
dalam labu ukur 10,0 mL dan diencerkan dengan fase gerak hingga tanda. Larutan
sejumlah tiga kali sehingga diperoleh sembilan data. Berdasarkan kromatogram dilihat
AUC pada waktu retensi sesuai dengan dengan waktu retensi aspartam. Kadar aspartam
dihitung dengan memasukkan nilai AUC ke persamaan kurva baku yang diperoleh
yang memenuhi syarat linearitas.
F. Analisis Hasil
Validasi metode analisis yang digunakan dalam penetapan kadar aspartam
pada penelitian ini dapat ditentukan berdasarkan parameter selektivitas, linearitas dan
rentang, akurasi, presisi,limit of detectiondanlimit of quantitation. 1. Selektivitas
Selektivitas dinyatakan dengan mengukur kemampuan memisah antara peak
aspartam dengan peak senyawa lain yang paling dekat dengan peak aspartam.
2. Linearitas dan rentang
Linearitas dilihat dari nilai koefisien korelasi (r) hasil pengukuran seri larutan
baku aspartam. Suatu metode dikatakan memiliki linearitas yang baik jika nilai r >
0,99 atau r2> 0,997 (Chan et al., 2004). Rentang ditentukan dari kadar aspartam yang
digunakan dalam analisis, mulai dari kadar terendah hingga tertinggi yang dapat
3. Akurasi
Akurasi metode analisis dinyatakan dengan % perolehan kembali (recovery) yang dihitung dengan cara sebagai berikut:
% perolehan kembali (recovery) = x100%
sebenarnya
metode analisis dikatakan memiliki akurasi yang baik apabila % perolehan kembali
aspartam baku berada pada rentang 98-102% (Yuwono dan Indrayanto, 2005) dan %
perolehan kembali sampel aspartam berada pada rentang 95-105% (Mulja dan Hanwar,
2003).
4. Presisi
Presisi metode analisis dinilai berdasarkan Coefficient of Variation yang dihitung dengan cara sebagai berikut:
x100%
dengan SD adalah simpangan baku, metode analisis dikatakan baik jika nilai CV
5. LOD (Limit Of Detection) dan LOQ (Limit Of Quantitation) LOD dan LOQ dapat dihitung menggunakan rumus:
LOD =
b x Sy
3
LOQ =
b x Sy
10
Dimana Sy merupakan simpangan baku residual yang diperoleh melalui akar
36
A. Optimasi Metode 1. Penentuan Panjang Gelombang Pengamatan
Aspartam memiliki gugus kromofor yang pendek dan tidak memiliki gugus
auksokrom yang bertanggung jawab dalam memberikan absorbansi gelombang
elektromagnetik seperti yang ditunjukkan pada gambar 6.
O
O
NH H2N
HO
O
O
Kurva serapan
Gamba
Gambar 7 menunjuk
adalah 206 nm. N
pengamatan pada 21
dilakukan Nollet (20
gelombang 214 nm,
menurut Snyder at
gelombang maksimum
2. Optimasi Fase G
Fase gerak y
pemisahan aspartam d
an aspartam dapat terlihat seperti pada gambar b
bar 7. Spetrum panjang gelombang maksimum aspar
jukkan bahwa panjang gelombang serapan m
Namun, pada penelitian ini digunakan pa
214 nm untuk menyamakan dengan peneli
(2000). Hal ini tidak menjadi masalah kar
, aspartam masih memberikan serapan yang
at al. (1997) pengukuran bisa dilakukan ti
um dengan syarat nilai koefisien korelasi meme
e Gerak
yang digunakan pada penelitian ini meng
dan produk degradasinya secara KCKT men
r berikut ini:
ng tinggi. Selain itu,
tidak pada panjang
menuhi persyaratan.
engacu pada metode
by HPLC (Nollet, 2000). Dalam metode tersebut fase gerak yang digunakan adalah bufer fosfat 12,5 mM pH 3,5 : asetonitril dengan perbandingan 90 : 10 dan 85 : 15.
Pada penelitian ini, fase gerak yang dioptimasi adalah bufer fosfat : asetonitril
dalam beberapa perbandingan yaitu 95 : 5; 90 : 10; 85 : 15 dan 80 : 20. Selain itu,
juga dilakukan optimasiflow ratefase gerak yang digunakan. Optimasi ini dilakukan untuk mengetahui komposisi mana yang paling baik digunakan dalam penetapan
kadar aspartam.
Dalam penelitian ini bufer fosfat dibuat dari campuran garam kalium
dihidrogen fosfat dan asam fosfat, dan diatur pada pH 4 karena aspartam dalam
bentuk larutan stabil pada range pH 3-5 dan stabilitas yang paling optimum adalah ketika berada pada pH 4,3 (Nollet, 2000). Campuran fase gerak bufer fosfat dan
asetonitril bersifat polar sedangkan fase diam yang digunakan yaitu Kromasil-C18
bersifat non polar. Dengan demikian sistem kromatografi dalam penelitian ini adalah
kromatografi pasrtisi fase terbalik dimana fase gerak yang digunakan bersifat lebih
polar dari fase diamnya.
Pada kromatografi fase terbalik, ionisasi analit harus diminimalkan. Analit
yang terion (gambar 8) menyebabkan waktu retensinya menjadi sangat singkat karena
analit bersifat terlalu polar dan tidak berinteraksi dengan fase diam yang bersifat non
polar. Oleh karena itu, pada penelitian ini digunakan bufer fosfat sebagai pelarut dan
fase gerak untuk mencegah agar aspartam tidak mengalami ionisasi dan tetap dapat
O
Gambar 8. Reaksi ionisasi aspartam dengan pengaruh asam (a) dan reaksi ionisasi aspartam
dengan pengaruh basa (b)
Stabilitas aspartam dalam larutan dipengaruhi oleh pH. Pada kondisi pH yang
tidak sesuai aspartam akan terdegradasi menjadi produk-produk lain yang memiliki
sifat yang berbeda dengan senyawa induknya sehingga dapat mempengaruhi
analisisnya. Oleh karena itu, penggunaan bufer fosfat pH 4 juga dimaksudkan untuk
mempertahankan pH pelarut dan fase gerak sehingga aspartam tetap dalam kondisi
yang stabil dan tidak mengalami degradasi. Mekanisme degradasi aspartam dapat
O
Gambar 9. Degradasi aspartam (Anonim, 1988)
Optimasi yang dilakukan pertama kali adalah optimasi perbandingan
komposisi fase gerak bufer fosfat pH 4 : asetonitril. Perbandingan yang dioptimasi
adalah 95 : 5; 90 : 10; 85 : 15 dan 80 : 20 (v/v). Pada optimasi perbandingan fase
Gambar 10. K
Gambar 11. K
. Kromatogram optimasi fase gerak bufer fosfat pH 4
. Kromatogram optimasi fase gerak bufer fosfat pH 4
pH 4 : asetonitri (95 : 5)
Gambar 12. K
. Kromatogram optimasi fase gerak bufer fosfat pH 4
: Varian Shimadzu LC-10 AD
: Kromasil-100 C18 250 x 4,6 mm, 5μm : bufer fosfat pH 4 : asetonitril (80:20) : 1,4 mL/menit
: 0,01/7
: baku 4,0 mg/100 mL : UV 214 nm
. Kromatogram optimasi fase gerak bufer fosfat pH 4
n kromatogram pada gambar 10-13 dapat d
erbandingan komposisi fase gerak 95 : 5; 90
4 : asetonitri (85 : 15)
4 : asetonitri (80 : 20)
dilihat bahwa peak
mengalami tailing. P
. Pada komposisi fase gerak dengan perbandin
gga dapat disimpulkan bahwa fase gerak denga
memberikan hasil yang paling baik. Namun s
cepatan alir terlebih dahulu dilihat hasil pemisa
enggunakan fase gerak tersebut. Hasilnya
: Varian Shimadzu LC-10 AD
: Kromasil-100 C18 250 x 4,6 mm, 5μm : buffer fosfat pH 4 : asetonitril (95:5) : 1,4 ml/menit
: 0,01/7 : sampel : UV 214 nm
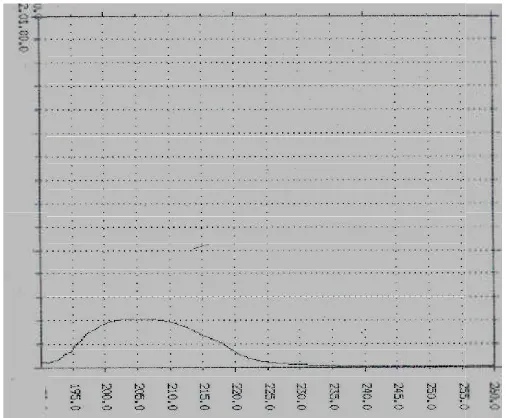
Kromatogram pemisahan aspartam pada sampel deng fosfat pH 4 : asetonitril (80 : 20)
dingan 80 : 20 tidak
ngan perbandingan 80
sebelum dilanjutkan
isahan aspartam pada
a dapat dilihat pada
Dari gambar di atas dapat dilihat bahwa pemisahan aspartam pada sampel
dengan fase gerak buffer fosfat pH 4 : asetonitril (80 : 20) memberikan hasil yang
baik dengan nilai resolusi 3,8875. Nilai tersebut memenuhi syarat dimana pemisahan
dikatakan baik jika nilai resolusi lebih besar atau sama dengan 2. Sehingga komposisi
tersebut yang akan dipilih sebagai fase gerak yang paling baik yang selanjutnya akan
dioptimasiflow ratenya.
Flow rate yang dioptimasi adalah 0,6; 0,8; 1,0; 1,2 dan 1,4 mL/menit. Hasil optimasi flow rate ini akan digunakan untuk menghitung nilai lempeng teoritis (N) danHETPsehingga dapat diketahuiflow rateyang paling optimum berdasarkan teori
Van Deemter yaitu yang memiliki nilai HETP paling kecil dan nilai N yang paling besar. Contoh perhitungan nilai N dan HETP dapat dilihat pada lampiran 2.
Hasil optimasi flow rate fase gerak bufer fosfat pH 4 : asetonitril (80 : 20) dapat dilihat dalam tabel IV.
Tabel IV. Hasil optimasiflow ratefase gerak bufer fosfat pH 4 : asetonitril (80 : 20)
Flow rate
(mL/menit)
Lempeng teoritis
(N) HETP (10
-3) tR (menit)
0,6 13.363,21 1,87 12,077
0,8 10.460,69 2,40 9,180
1,0 9.951,86 2,50 7,479
1,2 6.943,2 3,60 6,247
1,4 8.147,04 3,07 5,414
Gambar 15. Kurvaflow rate vs HETPfase gerak bufer fosfat pH 4 : asetonitril (80 : 20)
Berdasarkan data optimasi flow rate diperoleh flow rate 0,6 mL/menit yang memiliki nilai N paling besar dan nilaiHETPyang paling kecil. Namun karena waktu retensinya relatif panjang yaitu 12,077 menit, maka flow rate tersebut tidak dipilih karena kurang efisien. Dengan demikian, untuk mendapatkan pemisahan yang efisien,
flow rate yang dipilih adalah 1,4 mL/menit karena waktu retensinya yang paling singkat namun masih memberikan pemisahan yang baik dimana peak aspartam
terpisah dengan peak senyawa lain pada sampel (gambar 14). Selanjutnya, analisis
aspartam pada penelitian ini menggunakan fase gerak bufer fosfat pH 4 : asetonitril
(80 : 20) denganflow rate1,4 mL/menit.
0
3. Pengamatan Wa
Karena waktu
waktu retensi dapat d
waktu retensi senyaw
tu retensi untuk masing-masing senyawa bers
t digunakan untuk analisis kualitatif dengan ca
awa pada sampel dengan waktu retensi seny
hasil pengamatan waktu retensi baku aspartam
: Varian Shimadzu LC-10 AD
: Kromasil-100 C18 250 x 4,6 mm, 5μm : bufer fosfat pH 4 : asetonitril (80:20) : 1,4 mL/menit
: 0,01/7
: baku aspartam 4,0 mg/100 mL : UV 214 nm
togram waktu retensi baku aspartam dengan fase ger fosfat PH 4 (20 : 80) danflow rate1,4
atogram tersebut aspartam ditunjukkan oleh
nsi 5,118 menit. Berdasarkan data presisi injek
A
ersifat spesifik, maka
cara membandingkan
nyawa baku. Berikut
tam:
gerak asetonitril : bufer
leh peak nomor 13 jeksi diperoleh waktu
retensi (tR) dari asp
bahwa peak yang b aspartam. Berdasarka
spartam berada pada range 5,009-5,237. Ma berada pada range waktu tersebut diyakin kan gambar 15 dapat dilihat bahwa waktu ret
jukkan dengan peak nomor 13, masuk ke d .
bahwa dalam sampel minuman serbuk yang d
maka dilakukan pembandingan waktu reten
tu retensi baku aspartam.
: Varian Shimadzu LC-10 AD
: Kromasil-100 C18 250 x 4,6 mm, 5μm : buffer fosfat pH 4 : asetonitril (80:20) : 1,4 ml/menit
: sampel : UV 214 nm
kromatogram waktu retensi sampel dengan asetonitr (20 : 80) danflow rate1,4
tensi dari peak pada
nitril : bufer fosfat PH 4
Dari kromatogram di
ke dalam range waktu
merupakan peak dari bahwa peak tersebu penambahan baku asp
yang nantinya mengala
dari aspartam dalam s
Instrumen
di atas, terdapat satupeakdengan waktu retensi ktu retensi aspartam. Sehingga dapat diyakini b
ari aspartam di dalam sampel. Namun untuk
ebut benar merupakan peak dari aspartam aspartam ke dalam sampel yang akan diinjeks
galami penambahan luas area maupun tinggipe
sampel. Hasil yang diperoleh dapat dilihat pad
: Varian Shimadzu LC-10 AD
: Kromasil-100 C18 250 x 4,6 mm, 5μm : buffer fosfat pH 4 : asetonitril (80:20) : 1,4 ml/menit
: 0,01/7
: sampel + baku aspartam : UV 214 nm
romatogram waktu retensi sampel + baku aspartam
Asp
nsi 5,030 yang masuk
Pada kromatografi fase terbalik, pemisahan terjadi karena adanya perbedaan
koefisien distribusi masing-masing senyawa dalam fase gerak dan fase diam. Suatu
senyawa yang memiliki koefisien distribusi kecil akan terikat lebih kuat pada fase
geraknya dibandingkan pada fase diam. Dengan demikian, senyawa akan keluar lebih
dahulu dari kolom dan waktu retensinya relatif lebih cepat. Nilai koefisien distribusi
dipengaruhi oleh interaksi senyawa terhadap fase gerak dan fase diam. Pada
strukturnya, aspartam memiliki gugus polar dan non polar yang dapat berinteraksi
dengan fase gerak dan fase diam. Gugus polar pada aspartam akan berinteraksi
dengan fase gerak melalui ikatan hidrogen sedangkan gugus non polarnya akan
berinteraksi dengan fase diam melalui ikatan Van der Waals. Interaksi antara aspartam dengan fase gerak dapat dilihat pada gambar 19 berikut:
O
Gugus polar pada aspartam akan berinteraksi dengan fase diam melalui ikatan
Van der Waals. Ikatan ini secara sederhana dapat digambarkan seperti terlihat pada gambar 20.
Gambar 20. Ikatan aspartam dengan fase diam
B. Verifikasi Sistem KCKT
Untuk memastikan bahwa sistem yang digunakan dapat memberikan data
yang reprodusibel pada saat analisis, maka perlu dilakukan verifikasi sistem KCKT.
Beberapa uji yang biasa dilakukan untuk memastikan bahwa sistem sesuai untuk
suatu analisis antara lain verifikasi akurasi pompa dan verifikasi presisi sistem
injeksi.
1. Verifikasi akurasi pompa
Syarat kinerja dari pompa KCKT adalah kemampuannya untuk
mempertahankan ketepatan dan konsistensi laju fase gerak, yang penting untuk
menjaga stabilitas dan keterulangan pada reaksi antara analit dengan fase diam.
flow rate pompa ini dilakukan dengan menghitung waktu yang dibutuhkan untuk menampung 10 mL fase gerak. Flow rate yang terhitung kemudian dibandingkan dengan flow rate yang diatur pada sistem. Penyimpangan flow rate yang diterima adalah1% (Lam, 2004).
Tabel V. Data penyimpanganflow ratepada uji akurasi pompa
Berdasarkan data pada tabel V dapat dilihat bahwa % penyimpangan dari 5 kali
replikasi tidak ada satupun yang melebihi persyaratan. Dengan demikian dapat
disimpulkan bahwa sistem pompa yang digunakan memiliki akurasi yang baik.
2. Verifikasi presisi sistem injeksi
Uji ini dilakukan untuk mengetahui kemampuan suatu injektor untuk
menggambarkan jumlah yang sama dari suatu sampel yang diinjeksikan secara
berulang. Ketelitian injektor dapat ditunjukkan dengan membuat sedikitnya 6
replikasi sampel yang diinjeksikan. Coefficient of Variation (CV) data hasil injeksi kemudian dihitung untuk mengevaluasi ketelitian. CV yang diterima adalah ≤1%
untuk replikasi lebih dari 5 kali.
Berdasarkan uji presisi sistem injeksi yang dilakukan diperoleh data sebagai
Tabel VI. Hasil uji presisi sistem injeksi
Berdasarkan tabel VI dapat dilihat bahwa dari lima kali injeksi aspartam diperoleh
nilai CV yang memenuhi syarat yaitu ≤1%. Dengan demikian, dapat disimpulkan
bahwa sistem yang digunakan memiliki presisi injeksi yang baik dan dapat digunakan
untuk analisis. Perdasarkan perhitungan, range tR yang diperbolehkan yaitu 5,009-5,237. Dari lima kali injeksi tidak ada satupun tR yang keluar dari range yang dihasilkan. Range tR ini nantinya juga digunakan sebagai pedoman dalam langkah selanjutnya yaitu dalam pembuatan kurva baku dan penetapanrecoverybaku, nilai tR aspartam seharusnya tidak boleh keluar darirangeyang diijinkan.
C. Pembuatan Kurva Baku
Tujuan pembuatan kurva baku adalah untuk mendapakan persamaan regresi
linear yang selanjutkan digunakan untuk menghitung kadar aspartam untuk penetapan
recovery dan penetapan kadar sampel. Persamaan regresi linear diperoleh dari korelasi antara konsentrasi aspartam dengan luas areapeak (AUC). Agar kurva baku yang diperoleh layak ditampilkan dan memiliki sensitivitas yang baik maka nilai
AUC dibagi dengan 100000 sebagai faktor koreksi. Data kurva baku aspartam dari
Tabel VII. Data kurva baku aspartam
Pada penelitian ini digunakan 5 seri konsentrasi aspartam dengan 3 kali
replikasi. Replikasi dilakukan untuk mendapatkan nilai koefisien korelasi yang paling
baik. Persamaan kurva baku yang dipilih yaitu persamaan yang memiliki koefisien
korelasi paling mendekati 1. Suatu metode dikatakan memiliki linearitas yang baik
jika nilai r > 0,99 atau R2 > 0,997 (Chan et al., 2004). Dengan demikian dapat
dikatakan bahwa ketiga persamaan tersebut memiliki linearitas yang baik dan dapat
digunakan untuk penetapan kadar aspartam dalam sampel. Persamaan kurva baku
yang dipilih untuk penetapan kadar aspartam adalah persamaan kedua, y = 1,008x +
0,462 karena memiliki koefisien korelasi yang paling baik (r paling mendekati 1).
Hubungan antara konsentrasi aspartam dengan AUC/100000 dapat dilihat pada
gambar 21.
Replikasi 1 Replikasi 2 Replikasi 3 Kadar
Gambar 21. Kurva hubungan antara konsentrasi aspartam (mg/100mL) dengan AUC/100000
D. Hasil Validasi Metode Penetapan Kadar Aspartam
Validasi metode dilakukan untuk mengetahui apakah metode KCKT yang
digunakan dalam penelitian ini memiliki validitas yang baik atau tidak untuk
penetapan kadar aspartam dengan kondisi percobaan sesuai hasil optimasi. Suatu
metode dikatakan memiliki validitas yang baik jika parameter-parameter yang
diujikan memenuhi persyaratan yang telah ditentukan. Parameter-parameter yang
harus dipenuhi dalam penelitian kali ini adalah linearitas, presisi, akurasi, selektivitas,
LODdanLOQ.
Kadar aspartam baku (mg/100ml)