Case Report Session
STROKE-LIKE SYNDROME
Oleh:
Adriyan Sikumalay 110312056
Airena Niza Nugroho 1110313012
Azalia Karina 1310311035
Pembimbing: dr. Amilus Ismail, Sp. S dr. Edinirwan, Sp. S, M. Biomed
BAGIAN ILMU PENYAKIT SYARAF RSUD ACHMAD MOCHTAR BUKITTINGGI FAKULTAS KEDOKTERAN UNIVERSITAS ANDALAS
BAB 1 PENDAHULUAN
1.1 Latar Belakang
Mitokondria merupakan salah satu komponen di dalam sel yang penting untuk berbagai proses seluler. Salah satu mekanisme penting yang dilakukan mitokondria adalah fosforilasi oksidatif. Fosforilasi oksidatif ini bertujuan untukk memproduksi energi seluler dalam bentuk adenosine trifosfat (ATP). Gangguan yang terjadi pada mitokondria akan berupa sekelompok penyakit heterogen yang biasanya bermanifestasi pada jaringan yang membutuhkan energi tinggi seperti otot, otak, jantung, dan saraf, sehingga disebut dengan mitochondrial
encephalomyopathies (Selim, 2013).
Penyakit mitokondria pada anak semakin diakui keberadaannya. Penyakit ini telah dilaporkan sebagai penyakit neurometabolik paling sering yang dapat diturunkan pada populasi berbeda di Inggris. Prevalensi penyakit yang sebenarnya sulit dipastikan, mengingat penyakit mitokondrial jarang dilaporkan. Akan tetapi, bukti epidemiologis menunjukkan sebuah prevalensi 1 dari 7.634 kelahiran hidup dan resiko seumur hidup mengalami penyakit mitokondria sekitar 1 dari 5.000. Mutasi individual dilaporkan berada pada frekuensi karier lebih tinggi, mendekati 1 dari 400, namun hanya sebagian kecil dari individu tersebut akan mengalami penyakit tersebut (Kisler, 2010).
Bidang kedokteran mitokondria baru saja berkembang dalam 25 tahun terakhir. Para dokter memiliki keterbatasan bukti dalam membuat formulasi keputusan klinis mengenai diagnosis, terapi, dan manajemen keseharian pasien. Penyakit ini pun masih kekurangan penanda yang cukup sensitif dan spesifik. Hal ini dapat dikarenakan penyakit tersebut tergolong penyakit yang jarang sehingga pembiayaan untuk studi terbatas (Parikh, 2014;
Parikh, 2009). Oleh karena itu, perlu diketahui mengenai penyakit mitokondria pada anak, khususnya ensefalopati mitokondria.
1.2 Batasan Masalah
Penulis membatasi pembahasan pada case report ini pada definisi, epidemiologi, klasifikasi, etiologi, faktor risiko, patofisiologi, manifestasi klinis, pemeriksaan penunjang, dan terapi stroke-like syndrome.
1.3 Tujuan Penulisan
Penulisan case report ini bertujuan untuk memahami serta mengetahui tentang definisi, epidemiologi, klasifikasi, etiologi, faktor risiko, patofisiologi, manifestasi klinis, pemeriksaan penunjang, dan terapi pada stroke-like syndrome.
1.4 Metode Penulisan
Case report ini ditulis berdasarkan sebuah laporan kasus stroke-like syndrome disertai
BAB 2
TINJAUAN PUSTAKA
2.1 Mitokondria
Mitokondria adalah suatu organel yang terdapat pada semua sel berinti dan merupakan pembentuk utama ATP melalui reaksi oxidative phosphorylation (OXPHOS). Proses ini melibatkan tahapan transfer elektron pada rantai respirasi (kompleks I–IV) dan ATP synthase (kompleks V). Mitokondria mempunyai DNA sendiri, namun organel ini bekerja di bawah kendali genetik dari DNA inti maupun dari genome mitokondria (Chial, 2008; Solano, 2001).
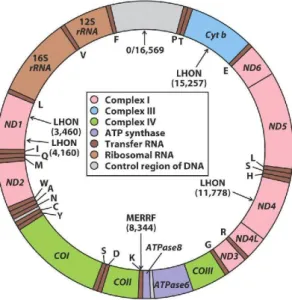
DNA mitokondria (mtDNA) merupakan molekul dengan ukuran 16,569-kb berbentuk sirkuler, double-stranded, yang mengandung 37 gen: 2 gen rRNA, 22 gen tRNA, dan 13 gen structural yang mengkode protein enzim yang bekerja pada respiratory chain (OXPHOS) di mitokondria, Rantai respirasi ini merupakan tahap akhir dari metabolism oksidatif dan terbentuknya ATP (Chial, 2008; Solano, 2001).
Gambar 2.1. DNA mitokondria berbentuk sirkuler dengan beberapa complex region
2.2 Mutasi DNA Mitokondria
Seperti halnya pada DNA inti, DNA mitokondria juga dapat mengalami mutasi. Point
mutation pada mtDNA dapat mengakibatkan penurunan jumlah enzim yang diperlukan pada respiratory chain sehingga produksi ATP juga akan sangat berkurang. Gangguan produksi
energi dalam sel pada akhirnya akan menimbulkan berbagai kelainan pada individu yang menderita. Kelainan ini terutama terjadi pada organ yang membutuhkan energi yang besar, yaitu otak dan otot. Oleh sebab itu, manifestasi klinis penyakit akibat kelainan mitokondria sebagian besar diakibatkan oleh kerusakan pada otak/sistem saraf pusat dan otot (Reeve, 2008). Lebih dari 150 mutasi titik dan kesalahan urutan mtDNA yang telah diketahui berhubungan dengan beberapa macam penyakit. Penyakit yang terkait dengan mutasi pada mtDNA dibagi menjadi dua kelompok besar: 1) kelompok gangguan yang disebabkan oleh mutasi pada gen yang terlibat pada sintesis protein (sindrom deplesi DNA mitokondria), dan 2) kelompok gangguan yang disebabkan mutasi pada gen yang mengkode protein individual yang terlibat dalam respiratory chain (sindrom delesi DNA mitokondria) (Chial, 2008; Solano, 2001; Reeve, 2008). Sindroma delesi mtDNA lebih bermanifestasi pada dewasa, sedangkan sindrom deplesi mtDNA biasanya terjadi pada anak, dengan karakteristik keterlibatan satu organ saja atau multisistem (Bonnen, 2013).Pembagiannya adalah sebagai berikut
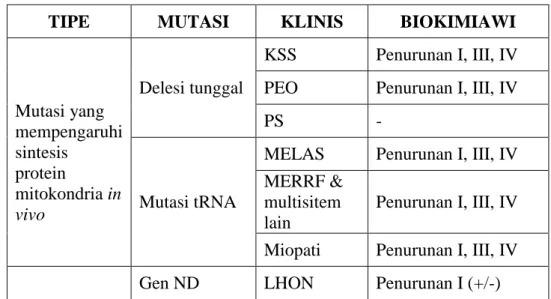
Tabel 2.1 Klasifikasi mutasi gen mtDNA
TIPE MUTASI KLINIS BIOKIMIAWI
Mutasi yang mempengaruhi sintesis protein mitokondria in vivo Delesi tunggal KSS Penurunan I, III, IV PEO Penurunan I, III, IV
PS -
Mutasi tRNA
MELAS Penurunan I, III, IV MERRF &
multisitem lain
Penurunan I, III, IV Miopati Penurunan I, III, IV Gen ND LHON Penurunan I (+/-)
Mutasi pada gen pengkode protein
MELAS, LS Penurunan I Miopati Penurunan I Cyt b Multisitem Penurunan III
Miopati Penurunan III Gen COX
Multisistem Penurunan IV Miopati Penurunan IV Gen ATPase 6 NARP/MILS Penurunan V
LA (lactic acidosis), ND (NADH dehydrogenase), KSS (Kearns-Sayre syndrome), PEO (progressive external ophtalmoplegia), PS (Pearson syndrome), MELAS (mitochondrial
encephalomyopathy,lactic acidosis, stroke-like episodes), MERRF (myoclonus epilepsy and raggered fibers), LHON (Leber hereditary optic neuropathy), LS (Leigh syndrome), NARP
(neuropathy, ataxia, retinitis pigmentosa), MILS (maternally inherited Leigh syndrome).
Studi tentang mutasi DNA mitokondria masih dilakukan untuk lebih memperjelas etiologi dari penyakit mitokondria ini. Bonnen et al pada tahun 2013 menemukan mutasi FBXL4 pada anak yang datang dengan ensefalopati, asidosis lakat, dan deplesi mtDNA berat pada ototnya. Mereka menunjukkan bahwa FBXL4 merupakan protein F-box yang terletak berdekatan dengan mitokondria dan mutasi serta hilangnya fungsi dari protein ini menyebabkan defisiensi rantai respiratori berat, hilangnya potensi membran mitokondria, dan gangguan jaringan mitokondria dinamis serta distribusi nukleoid pada fibroblas. Mereka menyimpulkan bahwa FBXL4 merupakan penyebab penyakit dan menetapkannya sebagai protein mitokondria dengan kemungkinan peran dalam mempertahankan integritas dan stabilitas mtDNA (Bonnen, 2013)
2.3 Diagnosis
Tidak ada parameter praktis berbasis konsensus yang dapat digunakan untuk klinis untuk membuat diagnosis atau memberikan terapi pada pasien terkait penyakit mitokondria. Kebanyakan ahli kedokteran mitokondria menggunakan sebuah pedoman yang didasarkan
pada konsep teoritis, rekomendasi yang terbatas, dan pengalaman pribadi. Variabilitas terjadi pada pendekatan diagnostik yang digunakan, luasnya pemeriksaan penunjang, interpretasi hasil, dan bukti penegakan diagnosis penyakit mitokondria tersebut (Parikh, 2014).
Sindroma ensefalopati mitokondria, asidosis laktat, dan episode seperti stroke (MELAS) adalah salah satu penyakit mitokondria paling sering yang ditemukan pertama pada tahun 1984. Pada tahun 1992, kriteria diagnostik klinis untuk sindrom MELAS meliputi episode stroke sebelum usia 40 tahun, ensefalopati yang ditandai dengan kejang dan/atau demensia, dan bukti miopati mitokondria yaitu asidosis laktat dan/atau ragged-red fibers (RRF). Diagnosis dapat diterima jika juga terdapat minimal dua dari kriteria berikut, perkembangan psikomotor awal normal, nyeri kepala rekuren, dan episode muntah rekuren. Lebih terkini, komite kelompokk studi MELAS di Jepang menerbitkan kriteria diagnosis lain yaitu kriteria definitif dengan minimal dua kriteria kategori A (nyeri kepala dengan muntah, kejang, hemiplegia, buta kortikal, dan lesi fokal akut pada pencitraan neurologi) dan dua kriteria kategori B (kadar laktat plasma atau cairan serebrospinal tinggi, abnormalitas mitokondrial pada biopsi otot, dan mutasi gen terkait MELAS) (Yatsuga, 2012; El-Hattab, 2015).
Peda prinsipnya, penurunan produksi ATP akibat gangguan mitokondria menyebabkan gejala klinis. Beberapa studi telah menunjukkan manifestasi klinis pada pasien dengan penyakit mitokondria yang mendukung istilah “semua jaringan dan semua tanda pada semua usia”. Akan tetapi, bayi dan anak-anak dengan penyakit mitokondria cenderung memiliki onset akut dari gejala dan perjalanan klinis jika dibandingkan dengan pasien dewasa, yang lebih perlahan dan progresif. Manifestasi klinis pada 103 pasien pediatrik yang didokumentasikan oleh Chi pada tahun 2014 adalah sebagai berikut (Chi, 2014).
Tabel 2.2 Manifestasi klinis penyakit mitokondrial
Diagnosis penyakit mitokondria memiliki sedikit sistem skor dalam kriteria diagnosisnya. Wolf et al mengajukan sistem skor kriteria diagnostik mitokondria (MDC) untuk bayi dan anak pada tahun 2002. Sistem ini mengevaluasi manifestasi klinis (I; skor klinis maksimum: 4 poin) dari gejala otot (IA; skor maksimum: 2 poin), abnormalitas sistem saraf pusat (IB; skor maksimum: 2 poin), dan keterlibatan multisistem (IC; skor maksimum: 3 poin), ditambah dengan abnormalitas metabolik dan neuroimaging (II; skor tambahan maksimum: 4 poin). Anomali histologis (III) dapat meningkatkan skor sebanyak 4 poin, menyebabkan skor maksimal sebesar 12 poin. Skor 8-12 didefinisikan sebagai definite, skor 5-7 didefinisikan sebagai probable, skor 2-4 didefinisikan sebagai possible, dan skor 1 didefinisikan sebagai
Tabel 2.3 Kriteria Klinis (Wolf, 2002)
Beragamnya manifestasi klinis yang dapat terjadi pada penyakit mitokondria membuat kepentingan pemeriksaan penunjang dalam menegakkan diagnosis. Pemeriksaan penunjang yang dapat dilakukan adalah pemeriksaan darah, survei metabolik, pencitraan, biopsi jaringan, pemeriksaan enzimatik, dan uji genetik mitokondria (Chi, 2014).
Tabel 2.4 Kriteria Biokimia (Wolf, 2002)
Sistem skor untuk Kriteria Biokimia: Unlikely: 0; Possible: 1–2; Probable: 2; Highly Probable: >2
Tabel 2.5 Kriteria Genetik (Wolf, 2002)
Sistem skor untuk Kriteria Genetik: Definite: kriteria 1 atau 2 abnormal; Probable: Kriteria 3 abnormal; Undetermined: kriteria 4 ada
2.4 Terapi
Seperti penyakit genetis lain, penyakit akibat mutasi mtDNA membutuhkan penanganan yang cukup kompleks. Secara garis besar, pendekatan terapi penyakit ini dapat dibagi menjadi tiga kelompok. Yaitu: 1) Terapi simptomatis sesuai gejala yang timbul pada
masing-masing penderita; 2) Terapi farmakologis untuk meningkatkan kualitas hidup penderita; dan 3) Terapi gen (Dimauro, 2005; Taylor, 2005; Parikh, 2014).
2.4.1. Terapi simptomatis
Telah dijelaskan sebelumnya bahwa manifestasi klinis utama dari penyakit akibat mutasi mtDNA adalah kelainan pada otot dan sistem saraf pusat. Oleh karena itu gejala-gejala yang terkait gangguan saraf dan otot seperti gangguan perkembangan mental dan motoris, kejang, atonia/distonia, dan kontraktur otot banyak ditemukan pada penderita. Masing-masing penderita dapat menunjukkan gejala yang berbeda, oleh karena itu pendekatan terapinya harus disesuaikan dengan gejala klinis yang ada, bila perlu penanganan oleh ahli dari masing-masing disiplin ilmu yang sesuai dapat dilakukan (Dimauro, 2005; Taylor, 2005; Parikh, 2014).
Sebagai contoh, gejala kejang yang timbul pada penderita Leigh Syndrom perlu penanganan dengan pemberian obat-obatan antikonvulsan. Obat psikotropika efektif pada pasien dengan gejala psikiatrik, khususnya depresi, Hambatan perkembangan mental dan motoris pada penderita PDH defisiensi memerlukan penanganan khusus antara lain terapi wicara dan terapi okupasi. Tindakan pembedahan oleh opthalmologist diperlukan untuk penanganan ptosis parah pada pasien dengan oftalmoplegia eksternal progresif. Katarak kongenital juga dapat ditangani dengan pembedahan (Dimauro, 2005; Taylor, 2005; Parikh, 2014).
Pada pasien dengan sindroma Kearns-Sayre disertai blockade konduksi jantung. Perlu dilakukan pemasangan pacemaker untuk menyelamatkan nyawa pasien. Pada pasien dengan kardiomiopati dan gangguan multisystem dapat dipertimbangkan tindakan transplantasi jantung (Dimauro, 2005; Taylor, 2005; Parikh, 2014).
Kehilangan pendengaran neurosensoris dapat ditangani dengan implantasi koklea. Gagal hepar yang sering terkait dengan sindroma deplesi mtDNA, dapat ditangani dengan
transplantasi liver. Myoglobinuria rekuren dapat ditemukan pada pasien dengan defisiensi coenzyme Q10 (CoQ10) primer atau dengan mutasi gen pengkode protein mtDNA. Selama episode akut, semua pasien harus dilakukan rehidrasi dan menjalani dialysis ginjal karena myoglobinuria dapat menyebabkan komplikasi gagal ginjal (Dimauro, 2005; Taylor, 2005; Parikh, 2014).
2.4.2 Terapi Farmakologis
Dalam makalah ini, yang dimaksud dengan terapi farmakologis adalah pemberian obat-obatan yang bekerja dengan menghambat metabolit toksik sebagai hasil dari metabolism alternative, maupun obat-obatan yang bekerja meningkatkan fungsi dan life time enzim pada rantai respirasi. Yang termasuk di sini adalah:
2.4.2.1 Pengeluaran Metabolit Noxius
Asidosis laktat adalah salah satu tanda khas dari penyakit akibat kelainan mitokondria. Karena metabolism aerob di mitokondria mengalami gangguan, maka jalur metabolisme anaerob meningkat dan menghasilkan metabolit asam laktat. Asam laktat bersifat neurotoksik, sehingga kadarnya perlu diturunkan. Obat penurun asam laktat yang umum digunakan adalah dikloroasetat (DKA) yang bekerja dengan menghambat piruvat dehydrogenase (PDH) kinase, menyebabkan PDH tetap defosforilasi, bentuk aktif dan membantu metabolism piruvat dan oksidasi laktat. DKA sendiri mempunyai efek samping neuropati perifer, sehingga DKA tidak boleh digunakan dalam jangka panjang pada kelainan mitokondria yang rentan terkena neurophaty perifer (Dimauro, 2005; Taylor, 2005; Parikh, 2014).
Sindroma ensefalopati neurogastrointestinal mitokondrial (MNGIE) mempunyai tanda khas adanya defek pada sinyal intergenomik. Pasien dengan kelainan multisistemik ini mengalami mutasi pada gen TP yang mengkode enzim timidin kinase, akibat yang ditimbulkan
adalah akumulasi timidin dalam darah. Strategi pengobatan yang dapat dilakukan adalah dengan menurunkan metabolit ini dengan hemodialisa. Pendekatan yang lebih radikal seperti transplantasi sel stem allogenik dapat dipertimbangakan karena hasilnya cukup menjanjikan (Dimauro, 2005; Taylor, 2005; Parikh, 2014).
Dari beberapa pengamatan, telah diketahui bahwa sel satelit dan mioblas mengandung jumlah mtDNA mutan lebih sedikit dibandingkan serabut otot matur. Dari data ini, beberapa peneliti berusaha mengembangkan strategi pengobatan dengan menggunakan agen miotoksik, bupivocaine, untuk menyebabkan nekrosis otot terbatas, yang dapat menghasilkan jaringan otot dengan jumlah mutan mtDNA lebih sedikit. Sayangnya, percobaan dengan injeksi unilateral bupivocaine pada otot levator palpebral pada lima pasien dengan oftalmoplegia eksternal progresif atau KSS tidak menghasilkan perbaikan apapun. Pendekatan lain berdasar pada fakta bahwa latihan isometric menyebabkan mikrotrauma dan nekrosis otot terbatas. Salah satu penelitian pada 1 pasien dengan mioglobinuria rekuren karena mutasi nonsense gen COX I mtDNA, mengindikasikan bahwa ada sebuah proses regenerasi. pada beberapa serabut otot yang dibiopsi. Fakta ini mendukung pendekatan terapi dengan menginduksi myoglobinuria pada pasien yang mengalami intoleransi latihan atau kelemahan karena mutasi mtDNA spesifik otot. Selain itu, baik latihan ketahanan dan resistance dapat digunakan sebagai kombinasi untuk penanganan miopati mitokondria (Dimauro, 2005; Taylor, 2005; Parikh, 2014).
2.4.2.2 Pemberian Akseptor Elektron
Encephalomiopati mitokondrial dapat disebabkan oleh defisiensi CCoQ10 primer. Kekurangan CoQ10 di mitokondria akan mengganggu aliran eklektron dari kompleks I dan II ke kompleks III, dimana akan mengurangi sintesis ATP. Sebagai tambahan, CoQ10 berfungsi sebagai antioksidan sehingga defisiensinya akan berdampak pada sel post-mitotik seperti neuron dan otot yang rentan terhadap kerusakan oksidatif Pendekatan terapi yang dapat
dilakukan adalah pemberian 2 akseptor elektron buatan, yaitu menadiol difosfat 40 mg/hari dan vitamin C 4 g/hari (Dimauro, 2005; Taylor, 2005; Parikh, 2014).
Beberapa clinical trial dengan kedua obat itu menunjukkan kembalinya kekuatan dan ketahanan otot, biopsy otot setelah 8 bulan terapi menunjukkan bahwa level CoQ10 kembali normal, penumpukan lipid menghilang, aktivitas enzim rantai pernafasan meningkat, dan penurunan proporsi miofibril yang terapoptosis. Meskipun demikian, pasien yang mengalami ataksia membutuhkan dosis yang lebih besar (hingga 1.000 mg/hari) dan berespon lambat, hal ini mungkin diakibatkan kerusakan serbelar yang ireversibe (Dimauro, 2005; Taylor, 2005; Parikh, 2014).
2.4.2.3 Pemberian Vitamin dan Kofaktor
Berbagai macam vitamin dan kofaktor telah digunakan pada pasien ensepalomiopati mitokondrial dan menunjukkan hasil yang positif, antara lain adalah riboflavin (vitamin B2), thiamine (Vitamin B1), Asam folat, CoQ10, I-Carnitine, dan creatine. Beberapa diantaranya seperti riboflavin dan CoQ10 adalah komponen dari rantai respiratori. Sedangkan kofaktor yang lain menurun pada beberapa kondisi seperti asam folat yang lebih rendah dari normal pada darah dan CSF pasien dengan KSS. Pada beberapa pasien, kadar carnitin bebas cenderung menurun karena carnitine teresterifikasi meningkat. Pergeseran ini mencerminkan kerusakan parsial dari β-oksidasi disertai gangguan CoQ10. Gabungan vitamin yang dapat digunakan untuk kasus ini adalah L-carnitine (1000 mg 3x/hari) dan CoQ10 (minimal 400 mg/hari) dengan tujuan mengembalikan level carnitine bebas dan meningkatkan aktivitas oxygen radical
scavenger CoQ10 (Dimauro, 2005; Taylor, 2005; Parikh, 2014).
Pada pasien MELAS, terjadi defisiensi carnitin primer, pendekatan terapi yang dapat dilakukan adalah pemberian Carnitine Monohidrat. Pada suatu percobaan, pemberian Carnitine monohidrat pada 6 pasien dengan MELAS dan 1 dengan kelainan mitokondrial yang tidak
jelas menunjukkan adanya perbaikan pada aktifitas yang berat tetapi tidak pada latihan aerobik yang ringan (Dimauro, 2005; Taylor, 2005; Parikh, 2014).
Suplementasi tembaga memberikan hasil yang positif pada beberapa penelitian in vitro untuk terapi ensepalomiopati infantile yang terkait dengan defisiensi COX. Pada kelainan ini terjadi mutasi gen SCO2, yang mengkode protein COX-assembly yang dibutuhkan untuk insersi cooper ke holoenzim. Ketika tembaga ditambahkan pada medium kultur dari mioblast defisit COX yang memiliki mutasi SCO2, aktivitas COX dapat kembali normal. Hal ini menunjukkan bahwa suplementasi tembaga kemungkinan besar bermanfaat untuk bayi yang menderita cardiomiopati dan mutasi SCO2 (Dimauro, 2005; Taylor, 2005; Parikh, 2014).
Deplesi mtDNA juga dapat dicegah dengan penambahan produk defektif enzim (deoksiguinosine monofosfat dan deoxyadenosin monofosfat), deoxiguinase kinase, pada fibroblas dari pasien dengan sindrom hepatoserebral dan mutasi homozigus pada gen DGUOK (Dimauro, 2005; Taylor, 2005; Parikh, 2014).
2.4.2.4 Pemberian Oxygen Radical Scavengers (ORS)
Defek rantai respirasi memiliki efek yang berbahaya yaitu kerusakan produksi ATP, gangguan intracellular calcium buffering, produksi ROS berlebih, dan peningkatan apoptosis. Peningkatan produksi ROS merusak membran sel melalui peroksidasi lipid dan juga dapat memperparah jumlah mutasi mtDNA (Dimauro, 2005; Taylor, 2005; Parikh, 2014).
Dalam percobaan menurunkan ROS, beberapa Oxygen radical Scavengers telah digunakan pada sebagian besar kelainan yang disebutkan termasuk vitamin E, CoQ10, idebenone, dan dihidrolifoat. CoQ10 telah digunakan untuk penyakit mitokondria dengan hasil positif dan efek negatif yang minimal. CoQ10 dan analognya, idebenone, telah digunakan secara luas sebagai terapi kelainan neurodegenerative yang terkait dengan peningkatan ROS (Dimauro, 2005; Taylor, 2005; Parikh, 2014).
2.4.3 Terapi Gen
Seperti halnya pada kelainan akibat mutasi DNA inti, pendekatan terapi gen dapat dilakukan pada kelainan akibat mutasi DNA mitokondria. Untuk penyakit mitokondria yang disebabkan mutasi pada gen inti, masalahnya tidak berbeda dengan terapi gen untuk gangguan genetic mendelian, termasuk pemilihan vector viral atau non-viral yang sesuai, pengiriman ke jaringan yang sakit, dan reaksi imunologis potensial (Dimauro, 2005; Taylor, 2005; Parikh, 2014).
Namun pada penyakit akibat mutasi pada mtDNA murni, masalahnya jauh lebih kompleks. Genome mitokondria yang bersifat poliploid, fenomena heteroplasmy, ketidakmampuan untuk mentransfeksikan asam nukleat yang potensial untuk terapi ke mitokondria serta belum adanya realisasi model hewan coba dengan penyakit mtDNA menjadi masalah utama dalam pengembangan terapi berbasis gen. Walaupun demikian, beberapa strategi untuk menyelamatkan fungsi mitokondria melalui komplementasi defek genetic atau manipulasi langsung level mtDNA mutan telah dipertimbangkan. Salah satu cara melengkapi protein mitokondria disfungsional adalah melalui allotopic expression. Dengan menggunakan pendekatan ini, menjadi mungkin untuk mengekspresikan protein ATPase 6 wild-type secara allotopic dari suatu konstruksi nucleus-transfected pada sel cybrid transmitochondrial yang bersifat homoplasmy untuk mutasi pada 8993T>G MTATP6 (subunit 6 dari ATP synthase mitokondria) dimana dapat menyebabkan kelemahan neurogenik, ataxia dan retinitis pigmentosa (NARP syndrome). Intervensi ini ternyata menyebabkan pemulihan parsial pada defek biokimia akibat mutasi mtDNA. Strategi yang sama juga digunakan untuk mengekspresikan gen modifikasi subunit 4 NADH dehydrogenase (ND4) untuk melengkapi (complementation) mutasi 1778G>A yang menyebabkan LHON (Dimauro, 2005; Taylor, 2005; Parikh, 2014).
Strategi terapi selanjutnya adalah melakukan pergeseran pada heteroplasmy, yaitu menurunkan rasio genom mutan terhadap wild-type (gene shifting). Pendekatan ini dapat dicapai melalui beberapa cara: (1) menghambat replikasi genome mutan secara selektif dengan peptide asam nukleat; (2) memasukkan RNAs ke mitokondria; (3) memasukkan polipeptida ke mitokondria; (4) memilih untuk fungsi respirasi; (5) induksi regenerasi otot; dan (6) induksi fusi mitokondria (Dimauro, 2005; Taylor, 2005; Parikh, 2014).
Hibridisasi selektif derivate asam nukleat mtDNA mutan dapat menghambat replikasinya selama propagasi genome wild-type, sehingga menyebabkan proporsi genome mutan turun di bawah ambang patogenik (pathogenic threshold). Beberapa percobaan in vitro berhasil menurunkan rasio mutan A8344G MERRF (Dimauro, 2005; Taylor, 2005; Parikh, 2014).
Strategi terapi selanjutnya adalah memasukkan polipeptida ke mitokondria. Strategi terapi ini mempunyai implementasi logistic yang berbeda, meliputi ekspresi allotopik, ekspresi xenotopik, dan impor endonuclease restriksi. Ekspresi allotopik mengacu pada strategi yang ditujukan untuk menurunkan beban protein mutan dengan mengimpor versi normal dari protein mutan yang dikode mtDNA dari gen snuck ke dalam nucleus. Sebagai contoh, gen ATPase 6 mtDNA dapat diubah dari mitokondria ke kode genetic inti. Untuk meyakinkan bahwa protein inti baru dikode oleh gen yang telah diubah dikenali, dan ditranspor ke mitokondria, maka pada protein tersebut diberi leader peptide, dimana sekuen genetiknya dipinjam dari protein yang dikode mtDNA yang lain. Setelah genetic Trojan horse dibawa ke nucleus, produk translasinya di sitoplasma ditranspor ke mitokondria, dilepaskan iktan leader peptide-nya, dan bergabung dengan komponen F0 dari kompleks V bersama dengan ATPase 8 yang disintesis di mitokondria. Pendekatan ini telah direalisasikan in vitro untuk mengkoreksi defek biokimia sel cybrid yang mengalami mutasi T8993G NARP/MILS dan cybrid yang mengalami mutasi
G11778A LHON (Leber’s hereditary optic neuropathy) (Dimauro, 2005; Taylor, 2005; Parikh, 2014).
Trik molekuler lain yang digunakan untuk mengkoreksi defek rantai respirasi yang disebabkan mutasi mtDNA dengan transfeksi sel mamalia yang sakit adalah dengan gen mitokondria maupun gen inti dari organisme lain tetapi mengkode cognate protein ‘xenotopic
expression’. Pendekatan molekuler langsung adalah dengan mengimpor endonuclease restriktif
spesifik sebagai magic bullets untuk merusak mtDNA mutan secara selektif. Pendekatan ini telah dilakukan pada sel cybrid model mutasi pada gen ATPase 6 T8993G NARP/MILS dengan membuat site Smal unik pada mtDNA manusia. Gen untuk Smal fusi ke sekuen target mitokondria dan secara transien diekspresikan pada cybrid heteroplasmik yang kehilangan mtDNA mutan (Dimauro, 2005; Taylor, 2005; Parikh, 2014).
Pendekatan lain yang cukup efektif untuk menurunkan beban mutasi mtDNA secara in vitro adalah pemaparan terhadap metabolit keton. Sel hybrid yang mengalami delesi tunggal mtDNA dibiakkan dalam medium yang mengandung keton sebagai sumber karbon. Sel yang homoplasmik untuk delesi mtDNA mati, sedangkan sel homoplasmik untuk mtDNA wild-type hidup. Pada heteroplasmic cell lines, proporsi mtDNA wild-type meningkat dari 13% menjadi 22% setelah 5 hari perlakuan dalam medium ketogenik, dan pada percobaan ini didapatkan perbaikan sintesis protein mitokondria. Pergeseran heteroplasmik (heteroplasmic shifting) tidak hanya terjadi diantara sel (seleksi interseluler) tetapi juga di dalam sel (seleksi intraseluler). Hasil ini dapat dijadikan rujukan kemungkinan dilakukannya percobaan in vivo pada pasien menggunakan diet ketogenik (Dimauro, 2005; Taylor, 2005; Parikh, 2014).
Dalam kaitannya dengan heteroplasmic shifting, mungkin tidak perlu lagi menurunkan jumlah mutasi jika kita dapat mendistribusikan ulang proporsi mtDNA mutan dengan wild-type dalam mitokondria, mengubah organel mutan homoplasmi dan organel wild-type homoplasmi menjadi organel heteroplasmi. Dengan cara ini, maka dapat dimanipulasi populasi mitokondria
mutan di bawah threshold untuk mengobati disfungsi mitokondria (Dimauro, 2005; Taylor, 2005; Parikh, 2014).
Pendekatan terapi selanjutnya adalah mengurangi produksi ROS. Pendekatan genetic dengan menangkap ROS telah berhasil dilakukan pada hewan coba tikus. Pertama, defisiensi kompleks I disebabkan oleh ekspresi anti-NDUFA1 ribozyme (NDUFA1 merupakan subunit katalitik kompleks I) menginduksi lesi retina dan saraf optic sama dengan LHON pada manusia. Kedua, regulasi penurunan MnSOD oleh ekspresi anti-SOD2 ribozyme pada tikus juga menginduksi lesi menyerupai LHON. Data ini mendukung konsep bahwa produksi ROS berlebihan merusak retina dan saraf optic. Ketika peneliti melakukan ko-ekspresi anti-NDUFA1 rybozyme (racun) dan SOD2 (antidot) pada tikus yang sama, lesi tidak muncul. Fakta ini menjelaskan bagaimana potensi terapi suatu SOD scavenging (Dimauro, 2005; Taylor, 2005; Parikh, 2014).
BAB 3
LAPORAN KASUS
Rontgen Vertebrae Lumbosakral AP/Lat: Tak tampak kelainan pada tulang belakang.
BAB 4 DISKUSI
Penyakit mitokondria merupakan sekelompok penyakit heterogen yang biasanya bermanifestasi pada jaringan yang membutuhkan energi tinggi seperti otot, otak, jantung, dan saraf. Manifestasi klinis nya dapat terjadi dengan gejala apa saja, pada organ mana saja, dan pada usia mana saja. Penyakit mitokondria pada anak memiliki onset yang lebih akut dan lebih berat dibandingkan penyakit mitokondria pada dewasa. Pemberian terapi untuk penyakit mitokondria tersebut meliputi terapi simtomatik, terapi farmakologis, dan terapi genetik.
DAFTAR PUSTAKA
1. Chial H, Craig J. 2008. mtDNA and Mitochondrial Diseases. Nature Education 1(1). http://www.nature.com/scitable/topicpage/mtdna-and-mitochondrial-diseases-903. Diakses tanggal 1 Januari 2013.
2. Solano A, Playán A, López-Pérez MJ, Montoya J. 2001. Genetic Diseases of Human Mitochondrial DNA. Salud Publica Mex 2001;43:151-161
3. Reeve AK, Krishnan KJ, Turnbull D. 2008. Mitochondrial DNA Mutations in Disease, Aging, and Neurodegeneration. Annals of the New York Academy of Sciences; 1147: 21-29
4. Dimauro S, Dacidzon G. 2005. Mitochondrial DNA and Disease. Annals of Medicine. 2005; 37: 222-232
5. Taylor RW, Turnbull DM. 2005. Mitochondrial DNA Mutations in Human Disease. (Online) http://www.nature.com/reviews/genetics. Diakses tanggal 1 Januari 2013. 6. Wolf NI, Smeitink JA. Mitochondrial disorders: a proposal for consensus diagnostic
criteria in infants and children. Neurology. 2002 Nov 12;59(9):1402-5.
7. Parikh S, Goldstein A, Koenig MK, Scaglia F, Enns GM, Saneto R, Anselm I, Cohen BH, Falk MJ, Greene C, Gropman AL, Haas R, Hirano M, Morgan P, Sims K, Tarnopolsky M, Van Hove JL, Wolfe L, DiMauro S. Diagnosis and management of mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med. 2015 Sep 2;17(9):689-701.
8. Kisler JE, Whittaker RG, McFarland R. Mitochondrial diseases in childhood: a clinical approach to investigation and management. Dev Med Child Neurol. 2010 May;52(5):422-33.
9. Bonnen PE, Yarham JW, Besse A, Wu P, Faqeih EA, Al-Asmari AM, Saleh MA, Eyaid W, Hadeel A, He L, Smith F, Yau S, Simcox EM, Miwa S, Donti T, Abu-Amero KK, Wong LJ, Craigen WJ, Graham BH, Scott KL, McFarland R, Taylor RW. Mutations in FBXL4 cause mitochondrial encephalopathy and a disorder of mitochondrial DNA maintenance. Am J Hum Genet. 2013 Sep 5;93(3):471-81.
10. Parikh S, Saneto R, Falk MJ, Anselm I, Cohen BH, Haas R, Medicine Society TM. A modern approach to the treatment of mitochondrial disease. Curr Treat Options Neurol. 2009 Nov;11(6):414-30.
11. Chi CS. Diagnostic approach in infants and children with mitochondrial diseases. Pediatr Neonatol. 2015 Feb;56(1):7-18.
12. Selim L, Mehaney D. Mitochondrial encephalopathy with lactic acidosis and stroke-like episodes in a Japanese child: Clinical, radiological and molecular genetic analysis. The Egyptian Journal of Medical Human Genetics (2013) 14, 317–322
13. El-Hattab AW, Adesina AM, Jones J, Scaglia F. MELAS syndrome: Clinical manifestations, pathogenesis, and treatment options. Molecular Genetics and Metabolism (2015) 116, 4–12