514 PERUBAHAN SELULER DAN MOLEKULER PADA NEFROPATI DIABETIK
Evy Sulistyoningrum1
1 Fakultas Kedokteran dan Ilmu-ilmu Kesehatan, Universitas Jenderal Soedirman Purwokerto
E-mail:[email protected]
ABSTRACT
Diabetic nehropathy is the most prevalent complication of diabetes mellitus. Hiperglycemia in diabetes mellitus activates several pathways leading to PKC activation. This activation then induce molecular changes involving many growth factors and cytokines. Various growth factors and cytokines further induce extracellular matrix accumulation and mesangial cell expansion leading to glomerular hypertrophy and sclerosis. This changes affects renal function in filtering albumin and further damage can cause abnormal renal function and lead to renal failure
Key Words: diabetic nephropathy, cellular changes, molecular changes
PENDAHULUAN
Nefropati diabetik (ND)
didefinisikan sebagai sindrom klinis pada
pasien DM yang ditandai dengan
albuminuria menetap > 300 mg/24 jam pada minimal dua kali pemeriksaan dalam kurun waktu 3 sampai 6 bulan pada minimal dua kali pemeriksaan dalam kurun waktu tiga
sampai enam bulan1 atau penurunan
kecepatan filtrasi glomerulus dan
peningkatan tekanan darah arterial tetapi tanpa penyakit ginjal lainnya atau penyakit
kardiovaskuler2. Kondisi ini merupakan
salah satu komplikasi Diabetes Mellitus (DM) yang paling serius dan paling sering menyebabkan gagal ginjal stadium terminal (end stage renal disease) hampir di seluruh
dunia3. Sekitar 40% penderita ESRD adalah
pasien DM dengan komplikasi nefropati
diabetik4. Nefropati diabetik merupakan
komplikasi mikrovaskuler tersering yang terjadi pada penderita DM di Amerika dan
Eropa5. Sekitar 20% sampai 30% penderita
kasus DM akan berkembang menjadi kasus nefropati diabetik. Setelah 20 tahun onset
nefropati, 20% penderita DM akan
mengalami gagal ginjal terminal4. Seiring
dengan meningkatnya prevalensi DM, diperkirakan prevalensi ND pun akan semakin meningkat. Artikel ini akan membahas mengenai perubahan struktural dan fungsional pada nefropati diabetik dan mekanisme molekuler yang mendasarinya.
Perubahan Struktur Ginjal Pada
Nefropati Diabetik
Nefropati diabetik menimbulkan
beberapa kelainan pada struktur histologis ginjal. Karakteristik histologis nefropati diabetik adalah perubahan struktur pada glomerulus. Glomerulus pada keadaan nefropati diabetik mengalami penambahan volume (hipertrofi) yang disebabkan karena
515 adanya penumpukan matriks ekstraseluler,
penebalan membrana basalis glomerulus,
dan glomerulosklerosis6. Gambaran awal
dari perubahan struktur ginjal diabetik adalah hipertrofi glomerular dan renal.
Perubahan ini akan diikuti dengan
peningkatan ketebalan membrana basalis glomerular, eskpansi mesangial dengan akumulasi protein matriks ekstraselular seperti kolagen, fibronektin dan laminin
disertai proliferasi sel mesangial
intraglomerular7. Nefropati diabetik tingkat
lanjut ditandai dengan glomerulosklerosis
dan fibrosis interstitial8. Glomerulosclerosis
pada ginjal dapat terjadi secara noduler ataupun difus (badan Kiimestiel dan
Wilson) yang ditandai dengan gambaran eosinofilik. pada bagian perifer glomerulus.
Nodulus ini terjadi akibat ekspansi
mesangial dan penebalan membrana basalis pada kapiler sekelilingnya dengan oklusi progresif kapiler glomerulus. Terjadinya glomerulosklerosis mengakibatkan ginjal tidak dapat bekerja sesuai fungsinya, karena jaringan sklerosis ini akan menekan kapiler yang pada keadaan normal kapiler ini berfungsi untuk filtrasi darah menjadi urin sehingga dapat menyebabkan laju fitrasinya
sangat menurun9. Perubahan gambaran
histologis glomerulus pada nefropati
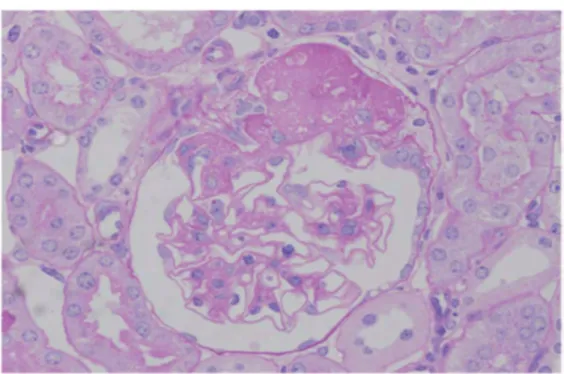
diabetik ditampilkan pada Gambar 1.
Gambar 1. Gambaran glomerulus pada nefropati diabetik10
Keterangan: Glomerulus mengalami sklerosis segmental (A) dengan proliferasi sel mesangial (B) dan ekspansi mesangial difus (C)
Bukti-bukti ilmiah melaporkan peran utama
sel mesangial intraglomeruler pada
kerusakan glomerulus pada nefropati
diabetik. Sel mesangial intraglomeruler
adalah sel epiteloid yang terletak di antara kapiler glomerulus dan berperan sebagai sel penyokong. Sel ini mengalami modifikasi struktur dengan akumulasi granula basofilik
516 pada sitoplasma. Sel ini juga mempunyai
fungsi menyerupai makrofag karena
mempunyai kemampuan fagositosis11.
Keadaan hiperglikemia pada diabetes akan menginduksi proliferasi dan aktivasi sel mesangial intraglomerular. Aktivasi sel ini akan menyebabkan pelepasan berbagai faktor pertumbuhan seperti TGF-β (Tumor Growth Factor-β), PDGF (Platelet-Derived Growth Factor), CTGF (Connective Tissue Growth Factor, bFGF (basic Fibroblast Growth Factor) dan berbagai sitokin seperti Il-1, TNF-α, komplemen. Pelepasan faktor
pertumbuhan dan sitokin ini akan
menyebabkan akumulasi protein matriks ekstrasel yang bertanggungjawab pada
glomerulosklerosis9.
Perubahan Fisiologi Ginjal Pada
Nefropati Diabetik
Nefropati diabetik juga
menimbulkan abnormalitas fungsi ginjal. Gambaran awal dari perubahan fungsi
ginjal diabetik adalah hiperfiltrasi
glomerular dan peningkatan urinary
albumin excretion (UAE)8. Pada ND, akan
terjadi perubahan ukuran pori dan
penurunan proteoglikan heparan sulfat yang merupakan barier anion pada membran basalis glomerulus. Kondisi ini beserta
pelepasan berbagai stitokin yang
mempengaruhi aliran dan permeabilitas
vaskular menyebabkan penurunan
kemampuan seleksi dalam fungsi
penyaringan glomerulus dan terjadi
mikroalbuminuria7. Kerusakan lanjut
glomerulus mengakibatkan ekskresi protein yang berlebihan serta penurunan fungsi ginjal12.
Hiperfiltrasi pada nefropati dianggap sebagai awal dari mekanisme patogenik dalam laju kerusakan ginjal. Hal ini terjadi pada saat jumlah nefron mengalami pengurangan progresif, glomerulus akan
melakukan kompensasi dengan
meningkatkan filtrasi nefron yang masih sehat dan pada akhirnya nefron yang sehat menjadi sklerosis. Peningkatan laju filtrasi
glomerulus pada nefropati diabetik
kemungkinan disebabkan oleh dilatasi arteriol aferen oleh efek yang tergantung
glukosa, yang diperantarai hormon
vasoaktif, IGF-1 (Insulin Growth Factor-1),
Nitric Oxide, prostaglandin, dan glukagon1.
Kondisi hiperglikemia akan
mengaktifkan Protein Kinase C (PKC) melalui diasilgliserol. Protein Kinase C selanjutnya menstimulasi aktivitas kerja angiotensin sehingga laju filtrasi glomerulus
(LFG) terganggu. Insulin growth factor-1
(IGF-1), VEGF dan endhotelin-1 (ET-1)
memicu hipertrofi tubulus ginjal. Protein Kinase C dapat menstimulasi TNF-α dan NFκB serta meningkatkan aktivitas fibrotik
yaitu CTGF dan TGF-β13. Kedua faktor
fibrosis tersebut akan meningkatkan
proliferasi jaringan ikat dan menyebabkan
hipertrofi glomerulus dan ekspansi
mesangial. Penurunan fungsi enzim
antioksidan juga diperparah PKC yang menstimulasi pengeluaran sitokin inflamasi
517 glomerulosklerosis. Kerusakan glomerulus
dan tubulus mengakibatkan ekskresi protein
yang berlebihan (albuminuria dan
proteinuria) serta peningkatan ureum dan kreatinin dalam darah yang pada akhirnya
berujung pada gagal ginjal7.
Pendekatan Molekular Nefropati
Diabetik
Perubahan struktural dan fungsional pada ginjal akibat DM disebabkan karena tingginya kadar glukosa dalam darah
(hiperglikemia). Hiperglikemia
menyebabkan terjadinya peningkatan
ekspresi GLUT-1 pada sel mesangial dan
glomerulus ginjal sehingga terjadi
peningkatan ambilan glukosa. Beberapa jalur yang telah teridentifikasi menjadi aktif akibat peningkatan ambilan glukosa antara lain: AGEs (Advance Glicosylated End-products) jalur polyol, Renin Angiotensin System (RAS), dan stress oksidatif yang akan mengaktifkan jalur PKC (Protein Kinase C)14.
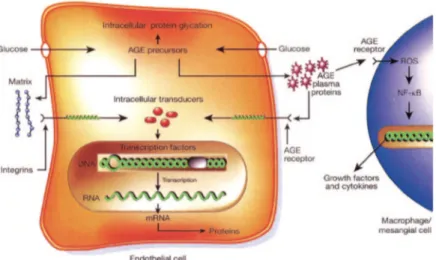
Hiperglikemia akan menyebabkan peningkatan produk glikosilasi dengan proses non enzimatik yang disebut AGEs (Advanced Glicosylation End Products). Proses ini dapat berlangsung di intraseluler dan ekstraseluler. AGEs intraseluler akan mengaktivasi protein kinase C, MAPK, serta NF-κB. Aktivasi ketiga zat ini akan meningkatkan ekspresi TGF-β pada sel mesangial dan sel endotel glomerulus.
TGF-β akan memacu aktivasi dari connective
tissue growth factor (CTGF). TGF- β dan
CTGF akan memacu sintesis kolagen tipe I dan IV serta peningkatan akumulasi
fibronektin di mesangial14.
Glukosa di ekstraseluler akan
tersintesis menjadi AGEs dan berikatan dengan reseptornya yaitu RAGEs (Receptor for Advanced Glicosylation End Products). Reseptor ini terletak pada membran sel
endotel dan sel mesangial15. Ikatan AGEs
dengan RAGE ini akan menyebabkan
terbentuknya ROS (Reactive Oxygen
Species), penurunan aktivasi eNOS (endothelial Nitrit Oxyde Synthetase), inaktivasi NO (Nitrit Oxide), memacu timbulnya jalur PKC, dan berikatan dengan NF-kB (Nuclear Factor-KappaB) di nukleus sel sehingga meningkatkan transkripsi berbagai protein seperti endothelin, ICAM-1 (Cellular Adhesion Molecule-ICAM-1), VEGF,
IL-1, IL-6, dan TNF-α 15,16 seperti yang
ditampilkan pada Gambar 3. Peningkatan
protein ini akan memacu terjadinya
infiltrasi sel-sel radang seperti makrofag,
monosit, dan leukosit13. Peningkatan sitokin
dan adanya infiltrasi sel radang ini selanjutnya akan menyebabkan terjadinya
abnormalitas pada glomerulus16.
Pembentukan AGEs pada protein seperti kolagen akan membentuk ikatan silang di antara berbagai polipeptida yang dapat
menyebabkan terperangkapnya protein
intersisium dan plasma yang tidak
terglikosilasi. AGEs juga dapat
mempengaruhi struktur dan fungsi kapiler,
termasuk glomerulus ginjal yang
518 fungsi penyaringan menjadi gagal.AGEs
berikatan dengan reseptor pada berbagai tipe sel misalnya sel endotel, monosit, makrofag, limfosit dan sel mesangial. Pengikatan tersebut menyebabkan berbagai aktivitas biologi termasuk migrasi sel
neutrofil, pengeluaran sitokin, peningkatan
permeabilitas endotel, peningkatan
proliferasi fibroblas serta sintesis matriks ekstraseluler17.
Gambar 2 Mekanisme aktivasi jalur AGEs (Brownlee, 2005)
Hiperglikemia juga dapat
mengakivasi jalur poliol. Jalur poliol ini terjadi melalui dua reaksi, yaitu reduksi glukosa menjadi sorbitol dan oksidasi sorbitol menjadi fruktosa. Reduksi glukosa menjadi sorbitol diperantarai oleh enzim aldosa reduktase dan kofaktor NADPH.
Oksidasi sorbitol menjadi fruktosa
diperantarai oleh enzim sorbitol
dehidrogenase dan kofaktor NAD+.
Penurunan NADPH akan mengakibatkan penurunan produksi NO pada sel endotel, peningkatan produksi prostaglandin E2, dan
peningkatan rasio NADH : NAD+ di sitosol.
Peningkatan rasio ini akan menghambat jalur glukolitik sehingga menyebabkan terjadinya stress oksidatif, peningkatan
sintesis DAG, dan jalur AGEs7.
Aktivasi jalur poliol dan AGEs dapat menstimulasi aktivasi jalur PKC. Aktivasi
PKC akan mengakibatkan penurunan
produksi NO, penurunan produksi eNOS, aktivasi VEGF, TGF-β, PAI-1 (Platelet Activator Inhibitor-1), NF-kB, oksidasi
NADPH, dan peningkatan ekspresi
endothelin serta VEGF. Hal ini
menyebabkan terjadinya penurunan aliran darah ke ginjal, peningkatan akumulasi
matriks ekstraseluler, penurunan
fibrinolisis, peningkatan produksi dan penurunan permeabilitas vaskuler yang bisa memacu terjadinya kerusakan pada vaskuler
glomerulus18.
Hiperglikemia pada DM juga dapat
memicu peningkatan reactive oxygen
519 keadaan penurunan fungsi enzim-enzim
antioksidan karena produksi ROS yang berlebihan. Kadar ROS yang berlebihan
menstimulasi pengeluaran sitokin pro
inflamasi. Sitokin pro inflamasi tersebut
adalah interleukin IL-1, IL-6, IL-8, tumor
necrosis factor-α (TNF-α), nuclear factor kappa B (NFκB), monocyte chemoattractant protein-1 (MCP-1),
cellular adhesion molecules (CAMs), nitric oxide (NO), peroksinitrit (ONOO-), connective tissue growth factor (CTGF), dan transforming growth factor-ß (TGF-ß) yang mengakibatkan kerusakan struktural
dan fungsional ginjal13. Peran berbagai jalur
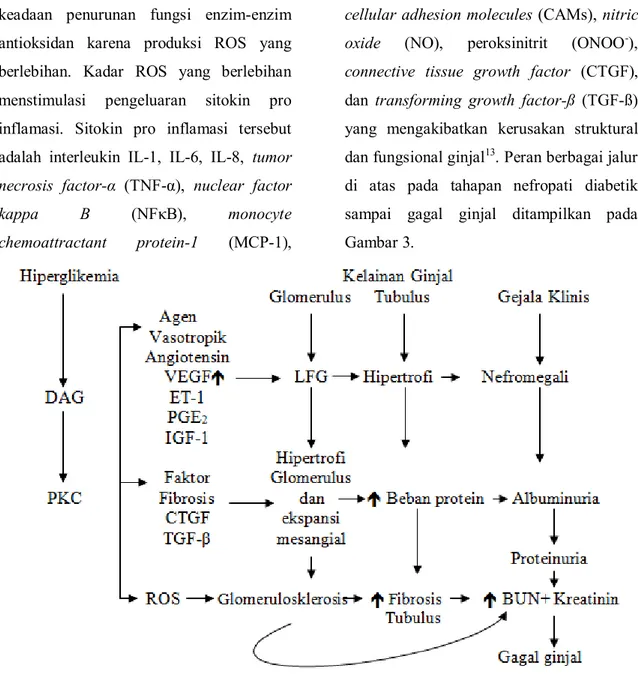
di atas pada tahapan nefropati diabetik sampai gagal ginjal ditampilkan pada Gambar 3.
Gambar 3. Skema Patogenesis Nefropati Diabetik sampai ESRD7
Keterangan: DAG: Diacyl Gliserol, PKC: Protein Kinase C, VEGF: Vascular Endothelial Growth Factor , ET-1: Endothelin-1, PGE2: Prostaglandin E2, IGF-1: Insulin Growth Factor-1, LFG: Laju Filtrasi Glomerulus, CTGF: Connective Tissue Growth Factor, TGF-β : Transforming Growth Factor-β, ROS: Reactive Oxygen Specie, BUN: Blood Urea Nitrogen
KESIMPULAN
Berbagai Nefropati diabetik
merupakan komplikasi terbanyak dari
diabetes mellitus. Kondisi hiperglikemia
pada diabetes mellitus mendasari
serangkaian proses yang menyebabkan
perubahan struktur glomerulus yang
selanjutnya menyebabkan abnormalitas
fungsi ginjal. Tahap lanjut proses ini adalah gagal ginjal.
DAFTAR PUSTAKA
1. Hendromartono. 2009. Nefropati Diabetik
dalam Buku Ajar Ilmu Penyakit Dalam.
520
2. Batuman, V. 2012. Diabetic Nephropathy. Medscape Reference. Diakses dari : http://emedicine.medscape.com/article/238 946-overview. pada tanggal 15 September 2012
3. Scrijvers BF, Vriese AS, Flyvbjerg A, 2004. From Hyperglycemia to Diabetic Kidney Disease: The Role of Metabolic, Hemodynamic, Intracellular Factors and Growth Factors/Cytokines. Endocrine Reviews 25: 971-1010
4. American Diabetic Association. 2010. Diagnosis and Classification of Diabetes Mellitus. Diabetes Care. 33: S62-S69
5. Dronavalli, S, Duka, I, Bakris, GL. 2008. The Pathogenesis of Diabetic Nefropathy. Nature Clinical Practice Endocrinology and Metabolism. Vol 4 : 445-452.
6. Scobie, IN. 2007. Atlas of Diabetes Mellitus Third Edition. London : Informa Healthcare
7. Ohshiro, Y, Lee, Y, King, GL. 2005. Mechanism of Diabetic Nephropathy : Role of Protein Kinase-C Activation. Advanced Studies in Medicine. Vol 5(1A) : S5-19.
8. Bloomgarden ZT, 2005, Diabetic Nephropathy. Diabetes Care; 28: 745-51
9. Qian, Y, Feldman, E, Pennathur, S, Kretzler, M, Brosius, FC. 2008. From Fibrosis to Sclerosis: Mechanism of Glomerulosclerosis in Diabetic Nephropathy. Diabetes Journal.,57
10. Tomino, Y. 2012. Lesson from the KK-Ay mouse, a spontaneous animal model for the treatment of human type 2 diabetic nephropathy. Nephro-Urol Mon. 4(3) : 524-9
11. Kierzenbaum, AL. 2007. Histology and Cell Biology, 2nd Edition. Canada: Mosby, Inc., 406
12. Obineche, AN dan Adem, A. 2005. Update in Diabetic Nephropathy. International Journal of Diabetes and Metabolism. Vol 13 : 1-9.
13. Elmarakby, AA. dan Sullivan , JC. 2010. Relationship Between Oxidative Stress and Inflammatory Cytokines. Cardiovascular Therapeutics. Vol 00 : 1–11.
14. Kanwar, YS., Wada, J, Sun, L, Xie, P, Elisabeth,W. 2012. Diabetic Nephropathy : Mechanisms of Renal Disease Progression. Experimental and Biology Medicine. Vol 233 : 4-11.
15. Brownlee, M. 2005. The Pathobiology of Diabetic Complications a Unifying Mechanism. Diabetes. Vol 54 : 1615 – 1625
16. Goh, S dan Cooper, ME. 2008. The Role of Advanced Glycation End Products in Progression and Complications of Diabetes. Journals Clinical Endocrinol Metabolism. Vol 93 : 1143-1152.
17. Tan, AL, Forbes, JM, Cooper, ME. 2007. AGE, RAGE and ROS in diabetic nephropathy. Semin Nephrol 27:130–143
18. Schena, FP dan Gesualdo, L. 2005. Pathogenetic Mechanism of Diabetic Nephropathy. Journal of the American Society of Nephrology. Vol 16 : S30-33.