REFRAT
SINDROM MYELODISPLASIA
Disusun oleh :
Hastin Mutiara Surga G0007084
Yessi Perlitasari G0007173
Denni Tri Hananto G0007190
Pembimbing
dr. Suradi Maryono, SpPD, KHOM-FINASIM
KEPANITERAAN KLINIK SMF ILMU PENYAKIT DALAM FAKULTAS KEDOKTERAN UNS/RSUD DR MOEWARDI
S U R A K A R T A 2011
BAB I PENDAHULUAN
Sindrom myelodisplasia (MDS) adalah gangguan sumsum tulang, ditandai dengan hematopoesis yang tidak efektif, berbagai tingkat sitopenia serta peningkatan risiko leukemia akut (Steensma, 2003). MDS mewakili spektrum gangguan neoplastik sel induk klonal yang ditandai oleh kegagalan sumsum tulang dengan sitopeni, dan persentase leukemia berkisar dari kurang dari 5% sampai 19% dan terjadi pada populasi lanjut usia. Kejadian MDS dalam data yang baru-baru ini diterbitkan oleh Surveillance, Epidemiology, and End Results (SEER) meningkat dari kurang dari 5 per 100.000 pasien di bawah usia 60 menjadi 36,2 per 100.000 pada pasien lebih dari 80 tahun. Dengan rata-rata usia diagnosis 76 tahun. Secara umum, pria dan kulit putih memiliki insiden yang lebih tinggi dari penyakit ini (Rami, 2009).
Seperti halnya penyakit kanker pada umumnya, penyebab MDS yang pasti belum diketahui. Studi epidemiologi menunjukkan MDS dihubungkan dengan paparan bahan kimia seperti benzen, halogenated hydrocarbon, hydrogen peroksida serta paparan radiasi. Beberapa hal dapat mendasari patologi fenotip dan biologi pada penyakit ini, termasuk kelainan kromosom dan genetik, perubahan epigenetic serta dearrangements sitokin dan sistem imun (Epling-Burnette, 2009). Onkogenesis pada MDS bersifat multistep dimana proses akumulasi perubahan genetik yang pada akhirnya menuju suatu neoplasma ganas setelah sebelumnya melewati fase pre maligna. Pada fase awal, sel induk normal dan abnormal sama-sama berfungsi, tetapi pada fase selanjutnya klon ganas lebih dominan. Ciri dari penyakit ini pada usia dini adalah apoptosis yang dipercepat pada sel induk hematopoietik disertai dengan peningkatan kompensasi dalam proliferasi (Steensma, 2003). .
Setelah diagnosis dibuat, hematologi / onkologi medis mencoba untuk mengklasifikasikan pasien ke kategori untuk memprediksi prognosis dan memutuskan strategi pengobatan yang akan dilakukan. Tujuan pengobatan pada
kelompok risiko rendah (kelompok dengan prognosis yang lebih baik) adalah untuk meningkatkan kualitas hidup dan mengurangi kebutuhan untuk transfusi, yang dapat dicapai melalui pilihan yang berbeda, termasuk faktor pertumbuhan
erythropoietic, lenalidomide, dan agen hypomethylating. Pada kelompok risiko tinggi (kelompok dengan prognosis buruk), tujuan pengobatan adalah untuk meningkatkan kelangsungan hidup dan memperlambat perkembangan penyakit. Pilihan pengobatan bagi kelompok ini termasuk transplantasi stem cell alogenik pada pasien yang memenuhi kriteria dan penggunaan agen hypomethylating (Rami, 2009). Meskipun tersedia berbagai pengobatan alternatif yang dapat dilakukan, sebagian besar pasien meninggal karena komplikasi dari penyakit atau transformasi menjadi leukemia myeloid akut (AML).
BAB II
TINJAUAN PUSTAKA
A. DEFINISI
Sindrom myelodisplasia atau myelodisplasia syndrome (MDS) adalah kelainan neoplastik hemopoetik klonal yang disebabkan oleh transformasi ganas sel induk myeloid sehingga menimbulkan gangguan maturasi dan diferensiasi seri myeloid, eritriod atau megakariosit, yang ditandai dengan hematopesis inefektif, sitopenia pada darah tepi dan sebagian akan mengalami transformasi menjadi leukemia myeloid akut (Rami, 2009).
B. ETIOLOGI
Etiologi MDS tidak diketahui secara pasti, namun dapat terjadi karena bertambahnya usia, perubahan genetik yang diwariskan atau disebabkan oleh paparan zat berbahaya. Faktor risiko meliputi pemaparan terhadap pelarut benzena atau bahan lainnya, halogenated hydrocarbon, tembakau dan asap rokok serta penurunan sistem imun. Kemoterapi dan radiasi yang berhubungan dengan terapi juga dapat terkait dengan MDS (Steensma, 2007).
1. Penuaan
Sebagaimana disebutkan di atas, penuaan tampaknya menjadi faktor risiko terpenting dalam perkembangan MDS karena risiko terjadinya mutasi meningkat sebanding dengan usia.
2. Kimia
Paparan tingkat tinggi dari beberapa bahan kimia lingkungan, terutama produk benzena dan minyak bumi, terkait dengan perkembangan MDS.
3. Rokok
Paparan bahan kimia dalam asap tembakau / rokok dapat meningkatkan risiko perkembangan MDS.
4. Sitotoksik kemoterapi
Pasien yang sebelumnya mengalami pengobatan kanker atau kondisi lain dengan kemoterapi, akan meningkatkan risiko untuk terjadinya MDS sekunder atau terkait pengobatan. Ini mewakili kurang dari 10 persen dari semua kasus MDS. Sekunder MDS dikaitkan dengan mutasi yang berbeda yang terjadi pada MDS spontan dan memiliki prognosis yang lebih buruk. Waktu antara paparan obat dan terjadinya MDS dapat 2-3 tahun hingga lebih dari 10 tahun.
5. Radiasi
Terapi radiasi sebelumnya, atau paparan radiasi lingkungan tingkat tinggi dikaitkan dengan peningkatan risiko MDS. Dalam beberapa kasus mungkin tidak terlihat sampai 40 tahun setelah paparan.
6. Kelainan Bawaan
Beberapa kelainan bawaan seperti sindrom Bloom, Down Syndrome, anemia Fanconi dan neurofibromatosis memiliki risiko lebih untuk terjadinya mutasi yang menyebabkan kanker atau MDS (Leukaemia Fondation, 2009)
C. PREVALENSI
Perkiraan terbaru dari American Cancer Society (2009), MDS di Amerika Serikat berkisar 12.000 kasus baru setiap tahun. Jumlah kasus baru nampaknya akan meningkat karena peningkatan usia rata-rata populasi. Sekitar 80% sampai 90% dari semua pasien dengan MDS umumnya lebih dari 60 tahun.
Sedangkan insidens MDS dalam data yang baru-baru ini diterbitkan oleh Surveillance, Epidemiology, and End Results (SEER) meningkat dari kurang dari 5 per 100.000 pasien di bawah usia 60 menjadi 36,2 per 100.000 pada pasien lebih dari 80 tahun. Dengan rata-rata usia diagnosis 76 tahun. Secara umum, pria dan kulit putih memiliki insiden yang lebih tinggi dari penyakit ini (Rami, 2009).
D. PATOFISIOLOGI
Penyebab MDS masih belum dikehui dengan pasti, dan sulit dipisahkan dari penyebab leukemia dan penyakit mieloproliferatif lainnya. Diajukan sebuah hipotesis bahwa pengaruh factor lingkungan, kelainan genetic dan interaksi sel menimbulkan mutasi pada tingkat selinduk sehingga menimbulkan ketidakseimbangan proses proliferasi dan diferensiasi. Variasi perubahan prose situ akan menyebabkan transformasi kea rah leukemia akut, MDS atau penyakit myeloproliferatif (MPD) (Uwe, 2007).
Pada MDS terjasi ketidakserasian antara proliferasi dengan diferensiasi, dimana daya proliferadi masih cukup tetapi terjadi gangguan diferensiasi atau maturasi sehingga terjasi hemopoesis inefektif, dengan kematian premature sel (eritroid, myeloid, megakariosit) dalam sumsum tulang sebelum sempat dilepaskan ke darah tepi. Hal ini berakibat terjadinya sumsum tulang hiperseluler, tetapi terjadi sitopenia pada darah tepi (Uwe, 2007)..
Bagan 1. Faktor-faktor yang berkontribusi terhadap terjadinya MDA (Uwe, 2007).
E. GEJALA KLINIS
Gejala MDS sering tidak jelas dan spesifik, dan diagnosis sering dibuat selama pemeriksaan untuk anemia, trombositopenia, atau neutropenia pada pemeriksaan darah rutin. Jika tampak tanda-tanda dan gejala, biasanya tergantung pada jenis sel yang terpengaruh.
Ketika eritrosit terpengaruh (situasi yang paling umum), pasien datang dengan tanda-tanda anemia, termasuk pucat, konjungtiva anemis, takikardi, hipotensi, kelelahan, sakit kepala, dan intoleransi latihan, atau dengan tanda dan gejala memburuknya kondisi atau penyakit yang mendasari seperti angina pectoris, gagal jantung, atau emfisema.
Ketika trombosit yang terpengaruh, kurang dari 20% dari pasien datang dengan gejala trombositopenia terisolasi sebagai perdarahan kecil (misalnya, perdarahan mukosa, petechie, mudah memar, epistaksis) atau perdarahan besar (misalnya, perdarahan gastrointestinal, perdarahan intrakranial).
Ketika neutrofil yang terpengaruh, terjadi neutropenia terisolasi misalnya infeksi bakteri yang sering terjadi pada sistem organ yang berbeda. Infeksi merupakan keluhan utama dari 10% kasus dan penyebab kematian dari 21% kasus.
Splenomegali dan limfadenopati jarang terjadi pada MDS. Jika terdeteksi, maka harus curiga terhadap adanya neoplasma myeloproliferatif atau limfoproliferatif (Barzi, 2010).
F. DIAGNOSIS
Langkah diagnosis MDS adalah sebagai berikut :
1. Diagnosis MDS sangat dicurigai apabila dijumpai gejala klinik yang sesuai, terutama pada orang tua, yang disertai sitopenia (anemia, leukopenia, trombositopenia) persisten atau monositosis yang tidak dapat diterangkan.
2. Kemudian dilakukan pemeriksaan teliti terhadap apusan darah tepi dan sumsum tulang untuk mencari tanda-tanda displastik. Abnormalitas morfologi pada penderita MDS dapat dilihat pada Tabel 1.
Tabel 1. Abnormalitas Morfologi pada Penderita MDS (List, 2009)
Jenis sel Apus darah tepi Sumsum tulang
Eritroid Ovalomakrosit Eliptosit Akantosit Stomatosit Teardrops Normoblas Basophilic stippling Howel-Jolly bodies Eritropoiesis megaloblastoid Nuclear budding Ringed sideroblast Internuclear bridging Karioreksis Fragmen nuclei Vakuolisasi sitoplasma Multinuklearitas
Mieloid Anomali Pseudo-Pelger- Huet Hipogranulasi Nuclear sticks Hipersegmentasi Ring-shaped nuclei Auer rods Defektif granulasi
Hambatan maturasi pada tingkat mielosit
Peningkatan bentuk monositoid
Lokasi abnormal prekursor imatur
Megakariosit Giant platelet
Trombosit hipogranuler/ Agranuler
Mikromegakariosit Hipogranulasi
Nukleus kecil multipel 3. Jika dijumpai tanda displastik pada satu alau lebih jenis sel, penyebab
dysplasia di luar MDS harus disingkirkan (dengan anamnesis, pemeriksaan klinik, laboratorium atau pemeriksaan lain). Penyebab dysplasia diluar MDS antara lain: defisiensi vitamin B12, defisiensi folat, infeksi virus seperti HIV, pemakaian antibiotika tertentu, agen kemoterapi, etanol, benzene, atau timah hitam. Apabila penyebab-penyebab ini telah dapat disingkirkan, diagnosis MDA sudah dapat ditetapkan.
4. Langkah selanjutnya adalah melakukan klasifikasi berdasarkan FAB atau WHO.
5. Jika fasilitas tersedia, pemeriksaan sitogenetik dikerjakan untuk menilai prognosis. Pemeriksaan sitokimia, imunofenotiping, imunokimia, pemeriksaan onkogen dan kultur jaringan dapat membantu dignosis, tetapi secara rutin tidak selalu diperlukan.
Sebenarnya untuk diagnosis SDM perlu dibantu dengan pemeriksaan pembiakan sel-sel sumsum tulang dan pemeriksaan sitogenetik. Sitogenetik sumsum tulang dapat memberikan informasi prognosis dan adanya abnormalitas kromosom yang merupakan kunci untuk membedakan MDS primer dan sekunder. Kromosom abnormal sumsum tulang ditemukan pada 30 – 50 % pasien MDS de novo. Berbagai kelainan sitogenetik pada MDS termasuk delesi, trisomi, monosomi dan anomali struktur.
Bagan 3. Algoritma Diagnosis MDS menurut kriteria WHO (Barbara, 2004) Has there been exposure to cytotoxic
drugs or irradiation ?
Are there 5-19% blast cells in the blood or 10-19% blast cell in the bone
marrow or Auer rods ?
Are there no more than 5% blast cells in the blood and 5-9% in the bone
marror ?
Is there an isolates 5q- ?
Is there multiliniage dysplasia ? N O N O N O N O N O RA or RARS YE S YE S YE S YE S YE S Therapy Related MDS RAEB-II RAEB-I RCMD or RCMD-RS 5q - syndrome
F. KLASIFIKASI
FAB membuat klasifikasi khusus untuk MDS yang diterima secara luas sampai saat ini. FAB membagi MDS menjadi 5 kategori berdasarkan jumlah blast dalam darah tepid an sumsum tulang, jumlah monosit dalam darah tepi, serta jumlah ringed sideroblast dalam sumsum tulang.
1. Refractory Anemia (RA)
Pada RA dijumpai sitopenia, paling sedikit pada satu turunan sel (cell lineage), pada umumnya pada seri eritroid. Sumsum tulang hiperseluler atau normoseluler dengan perubahan displastik terutama pada sistem eritroid, system granulosit dan megakariosit mengalami perubahan displastik dalam derajad yang lebih ringan.
Blast dalam darah tepi < 1 % dan dalam sumsum tulang < 5%.
2. Refractory Anemia with Ringed Sideroblast (RARS)
Pada RARS dijumpai sitopenia (hampir selalu disertai anemi), perubahan displastik, jumlah blast seperti dapa RA. Ring
sideroblast dijumpai > 15% dari sel eritroid berinti dalam sumsum
tulang.
3. Refractory Anemia with Excessive Blast (RAEB)
Pada RAEB dijumpai sitopenia dari dua atau lebih turunan sel pada darah tepi. Perubahan displastik pada ketiga lineage dalam sumsum tulang lebih nyata. Blast darah tepi < 5%, dan dalam sumsum tulang antara 5-20 %.
4. RAEB in Transformation to Leukemia (RAEBt)
Pada RAEBt gambaran hematologi sama dengan RAEB, tetapi blast darah tepi > 5% atau blast dalam sumsum tulang 21-30% atau adanya auer rod pada sel blast.
5. Chronic Myelo-Monocytic Leukemia (CMML)
Pada CMML dijumpai monositosis pada darah tepi (monosit > 1.109 per liter). Dalam darah tepi < 5%, sedangkan dalam sumsum
Tabel 2. Kelainan Darah Tepi dan Sumsum Tulang pada MDS Menurut Klasifikasi FAB (Brain, 2003)
Jenis MDS Darah Tepi Sumsum Tulang
Refractory Anemia (RA) Anemia <1% blasts
monocytes < 1.109 /l
< 5% blast
< 15% ring sideroblast of erythroblast
Refractory Anemia with Ringed Sideroblast (RARS) Anemia <1% blasts monocytes < 1.109 /l < 5% blast > 15% ring sideroblast of erythroblast
Refractory Anemia with Excess Blast
Anemia >1% blasts
monocytes < 1.109 /l
≥ 5% blast
Refractory Anemia with Excessive Blast in Transformation to Leukemia (RAEB-t) Anemia >5% blasts ≥ 20% blast Chronic MyeloMonocytic Leukemia (CMML) Monocytes < 1.109 /l Granulocytes often increased <5% blasts Blast up to 20% Promonocytes often increased
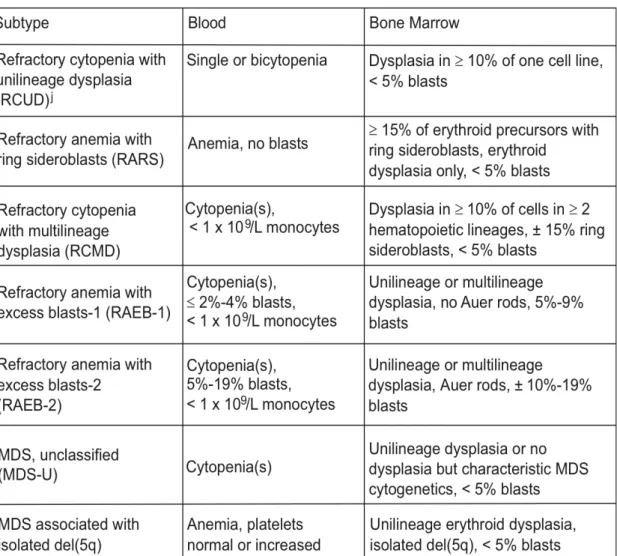
Pada tahun 2001 WHO membuat klasifikasi yang lebih detail yang memunyai hubungan yang lebih baik dengan prognosis. Penggolongan yang diusulkan WHO untuk MDS adalah :
1. Refractory Anemia (RA)
2. Refractory Anemia with Ringed Sideroblast (RARS)
3. Refractory Cytopenia with Multilineage Dysplasia (RCMD)
4. Refractory Anemia with Excessive Blast-1 (RAEB-1) 5. Refractory Anemia with Excessive Blast-2 (RAEB-2)
6. Myelodiaplasiasyndrome unclassified
Tabel 3. Kelainan Darah Tepi dan Sumsum Tulang pada MDS Menurut Klasifikasi WHO 2001 (Brunning et al, 2001)
Jenis MDS Darah Tepi Sumsum Tulang
Refractory Anemia (RA) Anemia
No or rare blast
Erythroid dysplasia only < 5% blast
< 5% ring sideroblast
Refractory Anemia with Ringed Sideroblast
(RARS)
Anemia No blast
≥ 5% ring sideroblast Erythroid dysplasia only < 5% blast Refractory Cytopenia with Multilineage Dysplasia (RCMD) Cytopenias (bicyto-penia/pancytopenia) No or rare blast No Auer rods Dysplasia in > 10% of the cell in two or more myeloid cell lines
≥ 5% ring sideroblast < 5% blast
No Auer rods
Refractory Anemia with Excessive Blast-1 (RAEB-1) Cytopenias <5% blasts No Auer rods < 1.109 monocytes Unliniage or multilineage dysplasia 5-9% blast No Auer rods
Refractory Anemia with Excessive Blast-2 (RAEB-2) Cytopenias 5-19% blast Auer rods ± Unliniage or multilineage dysplasia 10-19% blast Auer rods ± Myelodiaplasiasyndrome unclassified (MDS-U) Cytopenias No or rare blast No Auer rods
Unlineage dysplasia : one lyeloid cell line
< 5% blast No Auer rods MDS associated with del-5q syndrome Anemia Usually normal or increased platelet <5% blast Normal to increased megakaryocytes with hypolobulated nuclei <5% blast
Isolated del(5q) cyto-genetic abnormality No Auer rods
Tabel 5. Kelainan Darah Tepi dan Sumsum Tulang pada MDS Menurut Klasifikasi WHO 2008 (Peter L, 2011).
Perbedaan Klasifikasi MDS Menurut FAB dan WHO
Sebetulnya tidak terdapat perbedaan yang bermakna berdasarkan klasifikasi FAB dan WHO, tetapi klasifikasi WHO dimodifikasi untuk menajamkan prognosis, yaitu :
1. WHO memasukkan refractory cytopenia sebagai salah satu kategori MDS karena pada kenyataannya ada kasus-kasus MDS tanpa disertai anemia sehingga tidak dapat dimasukkan ke dalam klasifikasi FAB
2. FAB memakai istilah RAEB dan RAEB-t, sedangkan WHO menggunakan istilah RAEB-1 dan RAEB-2 dengan titik pemilah jumlah blast yang sedikit berbeda
3. WHO memasukkan satu kategori baru MDS associated with del(5q), di klinik dikenal sebagai 5q-syndrome yang mempunyai prognosis yang sangat baik.
MDS seharusnya dibedakan dengan myeloproliferative disorder yang lain dan beberapa variasi dari SDM sekunder termasuk defisiensi nutrisi, proses infeksi, efek obat dan toxic exposures (Hoffbrand, 2005).
G. TATA LAKSANA
Beberapa regimen terapi telah digunakan pada pasien SDM, tetapi sebagian besar tidak efektif di dalam merubah perjalanan penyakitnya. Karena itu pengobatan pasien SDM tergantung dari usia, berat ringannya penyakit dan progresivitas penyakitnya. Berbagai macam regimen terapi telah dan sedang dicobakan pada penderita MDS namun sampai saat ini transplantasi sumsum tulang masil merupakan satu-satunya terapi yang memberikan kepastian hingga terapi simtomatik masih memegang peranan yang penting bagi pasien MDS.
Transplantasi Sumsum Tulang (Bone Marrow Transplatation)
Cangkok sumsum tulang alogenik merupakan pengobatan utama pada SDM terutama dengan usia < 30 tahun, dan merupakan terapi kuratif, tetapi masih merupakan pilihan < 5% dari pasien. Pada pasien MDS dengan prognosis jelek, transplantasi sumsum tulang merupakan satu-satunya pilihan yang memberikan harapan. Transplantasi stem sel autologus akhir-akhir ini mulai mendapatkan perhatian mengingat pada beberapa kasus MDS dapat dijumpai hemopoesis poliklonal yang normal setelah kemoterapi. Satu studi mendapatkan angka disease free survival > 30% setelah 2 tahun pasca transplantasi stem sel autologus (Asharianti, 2007).
Kemoterapi
Pilihan kemoterapi pada MDS bervariasi dari kemoterapi intensif sampai terapi sitostatika dosis rendah. Penggunaan kemoterapi pada MDS biasanya diberikan pada tipe RAEB, RAEB-t dan CMML. Sejak tahun 1968 pengobatan ARA-C dosis rendah yang diberikan pada pasien SDM dapat memberikan response rate antara 50 – 75 % dan respons ini tetap bertahan 2 – 14 bulan setelah pengobatan. Dosis ARA-C yang direkomendasikan adalah 20 mg/m2/hari secara drip atau 10 mg/m2/hari
secara subkutan setiap 12 jam selama 21 hari. Komplikasi akibat terapi ditemukan sangat tinggi 13-30% pada beberapa studi yang berbeda, bahkan pada studi lainnya survival didapatkan lebih pendek dibandingkan penderita yang tidak mendapatkan terapi (Asharianti, 2007).
GM-CSF atau G-CSF
Sitokin dan hematopoietic growth factor (HGF) memainkan peranan penting sebagai bagian dari terapi simtomatik menderita MDS, baik GM-CSF atau G-CSF. Pada pasien MDS yang mengalami pansitopeni dapat diberikan GM-CSF atau G-CSF untuk merangsang diferensiasi dari hematopoetic progenitor cells. GM-CSF diberikan dengan dosis 30–500 mcg/m2/hari atau G-CSF 50–1600 mcg/m2/hari (0,1–0,3
mcg/kgBB/hari/subkutan) selama 7–14 hari. Studi multisenter membuktikan bahwa pemberian GM-CSF dapat meningkatkan granulosit dan tidak terbukti dapat meningkatkan kadar hemoglobin dan trombosit. Terapi dengan eritropoetin dapat meningkatkan hemetokrit 25% penderita sehingga kebutuhan akan transfusi menjadi jauh berkurang (Asharianti, 2007).
Lain-lain
Piridoksin, androgen, danazol, asam retinoat dapat digunakan untuk pengobatan pasien SDM. Piridoksin dosis 200 mg/hari selama 2
bulan kadang-kadang dapat memberikan respon pada tipe RAEB walaupun sangat kecil. Danazol 600 mg/hari/oral dapat memberikan
response rate 21 – 33 % setelah 3 minggu pengobatan (Asharianti, 2007).
Strategi Terapi
Hoffbrand (2005) mengkategorikan MDS menjadi dua kelompok, terdiri atas :
1. Low Risk MDS, yaitu penderita dengan blast <5% dalam sumsum tulang. Low Risk MDS dapat dikelola secata konservatif, dengan transfusi sel darah merah atau trombosit dan pemberian antibiotika bila terjadi infeksi. Dapat juga diberikan eritropoetin atau growth
factor seperti G-CSF untuk mengatasi leukopenia. Pemberian obat
imunosupresif, seperti siklosporin dan ATG dapat dipertimbangkan 2. High Risk MDS, yaitu penderita dengan blast sumsum tulang 5% atau
lebih. Untuk High Risk MDS dapat dipertimbangkan pemberian kemoterapi, baik tunggal maupun intensif disamping terapi suportif. Pada pendetita kurang dari 50 tahun, stem cell transplantation merupakan satu-satunya pengobatan yang sapat menberikan kemungkinan kesembuhan. Untuk High Risk MDS dengan umur tua (65 tahun) dianjurkan hanya pemberian terapi suportif karena manfaat kemoterapi tidak sebanding dengan efek sampingnya.
H. PROGNOSIS
MDS adalah kumpulan beberapa sindrom dengang sifat biologis yang berbeda-beda sehingga prognosis MDS sangat bervariasi. Banyak faktor yang berperan dalam prognosis MDS, antara lain klasifikasi, umur, jenis kelamin, kadar jemoglobin, jumlah netrofil, jumlah trombosit, jumlah monosit, adanya sel muda (blast) dalam sirkulasi, perubahan displastik dari sumsum tulang, persentase sel blast dalam sumsum tulang, sitogenetik dan kultur dari sumsum tulang (Itzykson, 2011).
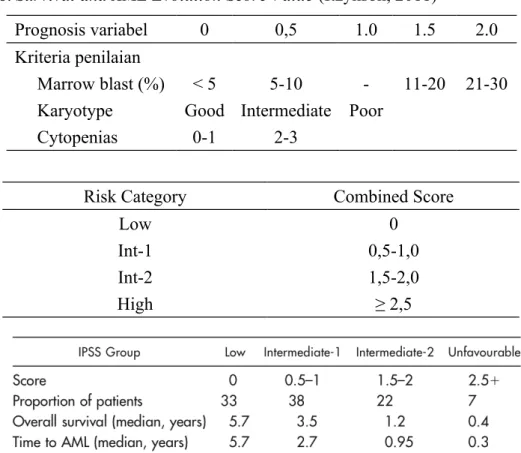
Sistem scoring menurut International Prognostic Scoring System menggunakan analisis multivariat dengan mengkombinasikan subklasifikasi dari sitogenetik, presentase sel blast sumsum tulang dan sitopenia sehingga model ini memunginkan penilaian prognosis lebih baik dibandingkan dengan beberapa sistem terdahulu (Itzykson, 2011).
Tabel 6. Survival and AML Evolution Score Value (Itzykson, 2011)
Prognosis variabel 0 0,5 1.0 1.5 2.0
Kriteria penilaian
Marrow blast (%) < 5 5-10 - 11-20 21-30 Karyotype Good Intermediate Poor
Cytopenias 0-1 2-3
Risk Category Combined Score
Low 0
Int-1 0,5-1,0
Int-2 1,5-2,0
BAB III KESIMPULAN
Myelodisplasia sindrom adalah suatu kelainan sumsum tulang yang berakibat pada ketidakefektifan hematopoesis, yang mana menimbulkan sitopenia serta cenderung berisiko pada akut leukimia. Gangguan ini diketahui dapat terjadi oleh sebab bertambahnya usia, perubahan genetik yang diwariskan atau disebabkan oleh paparan zat berbahaya. Gejala yang dapat ditimbulkan adalah berkaitan dengan kejadian sitopenia seperti anemia, bercak-bercak petechie atau perdarahan dari akibat rendahnya platelet serta infeksi. Sehingga pasien dengan kecurigaan gejala sindrom myelodisplasia ini perlu melakukan pemeriksaan darah tepi serta sitogenik. Penatalaksanaan yang dapat dilakukan adalah dengan pencangkokan sumsum tulang untuk usia di bawah 30 tahun, kemudian kemoterapi, serta pemberian GM-CSF atau G-CSF untuk perangsangan hematopoetic progenitor cells.
DAFTAR PUSTAKA
American Cancer Society. 2009. Myelodisplastic Syndrome Overview. Diunduh dari : www.cancer.org (2 September 2011).
Asharianti A. 2007. Sindrom Dismielopoetik. Dalam : Sudoyo AW, Setiyohadi B, Alwi I, Simadibrata MI (eds). Buku Ajar Ilmu Penyakit Dalam. Jilid 1. Edisi 4. Jakarta: Pusat Penerbitan Departemen Ilmu Penyakit Dalam Fakultas Kedokteran Universitas Indonesia, Hal: 710-2.
Attilio O and Magdalena B. 2009. MyelodiaplasiaSyndromes. Am J Clin Patho 132:290-305
Barbara B. 2004. The WHO Classification of the Myelodiaplasia Syndromes. Experimental Oncology (26) : 166-9.
Barzi A and Sekkeres MA. 2010. Myelodiaplasiasyndromes: A practical approach to diagnosis and treatment. Cleveland Clinical Journal of Medicines 77 (1):37-44.
Brain BJ. 2003. Leukaemia Diagnosis. 3rd edition. Oxford: Blackwell Publishing.
P: 117.
Brunning RD, Bennet JM, Flandrin G, et al. 2001. Myelodiaplasiasyndromes. IARC Press : 63-7.
Epling-Burnette PK and List AF. 2009. Advancements in the molecular pathogen- esis of myelodiaplasiasyndrome. Curr Opin Hematol (16):70-76.
Hoffbrand AV. 2001. Sindrom Myelodisplasia. Dalam : Moss P and Pettit JE (eds). At a Glance Hematologi. Edisi 2. Jakarta: Erlangga. Hal: 79-81.
Hoffbrand AV. 2005. Myelodisplasia Sindrom. Dalam : Pettit JE and Moss P (eds). Kapita Selekta Hematologi. Edisi 4. Jakarta: EGC. Hal: 139-41.
Itzykson R, Ades L and Fenaux P. 2011. Biology and Prognostic Factors of Myelodysplastic Syndrome. American Society of Clinical Oncology :251-5. Leukaemia Foundation. 2009. Understanding Myelodisplastic Syndrome (MDS) :
A guide for patiens and families. Diunduh dari : www.leukaemia.org.au ( 29 Agustus 2011)
List AF and Doll DC. 2009. The MyelodiaplasiaSyndromes. In : Lee GR, Foerster J, Lukens J, Parakevas F, Greer JP, Rodgers GM (eds). Wintrobe’s Clinical Hematology, tenth ed. Vol 2. Philadelphia : Lippincott Williams and Wilkins. Pp : 2320 – 33.
Peter L, Eyal A, John M et al. 2011. MyelodiaplasiaSyndromes : Clinical Practice Guidelines in Oncology. Journal of the National Comprehensive Cancer Network (9) 1 : 30-56.
Rami SK and Alan F. 2011. Management of MyelodiaplasiaSyndromes: Starting a New Decade. American Society of Clinical Oncology : 262-8.
Rami SK and John MB. 2009. What Is “WHO” ?: MyelodiaplasiaSyndrome Classification and Prognosis. American Society of Clinical Oncology:413-9. Steensma DP. 2007. The spectrum of molecular aberrations in
myelodiaplasiasyndromes : in the shadow of acute myeloid leukemia. Haematologica (92):723-727.
Steensma DP and Tefferi A. 2003. The myelodiaplasiasyndrome(s) : a perspective and review highlighting current controversies. Leuk Res (27):95–120.
Uwe P, Michelle M and Gerhard E. 2007. The pathogenesis of myelodiaplasiasyndromes (MDS). Cancer Treatment Reviews (33) : S53– S58.