Atherosclerosis 151 (2000) 443 – 450
The V73M mutation in the hepatic lipase gene is associated with
elevated cholesterol levels in four Dutch pedigrees with familial
combined hyperlipidemia
Marie¨tte J.V. Hoffer
a,1, Harold Snieder
b, Sebastian J.H. Bredie
c,
Pierre N.M. Demacker
c, Johannes J.P. Kastelein
d, Rune R. Frants
a,*,
Anton F.H. Stalenhoef
caMGC-Department of Human Genetics,Leiden Uni6ersity Medical Centre,PO Box 9503,2300 RA Leiden, The Netherlands bTwin Research Unit,St Thomas’Hospital,London, UK
cDepartment of Medicine,Di6ision of General Internal Medicine,Uni6ersity Hospital Nijmegen,Nijmegen, The Netherlands dDepartment of Vascular Medicine,Academic Medical Centre,Amsterdam, The Netherlands
Received 21 December 1998; received in revised form 20 September 1999; accepted 4 October 1999
Abstract
Familial combined hyperlipidemia (FCHL) is a heritable lipid disorder characterized by multiple lipoprotein phenotypes within a single family. Previously, we have shown an increased incidence of mutations in the LPL gene which was associated with elevated levels of very low density lipoprotein (VLDL) and decreased levels of high density lipoprotein among the families studied. Now, we report the results of our study on the hepatic lipase gene. We found the HL V73M variant to be present in four FCHL families. By means of a pedigree-based maximum log-likelihood method we analyzed the effect of this variant on the lipid levels in these families. Carriers of the HL V73M variant revealed significantly higher levels of total cholesterol (PB0.01) and apoB (PB0.01). These findings show that the HL V73M mutant explains another part of the variability in the phenotype observed among FCHL family members, compared with mutations in the LPL gene. Family analysis shows that in these FCHL families, carriers of mutations in the LPL or HL genes have an increased risk for FCHL compared with their non-carrier relatives. © 2000 Elsevier Science Ireland Ltd. All rights reserved.
Keywords:Hepatic lipase; Familial combined hyperlipidaemia; HLV73M mutation; apoB100; Cholesterol
www.elsevier.com/locate/atherosclerosis
1. Introduction
Familial combined hyperlipidemia (FCHL) is the most common genetic cause of lipid disorders and is associated with coronary artery disease (CAD) [1,2]. In FCHL families, affected relatives exhibit different com-binations of elevated plasma cholesterol and elevated triglyceride levels. In addition, characteristics such as elevated levels of plasma apolipoprotein (apo) B100, increased very low density lipoprotein (VLDL) produc-tion, predominance of small dense low density
lipo-protein (LDL), and insulin resistance have been reported [3 – 6].
So far, no unique metabolic defect has been identified in FCHL. Several kinetic studies have shown hepatic overproduction of VLDL apoB 100 [7 – 9]. In addition, impaired lipolysis of VLDL [8] and increased conver-sion of VLDL to LDL has been observed [10]. There-fore, in general, defective regulation of the apoB metabolism may be responsible for the development of FCHL.
Considering elevated levels of either VLDL, LDL or both as affected phenotype in family studies, FCHL was initially suggested to be an autosomal dominant disorder [1,2]. Using complex segregation analysis, Cul-len et al. [11] found evidence for a major gene acting on triglyceride levels in families with FCHL. Additional * Corresponding author. Tel.: +31-71-5276083; fax: +
31-71-5276075.
E-mail address: [email protected] (R.R. Frants)
1Present address: Department of Clinical Genetics, Academic
Medical Centre, Amsterdam, The Netherlands.
studies, using apoB levels and LDL subclass phenotype as quantitative traits in complex segregation analysis, showed that these traits appear to be influenced by genetic effects which are of use in the prediction of FCHL [12 – 15]. Taken together, FCHL can be consid-ered a complex disorder with a heterogeneous background.
Suggestive linkage results have been demonstrated between FCHL and several loci [16 – 18], but so far these results have not been confirmed [19,20]. It was also shown that specific mutations in the FABP2, APOA4, LPL and HL genes have higher frequencies among FCHL patients when compared with healthy control subjects [21 – 25]. We speculate that the lipo-protein lipase (LPL) and hepatic lipase (HL) genes could be good candidate genes causing FCHL because of their central roles in apoB and triglyceride metabolism. Previously, we have shown that the D9N and N291S mutations in the LPL gene are associated with elevated lipid levels of VLDL and decreased levels of HDL among carriers compared with their non-car-rier relatives in FCHL families [26].
In the present study, we investigated whether the HL gene is an additional genetic determinant of the vari-ability in phenotypic expression of FCHL. Therefore, 39 probands were screened for a HL protein isoform, V73M. Subsequently, using a pedigree-based maximum log-likelihood method, we investigated the influence of this HL variant on lipid and lipoprotein levels in four extended FCHL families.
2. Methods
2.1. Subjects
The FCHL pedigrees considered here have previously been used to investigate the inheritance of LDL sub-fraction profiles and plasma apoB levels [14,15]. The families were ascertained through probands selected from patients attending the University Lipid Research Clinics at the Universities of Nijmegen and Amsterdam. Probands fulfilled the following criteria:
1. elevated levels of both total cholesterol and triglyce-rides at first measurement;
2. cholesterol and triglyceride levels above the 90th percentile using the age- and sex-related percentile levels of the Prospective Cardiovascular Munster (PROCAM) study [27].
These values were consistent over several measurements in which probands had not been given any lipid lower-ing drugs. Families were included when a multiple-type hyperlipidemia with levels of total plasma cholesterol and/or triglycerides above the 90th percentile (single measurement) was present in at least one first-degree relative of the proband. Children were 15 years or older.
None of the FCHL probands had specific clinical signs, like tendon xanthomata, and none were ho-mozygous for the APOE*2 allele. For all probands, a secondary cause of hyperlipidaemia (i.e. diabetes melli-tus, hypothyroidism, and hepatic or renal impairment) was excluded by standard laboratory tests. The study protocol was approved by the ethical committee of the participating universities and each subject gave in-formed consent. Normolipidemic individuals, randomly selected from three different geographic areas in the Netherlands were used as a control population [28].
2.2. Measurement of lipids, lipoproteins and apolipoproteins
EDTA blood samples were obtained from the probands and family members after overnight fasting. No lipid lowering drugs were administered to the sub-jects for at least 4 weeks prior to the onset of the study. Plasma was separated from cells by centrifugation at 500×g for 10 min at room temperature, and used immediately for lipid and lipoprotein analysis. VLDL (dB1.006 g/ml) was isolated by ultracentrifugation for 16 h at 36 450 rpm in an fixed-angle TFT 45.6 rotor (Kontron, Zurich), in a Beckman L7-55 ultracentrifuge [29]. Plasma and lipoprotein cholesterol and triglyceride concentrations were determined by enzymatic, commer-cially available reagents (No. 237574; Boehringer – Mannheim, FRG; Sera-pak, No. 6639; Tournai, Belgium). HDL-cholesterol was determined in whole plasma using the polyethylene glycol 6000 method [30]. The apolipoproteins apoA1 and apoB were determined by immunonephelometry as previously described for apoB [31]. LDL subtraction pattern was determined as described by Bredie et al. [14].
2.3. DNA analysis
Genomic DNA was isolated from leucocytes accord-ing to Miller et al. [32]. Identification of carriers of the HL V73M variant was performed by PCR using the primers: 5%-AGC TGG AGA AGG AAG AAG GG-3%
(sense) and 5%-ATT CAC TCT CAG AGG AAG GG-3%
M.J.V.Hoffer et al./Atherosclerosis151 (2000) 443 – 450 445
ethidium bromide. In the case of the mutant allele, a restriction site is formed due to the GA change, and therefore digestion of the PCR product will reveal one fragment of 189 bp for the wildtype allele, and two fragments of 137 and 52 bp for the variant allele. Identification of the LPL N291S allele carriers in the two families originates from an earlier study [26].
2.4. Statistical analysis
To test for the statistical significance of the effect of the LPL N291S and HL V73M mutations on lipo-protein traits using all carriers in multiple families, a pedigree-based maximum likelihood method developed by Lange et al. [34,35], was used, as previously de-scribed [26,36].
For a given pedigree of n individuals a vector of observations (x) is defined and a vector of expected values [E(x)], that can depend on measured variables such as gender or genotype. The covariance between the residual part of the observations, i.e. the part that is not accounted for by the measured genotype or other variables, depends on the relationship between the pedi-gree members and on the genetic model assumed for the observations. Throughout, we have modelled the variances not accounted for by the measured genotype as consisting of additive genetic and random environ-mental variance, recognizing that the genetic part may also reflect environmental influences shared by family members. However, our main interest is to test for the influence of the measured genotype. For a given E(x) and expected covariance matrix S, the log likelihood of obtaining the observation vector x is: L= −1/2LnS−1/2[x−E(x)]%S−1[x−E(x)]+constant; where % denotes matrix transposition. The joint
log-likelihood of obtaining all pedigrees is the sum of the log-likelihood of the separate pedigrees. Estimation in-volves selection of parameter values under a specific model that maximizes the joint likelihood of all pedi-grees. The likelihood obtained for different models can be compared with chi-squared difference tests where X2
=2(L1−L0) and L1 and L0 denote the log likeli-hood for the general (H1) and the constrained (H0) hypothesis. The degrees of freedom (df) for this test are equal to the number of independent parameters be-tweenH1andH0[35]. The Fisher package [34] was used for genetic modelling. Preliminary analysis did not give any evidence for gene×gender interactions unlike our previous paper on LPL-93 GT/D9N [37]. The most general (full) model allowed for: age regression; gender-difference; differences between carriers and non-carriers of LPL N291S and/or HL V73M which were defined by four categories. For each of those four categories a different mean was estimated in the full model. By setting the regression for age or gender to zero or by constraining estimated means to be equal across carrier
status of LPL N291S or HL V73M in the specification of the submodels, it was possible to test for each of those effects separately. A significant decrease in log-likelihood of the submodel compared to the full model, as apparent from the chi-square difference test, indi-cates that a model which allows for the effect shows a better fit, i.e. the specific effect is significant.
In the model fitting to the pedigree data with the Fisher package, ascertainment correction was carried out by conditioning on the probands. The data from the probands were omitted from statistical calculations (e.g. the estimation of the means) to avoid possible ascertainment bias. Because the distribution of total triglyceride, VLDL-cholesterol and VLDL-triglyceride was skewed, these data were transformed by natural logarithm in order to obtain a normal distribution.
3. Results
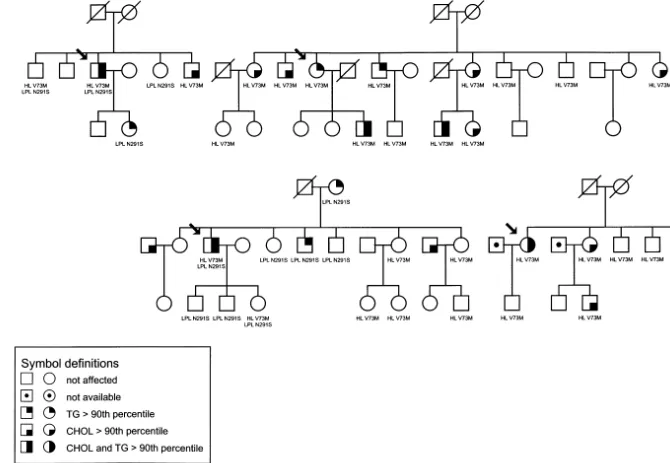
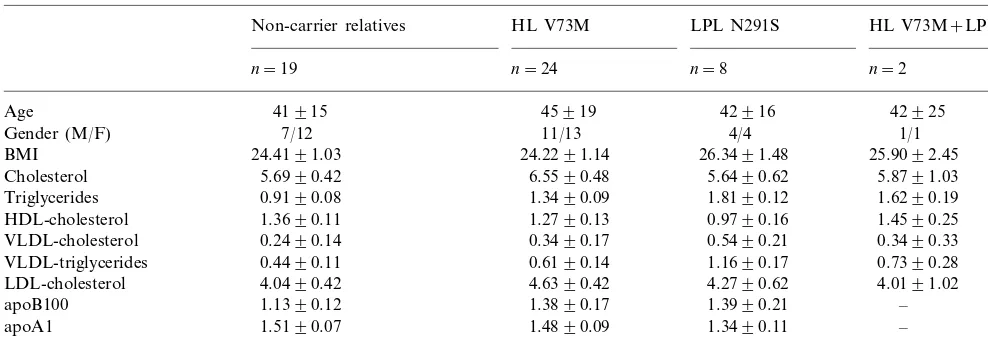
As a part of an ongoing study aiming at the identifi-cation of genetic risk factors underlying FCHL we screened 39 patients for the presence of a HL isoform, V73M. This revealed a substantially higher frequency of this variant in unrelated FCHL patients (allelic frequency 6.4%, 5 out of 39 probands) when compared with healthy controls (2.8%, 6/109), (OR 2.3, 95% CI 0.67 – 8.09). Subsequently, all relatives of four patients were analysed (Fig. 1). For one patient no relatives were available. Surprisingly, two of these families were previously identified for presence of the LPL N291 S variant [26]. Table 1 shows the descriptive biochemical parameters of all the relatives used in this study.
M
.
J
.
V
.
Hoffer
et
al
.
/
Atherosclerosis
151
(2000)
443
–
450
M.J.V.Hoffer et al./Atherosclerosis151 (2000) 443 – 450 447
Table 1
Characteristics of relatives of patients with familial combined hyperlipidemiaa
HL V73M LPL N291S HL V73M+LPL N291S Non-carrier relatives
n=19 n=24 n=8 n=2
Age 41915 45919 42916 42925
11/13 4/4
7/12 1/1
Gender (M/F)
24.4191.03
BMI 24.2291.14 26.3491.48 25.9092.45
5.6990.42
Cholesterol 6.5590.48 5.6490.62 5.8791.03
1.3490.09 1.8190.12
0.9190.08 1.6290.19
Triglycerides
HDL-cholesterol 1.3690.11 1.2790.13 0.9790.16 1.4590.25 0.3490.17 0.5490.21
0.2490.14 0.3490.33
VLDL-cholesterol
0.4490.11
VLDL-triglycerides 0.6190.14 1.1690.17 0.7390.28
4.0490.42
LDL-cholesterol 4.6390.42 4.2790.62 4.0191.02
1.3890.17 1.3990.21
1.1390.12 –
apoB100
1.4890.09 1.3490.11
apoA1 1.5190.07 –
−0.1790.11 −0.4790.15
0.0190.09 –
LDL subtraction
aAll levels are given as mean9SE in mmol/l except age; year, BMI; kg/m2and apolipoprotein levels: g/l.
Table 2
Log-likelihood for five different models testing the effects of age, gender, and carrier status for LPL N291S or HL V73M for lipid traits in FCHL pedigreesa
Model BMI Plasma Plasma VLDL- VLDL- LDL- HDL- apoB apoA1 LDL
CHOL TG CHOL TG CHOL CHOL subfraction
−24.86 52.72 31.4 37.76
1 Full model −73.18 −28.46 44.62 45.62 65.96 47.62
2 Age −80.49b −35.18b 50.79d 30.26 36.87 −35.51b 44.42 35.85b 64.75 46.85
−25.17 48.96c 26.36c 31.63b −28.79 32.71b
3 Gender −73.37 44.37 55.64b 40.03b
−24.86 49.14c 29.58 34.18c −28.49
−74.04 40.78c
4 LPLN291S 43.59d 64.46 41.05b
−28.56c
5 HLV73M −73.24 50.84 31.16 37.33 −30.21 44.59 40.59c 65.92 46.35
aLog-likelihoods for five models. Model definition; full: most general model allowing for: (i) age regression (ii) gender-difference (iii) difference
between carriers and non-carriers of LPL N291 S and (iv) HL V73M. The other (sub)models provide a test for each of the effects separately. Testing procedure: each model is tested against the full model. A significant decrease in log-likelihood of the submodel compared to the full model indicates a significant effect of the tested parameter. When twice the difference in log-likelihood of these models is higher than the x2
corresponding to df=1, this indicates a significant difference as indicated. Model 2–5: df=1.
bPB0.001 (
x2\10.83) cPB0.01 (
x2\7.88).
dPB0.05 (
x2\3.84).
VLDL- and HDL cholesterol, serum triglycerides and VLDL triglycerides, apoA1 levels and LDL subfrac-tion profile.
The HL V73M carriers exhibited significantly in-creased levels of total cholesterol and apoB compared with their non-carrier relatives, while the LPL N291S variant has a significant effect on total and VLDL triglyceride levels and also on the concentrations of HDL cholesterol and apoB and LDL subfraction profile (Table 2).
To find out whether the mutations in the HL and LPL genes are modifying or causal factors for FCHL we went back to the pedigrees of these families and counted the presence and absence of these mutations among affected and non-affected family members. If no association was present, one would expect simi-lar numbers of carriers among affected and non-af-fected individuals. However, counting affected
individuals as being carrier or non-carrier of the HL V73M or LPL N291S variant revealed that all af-fected individuals in these four families were carrier of either one or both of these two variants (Fig. 1, Table 3).
Table 3
Distribution of affected and non-affected individuals in four Dutch FCHL families
Totals Affected Non-affected
HL V73M and/or LPL 19 19 38
N291S
0
Non-carrier 12 12
4. Discussion
In this paper, using four FCHL families, we have shown that the HL V73M variant is associated with elevated levels of total cholesterol and apoB. Neither VLDL nor LDL cholesterol levels were elevated, sug-gesting that an increased number of VLDL remnant particles is responsible for the increase in cholesterol levels. Although it has previously been shown that allelic variation in the HL gene accounts for 25% of the variability in HDL cholesterol [38], no effect of HL V73M was found on HDL levels which may be masked by additional modifying genes present within these FCHL families.
Hepatic lipase is involved in the hydrolysis of triglyc-erides in IDL and HDL, as well as phospholipids in HDL. In addition, HL can facilitate chylomicron rem-nant uptake independently of lipolysis by acting as a bridging protein [39]. Individuals with HL deficiency may present with features like accumulation of triglyce-ride-rich lipoproteins as well as b-VLDL. In addition, patients have increased plasma apoB concentrations [40]. Recently, an association was found between a promotor variation and hepatic lipase activity [41]. Although no specific association with hyperlipidemia and the V73M variant was found in a previous study using patients with classical hyperlipoproteinemias, i.e. type I, III, IV and V hyperlipoproteinemia [33], our study revealed an increased frequency of HL V73M among patients with FCHL (type IIB hyperlipo-proteinemia) compared with control subjects. Similar results were found by Gehrisch et al. [25]. The amino acid at position 73 is not conserved in other lipases in human or other species but a mild effect on enzyme activity can not be excluded [33]. Functional studies supporting this hypothesis have not been performed so far.
The present study revealed that the LPL N291S mutation is significantly associated with a negative K value, indicating predominance of heavy, dense LDL particles. A previous segregation analysis in a large sample of Dutch FCHL families (including the families used in our study) [14] had already shown that the LDL subfraction profile can be used as an additional diag-nostic marker [4,14]. However, this association seems to be restricted to FCHL because recently, a study focus-ing on the identification of genetic loci underlyfocus-ing the small dense LDL particle phenotype in CAD families excluded the LPL gene as a candidate gene locus [42]. Recently, the HL gene has been excluded as a major candidate gene for FCHL using linkage analysis in Finnish families [43]. Due to non-affected carriers, the HL V73M variant was also not stringently linked with the expression of FCHL in our families. This suggests that the HL gene could be excluded as the principal genetic factor for FCHL. However, counting affected
individuals for being carrier or non-carrier of the HL V73M or LPL N291S variant revealed that almost all affected individuals in these four families were carrier of either one or both of these two variants. We found a similar result (OR 3.2, CI 1.6 – 6.6) in all FCHL families when we combined the LPL-93 GT/D9N, LPL N291S and HL V73M variants [26,37]. Together with the finding that carriers of these mutations have signifi-cantly higher lipid levels then their noncarrier relatives this strongly suggest that the phenotypic expression of FCHL within our families cannot be explained by one major gene but is due to a more complex genetic mode of inheritance. Because not all carriers of mutations in the LPL and HL genes have a FCHL phenotype our results suggest that these genes are more likely modify-ing than the major genes. This fits into the ‘double hit’ hypothesis first suggested by Kwiterovitz [44], i.e. a combination of mutations in two different genes pro-duces a full expression of the FCHL phenotype. How-ever, further investigation will be needed to explain these findings for the HL gene within our FCHL families.
All together, our study showed an association be-tween the V73M mutation in the HL gene and elevated cholesterol and apoB levels. The inheritance patterns of FCHL within our families suggest the involvement of multiple loci. Our results also indicate that both the LPL and HL gene are among the modifier genes of the FCHL phenotype but additional genes have to be identified to fully explain the observed inheritance pattern.
Acknowledgements
We are grateful to Janine Vogelaar (Nijmegen) for help in collecting blood samples from family members and for technical assistance in lipoprotein analysis. The authors acknowledge Dr P. de Knijff for his helpful discussions. This research was supported by the Praeventiefonds (project 28-2230) and by the Nether-lands Heart Foundation (NHS 92.056).
References
[1] Goldstein JL, Schrott HG, Hazzard WR, Bierman EL, Motulsky AG. Hyperlipidemia in coronary heart disease. II: genetic analy-sis of lipid levels in 176 families land delineation of a new inherited disorder, combined hyperlipidemia. J Clin Invest 1973;52:1544 – 68.
[2] Rose HG, Kranz P, Weinstock M, Juliano J, Haft JI. Inheri-tance of combined hyperlipoproteinemia: evidence for a new lipoprotein phenotype. Am J Med 1973;54:148 – 60.
M.J.V.Hoffer et al./Atherosclerosis151 (2000) 443 – 450 449
[4] Austin MA, Brunzell JD, Fitch WL, Krauss RM. Inheritance of low density lipoprotein subclass patterns in familial combined hyperlipidemia. Arteriosclerosis 1990;10:520 – 30.
[5] Brunzell JD, Albers JJ, Chait A, Grundy SM, Groszek E, McDonald GB. Plasma lipoproteins in familial combined hyper-lipidemia and monogenic familial hypertriglyceridemia. J Lipid Res 1983;24:147 – 55.
[6] Stalenhoef AFH, Demacker PNM, Lutterman JA, van ’t Laar A. Plasma lipoproteins, apolipoproteins, and triglyceride metabolism in familial hypertriglyceridemia. Arteriosclerosis 1986;6:387 – 94.
[7] Chait A, Albers JJ, Brunzell JD. Very low density lipoprotein overproduction in genetic forms of hypertriglyceridaemia. Eur J Clin Invest 1980;10:17 – 22.
[8] Sane T, Nikkila EA. Very low density lipoprotein triglyceride metabolism in relatives of hypertriglyceridemic probands: evi-dence for genetic control of triglyceride removal. Arteriosclerosis 1988;8:217 – 26.
[9] Venkatesan S, Cullen P, Pacy P, Halliday D, Scott J. Stable isotopes show a direct relation between VLDL apoB overpro-duction and serum triglyceride levels and indicate a metabolically and biochemically coherent basis for familial combined hyper-lipidemia. Arterioscler Thromb 1993;13:1110 – 8.
[10] Kissebah AH, Alfarsi S, Adams PW. Integrated regulation of very low density lipoprotein triglyceride and apolipoprotein-B kinetics in man: normolipemic subjects, familial hypertriglyce-ridemia and familial combined hyperlipidemia. Metabolism 1981;30:856 – 68.
[11] Cullen P, Farren B, Scott J, Farrall M. Complex segregation analysis provides evidence for a major gene acting on serum triglyceride levels in 55 British families with familial combined hyperlipidemia. Arterioscler Thromb 1994;14:1233 – 49. [12] Jarvik GP, Brunzell JD, Austin MA, Krauss RM, Motulsky AG,
Wijsman E. Genetic predictors of FCHL in four large pedigrees: influence of apoB level major locus predicted genotype and LDL subclass phenotype. Arterioscler Thromb 1994;14:1687 – 94. [13] Jarvik GP, Beaty TH, Gallagher PR, Coates PM, Cortner JA.
Genotype at a major locus with large effects on apolipoprotein B levels predicts familial combined hyperlipidemia. Genet Epi-demiol 1993;10:257 – 70.
[14] Bredie SJ, Kiemeney LA, de Haan AF, Demacker PN, Stalen-hoef AF. Inherited susceptibility determines the distribution of dense low-density lipoprotein subfraction profiles in familial combined hyperlipidemia. Am J Hum Genet 1996;58:812 – 22. [15] Bredie SJH, van Drongelen J, Kiemeney LA, Demacker PNM,
Beaty TH, Stalenhoef AFH. Segregation analysis of plasma apolipoprotein B levels in familial combined hyperlipidemia. Arterioscler Thromb Vasc Biol 1997;17:834 – 40.
[16] Wojciechowski AP, Farrall M, Cullen P, Wilson TM, Bayliss JD, Farren B, Griffin BA, Caslake MJ, Packard CJ, Shepherd J, et al. Familial combined hyperlipidaemia linked to the apolipo-protein AI-CII-AIV gene cluster on chromosome 11q23 – q24. Nature 1991;349:161 – 4.
[17] Dallinga Thie GM, Bu XD, Trip MV, Rotter JI, Lusis AJ, DeBruin TWA. Apolipoprotein A-I/C-III/A-IV gene cluster in familial combined hyperlipidemia: Effects on LDL-cholesterol and apolipoproteins B and C-III. J Lipid Res 1996;37:136 – 47. [18] Allayee H, Aouizerat BE, Cantor RM, Dallinga Thie GM,
Krauss RM, Lanning CD, Rotter JI, Lusis AJ, De Bruin TWA Families with familial combined hyperlipidemia and families enriched for coronary artery disease share genetic determinants for the atherogenic lipoprotein phenotype, Am J Hum Genet 1998; 63.
[19] Wijsman EM, Motulsky AG, Guo S, Yang M, Austin MA, Brunzell JD, Deep S. Evidence against linkage of familial com-bined hyperlipidemia to the AI-CIII-AIV gene complex. Circula-tion 1992;86:I420.
[20] Marcil M, Boucher B, Gagne´ E, Davignon J, Hayden M, Genest J Jr. Lack of association of the apolipoprotein A-I-C-III-A-IV gene XmnI and SstI polymorphisms and of the lipoprotein lipase gene mutations in familiar combined hyperlipoproteinemia in French Canadian subjects. J Lipid Res 1996;37:309 – 19. [21] Mailly F, Tugrul Y, Reymer PWA, Bruin T, Seed M,
Groene-meyer BF, et al. A common variant in the gene for lipoprotein lipase (Asp9- -\Asn): functional implications and prevalence in normal and hyperlipidemic subjects. Arterioscler Thromb 1995;15:468 – 78.
[22] Reymer PWA, Groenemeyer BE, Gagne E, Miao L, Appelman EEG, Seidel JC, et al. A frequently occurring mutation in the lipoprotein lipase gene (Asn291Ser) contributes to the expression of familial combined hyperlipidemia. Hum Mol Genet 1995;4:1543 – 9.
[23] Pihlajamaki J, Rissanen J, Heikkinen S, Karjalainen L, Laakso M. Codon 54 polymorphism of the human intestinal fatty acid binding protein two is associated with dyslipidemias but not with insulin resistance in patients with familial combined hyperlipi-demia. Arterioscler Thromb Vasc Biol 1997;17:1039 – 44. [24] Deeb SS, Nevin DN, Iwasaki L, Brunzell JD Two novel
apolipo-protein A-IV variants in individuals with familial combined hyperlipidemia and diminished levels of lipoprotein lipase activ-ity, Hum Mut 1996; 319 – 325.
[25] Gehrisch S, Tesche R, Kostka H, Julius U, Jaross W. Point mutations in the hepatic triglyceride lipase (HTGL) gene in familial combined hyperlipidemia. Circulation 1995;92:493. [26] Hoffer MJV, Bredie SJH, Boomsma DI, Reymer PWA,
Kastelein JJP, De Knijff P, Demacker PNM, Stalenhoef AFH, Havekes LM, Frants RR. The lipoprotein lipase (Asn291Ser) mutation is associated with elevated lipid levels in families with familial combined hyperlipidaemia. Atherosclerosis 1996;119:159 – 67.
[27] Assmann G, Schulte H. Results and conclusions of the prospec-tive cardiovascular munster (PROCAM) study. In: Lipid Metabolism Disorders and Cardiovascular Disease. Munich: MMV-Medizin, 1993:19 – 98.
[28] Smit M, De Knijff P, Rosseneu M, Bury J, Klasen E, Frants R, Havekes L. Apolipoprotein E polymorphism in The Netherlands and its effect on plasma lipid and apolipoprotein levels. Hum Genet 1988;80:287 – 92.
[29] Demacker PNM, Vos Janssen HE, Jansen AP, van ’t Laar A. Evaluation of the dual-precipitation method by comparison with the ultracentrifugation method for measurement of lipoproteins in serum. Clin Chem 1977;23:1238 – 42.
[30] Demacker PNM, Hijmans AG, Vos Janssen HE, van ‘t Laar A, Jansen AP. A study of the use of polyethylene glycol in estimat-ing cholesterol in high-density lipoprotein. Clin Chem 1980;26:1775 – 9.
[31] Bredie SJH, De Bruin TWA, Demacker PNM, Kastelein JJP, Stalenhoef AFH. Comparison of Gemfibrozil versus simvastatin in familial combined hyperlipidemia and effect on apolipo-protein-B-containing lipoproteins, low-density lipoprotein sub-fractions profile, and low-density lipoprotein oxidizability. Am J Cardiol 1995;75:348 – 53.
[32] Miller SA, Dykes DD, Polesky HF. A simple salting out proce-dure for extracting DNA from nucleated cells. Nucleic Acids Res 1988;16:1215.
[33] Hegele RA, Tu L, Connelly PW. Human hepatic lipase muta-tions and polymorphisms. Hum Mut 1992;1:320 – 4.
[34] Lange K, Weeks D, Boehnke M. Programs for pedigree analysis: MENDEL, FISHER, and dGENE. Genet Epidemiol 1988;5:471 – 2.
[36] Westendorp RG, Langermans JAM, Huizinga TWJ, Elouali AH, Verweij CL, Boomsma DI, Vandenbrouke JP. Genetic influence on cytokine production and fatal meningococcal disease. Lancet 1997;349:170 – 3.
[37] Hoffer MJV, Bredie SJH, Snieder H, Reymer PW, Demacker PNM, Havekes LM, et al. Gender-related association between the -93TG/D9N haplotype of the lipoprotein lipase gene and elevated lipid levels in familial combined hyperlipidemia. Atherosclerosis 1998;138:91.
[38] Cohen JC, Wang Z, Grundy SM, Stoesz MR, Guerra R. Varia-tion at the hepatic lipase and apolipoprotein AI/CIII/AIV loci is a major cause of genetically determined variation in plasma HDL cholesterol levels. J Clin Invest 1994;94:2377 – 84.
[39] Ji ZS, Lauer SJ, Fazio S, Bensadoun A, Taylor JM, Mahley RW. Enhanced binding and uptake of remnant lipoproteins by hepatic lipase-secreting cells in culture. J Biol Chem 1997;269:13429 – 36. [40] Demant T, Carlson LA, Holmquist L, Karpe F, Nilsson-Ehle P, Packard CJ, Shepherd J. Lipoprotein metabolism in hepatic
lipase deficiency: studies on the turnover of apolipoprotein B and on the effect of hepatic lipase on high density lipoprotein. J Lipid Res 1988;29:1603 – 11.
[41] Tahvanainen E, Syvanne M, Heikki Frick M, Murtomaki-Repo S, Antikainen M, Kesaniemi YA, et al. Association of variation in hepatic lipase activity with promotor variation in the hepatic lipase gene. J Clin Invest 1998;101:956 – 60.
[42] Rotter JI, Bu XD, Cantor RM, Warden CH, Brown J, Gray RJ, et al. Multilocus genetic determinants of LDL particle size in coronary artery disease families. Am J Hum Genet 1996;58:585 – 94.
[43] Pajukanta P, Porkka KVK, Antikainene M, Taskinen MR, Perola M, Murtomaki-Repo S, et al. No evidence of linkage between familial combined hyperlipidemia and genes encoding lipolytic enzymes in Finnish families. Arterioscler Thromb Vasc Biol 1997;17:841 – 50.
[44] Kwiterovich PO. Genetics and molecular biology of familial combined hyperlipidemia. Curr Opin Lipidol 1993;4:133 – 43.