1
CASE REPORT
EVANS SYNDROME IN 24 YEARS OLD WOMEN
Made Gita Ratnasari, Sianny Herawati, I Wayan Putu Sutirta Yasa, Anak Agung Wiradewi Lestari
Departement of Clinical Pathology, Medical Faculty of Udayana University Sanglah Hospital Denpasar
ABSTRACT
Evans syndrome is a rare case, a condition which occurs with simultaneously or sequentially Autoimmune Hemolytic Anemias (AIHA) and Idiopathic Trombocytopenias Purpura (ITP) and sometimes together with neutropenia, in the absence of etiology. This syndrome may be associated with other autoimmune or lymphoproliferative diseases. Course of the disease is very serious and, in rare cases, can even be life-threatening. First-line treatment consists of steroids and or immunoglobulin. We reported a woman, 24 years old came with complaints of weakness for 1 week, sometimes feel dizziness, jaundice, colour of urine is like tea. Of a complete blood count showed hemoglobin decreased, equal to 3,8 g/dL, and a decrease in platelets that is equal to 56x103/μl. Physical examination did not reveal any organ enlargement. The patient was given WRC (washed red cell) transfusion, TC (trombosit consentrate) transfusion, and methylprednisolone. In addition, patient were also given supportive theraphy such as fluid therapy IVFD NaCl 0,9% and oxygen therapy. Patient was hospitalized to establish diagnosis and was discharged after ten day of hospitalization. Prognosis is good, where no complication such as bleeding or sepsis.
Keywords : Evans syndrome, Autoimmune disease, AIHA, ITP
LAPORAN KASUS
EVANS SYNDROME PADA WANITA USIA 24 TAHUN
Made Gita Ratnasari, Sianny Herawati, I Wayan Putu Sutirta Yasa, Anak Agung Wiradewi Lestari
Bagian/SMF Patologi Klinik, Fakultas Kedokteran Universitas Udayana Rumah Sakit Umum Pusat Sanglah Denpasar
ABSTRAK
2 56x103/μl. Pada pemeriksaan fisik tidak ditemukan adanya pembesaran organ. Terapi yang diberikan yaitu transfusi WRC (washed red cell), tranfusi TC (trombosit consentrate), dan metilprednisolon. Selain itu pasien juga diberikan terapi suportif berupa terapi cairan IVFD NaCl 0,9% dan terapi oksigen. Pasien masuk rumah sakit untuk diagnosis dan dipulangkan setelah sepuluh hari perawatan. Prognosis pasien baik, dimana tidak terjadi komplikasi seperti pendarahan ataupun sepsis.
1
CASE REPORT
EVANS SYNDROME IN 24 YEARS OLD WOMEN
Made Gita Ratnasari, Sianny Herawati, I Wayan Putu Sutirta Yasa, Anak Agung Wiradewi Lestari
Departement of Clinical Pathology, Medical Faculty of Udayana University Sanglah Hospital Denpasar
ABSTRACT
Evans syndrome is a rare case, a condition which occurs with simultaneously or sequentially Autoimmune Hemolytic Anemias (AIHA) and Idiopathic Trombocytopenias Purpura (ITP) and sometimes together with neutropenia, in the absence of etiology. This syndrome may be associated with other autoimmune or lymphoproliferative diseases. Course of the disease is very serious and, in rare cases, can even be life-threatening. First-line treatment consists of steroids and or immunoglobulin. We reported a woman, 24 years old came with complaints of weakness for 1 week, sometimes feel dizziness, jaundice, colour of urine is like tea. Of a complete blood count showed hemoglobin decreased, equal to 3,8 g/dL, and a decrease in platelets that is equal to 56x103/μl. Physical examination did not reveal any organ enlargement. The patient was given WRC (washed red cell) transfusion, TC (trombosit consentrate) transfusion, and methylprednisolone. In addition, patient were also given supportive theraphy such as fluid therapy IVFD NaCl 0,9% and oxygen therapy. Patient was hospitalized to establish diagnosis and was discharged after ten day of hospitalization. Prognosis is good, where no complication such as bleeding or sepsis.
Keywords : Evans syndrome, Autoimmune disease, AIHA, ITP
LAPORAN KASUS
EVANS SYNDROME PADA WANITA USIA 24 TAHUN
Made Gita Ratnasari, Sianny Herawati, I Wayan Putu Sutirta Yasa, Anak Agung Wiradewi Lestari
Bagian/SMF Patologi Klinik, Fakultas Kedokteran Universitas Udayana Rumah Sakit Umum Pusat Sanglah Denpasar
ABSTRAK
2 56x103/μl. Pada pemeriksaan fisik tidak ditemukan adanya pembesaran organ. Terapi yang diberikan yaitu transfusi WRC (washed red cell), tranfusi TC (trombosit consentrate), dan metilprednisolon. Selain itu pasien juga diberikan terapi suportif berupa terapi cairan IVFD NaCl 0,9% dan terapi oksigen. Pasien masuk rumah sakit untuk diagnosis dan dipulangkan setelah sepuluh hari perawatan. Prognosis pasien baik, dimana tidak terjadi komplikasi seperti pendarahan ataupun sepsis.
Kata kunci: Sindrom Evans, Penyakit Autoimun, AIHA, ITP
PENDAHULUAN
Sindrom Evans didefinisikan sebagai kombinasi baik secara bersamaan atau berurutan dari Autoimmune Hemolytic Anemias
(AIHA) dan Idiopathic
Trombocytopenias Purpura (ITP), dan terkadang bersama-sama dengan neutropenia, dengan tidak adanya etiologi yang mendasari.1,2 Sindrom Evans adalah penyakit langka dimana hanya 0,8% - 3,7% yang terdiagnosis dari pasien yang menderita ITP dan AIHA. Kejadian familial jarang ditemukan.3
Di Malaysia tahun 1992, sindrom Evans ditemukan pada 12 dari 220 pasien dewasa dengan ITP dan 102 dengan AIHA.2 Sebuah penelitian oleh Silverstein dan Heck di Amerika, sebanyak 399 kasus AIHA dan 367 kasus ITP pasien dewasa, hanya enam dari 766 pasien ini mengalami sindrom Evans.1 Sindrom ini dominan terjadi pada ras kulit putih, dari 42 pasien yang dilaporkan dalam survei nasional terdapat 29 berkulit putih, 7 hitam, dan 6 memiliki latar belakang ras lainnya. Tidak ada predileksi pada sindrom Evans, pada suatu studi yang dilakukan oleh Liliana di Rumania tahun 2012 menyebutkan sindrom ini mempengaruhi anak laki-laki lebih sering daripada perempuan dengan rasio 1,4:1.4 Dan dalam suatu penelitian oleh Genty di Prancis tahun 2002, sebanyak 67% kasus sindrom evans terjadi pada wanita. Sindrom Evans terjadi pada
individu dari segala usia. Dalam sebuah survei tahun 1997 di Amerika Utara, melaporkan rata-rata usia saat diagnosis adalah usia 7,7 tahun.2
Etiologi sindrom Evans masih belum diketahui. Autoantibodi bereaksi terhadap antigen spesifik untuk sel darah merah, trombosit, atau neutrofil, tetapi autoantibodi ini tidak bereaksi silang.4 Sindrom Evans adalah diagnosis eksklusi. Gangguan seperti infeksi, penyakit rematologi, dan keganasan harus dikesampingkan. Hasil pemeriksaan darah lengkap dapat dijumpai anemia, trombositopenia, neutropenia, atau sitopenia pada pasien dengan sindrom Evans.1,2
Terapi lini pertama yang paling umum digunakan adalah kortikosteroid dan atau imunoglobulin intravena (IVIG).1,2 Sindrom Evans kadang-kadang fatal dan memiliki tingkat kematian sebesar 7%. Penyebab kematian terutama disebabkan oleh perdarahan dan sepsis.4
ILUSTRASI KASUS
3 mendenging. Satu minggu sebelumnya
pasien berobat ke dokter swasta didiagnosis anemia dan sakit kuning lalu diberikan vitamin. Sehari sebelum pasien datang ke RS. Sanglah, pasien berobat ke RS. Swasta. Kadar hemoglobin saat itu adalah 3,8 g/dL, dan kadar trombosit 56x103/µl. Akhirnya RS. Swasta tersebut merujuk pasien ke RS. Sanglah. Dari hasil anamnesis didapatkan pasien mengeluh lemas, terdapat riwayat panas badan, tidak ada riwayat ke wilayah timur, BAK (buang air kecil) dikatakan seperti teh, serta tidak terdapat rambut rontok.
Dari riwayat penyakit dahulu, pasien mengatakan tidak pernah menderita hipertensi, kencing manis, penyakit jantung, asthma, stroke, liver, ginjal TBC Paru. Riwayat keluhan yang sama dalam keluarga disangkal. Tidak ada riwayat penyakit jantung, hipertensi, kencing manis dan asthma di keluarga pasien. Tidak ada kebiasaan merokok dan mengkonsumsi alkohol. Pada pemeriksaan fisik, keadaan umum baik, tekanan darah 110/80 mmHg, nadi 100 kali/menit, respirasi 22 kali/menit, suhu aksila 37,8C.
Pada pemeriksaan mata ditemukan tanda anemia serta ikterus,
refleks pupil normal, dan tidak ditemukan adanya edema palpebral. Pemeriksaan THT dan leher dalam batas normal, tidak ditemukan pembesaran kelenjar dan kaku kuduk. Torak terlihat simetris, paru serta jantung berada dalam batas normal, suara jantung S1S2 tunggal reguler, serta
tidak ditemukan murmur, rhonki, dan ‘wheezing’. Pada pemeriksaan abdomen tidak ditemukan adanya kelainan seperti distensi, meteorismus, ascites, nyeri tekan, dan tidak ada pembesaran organ. Pada keempat ekstremitas teraba hangat serta tidak ditemukan edema.
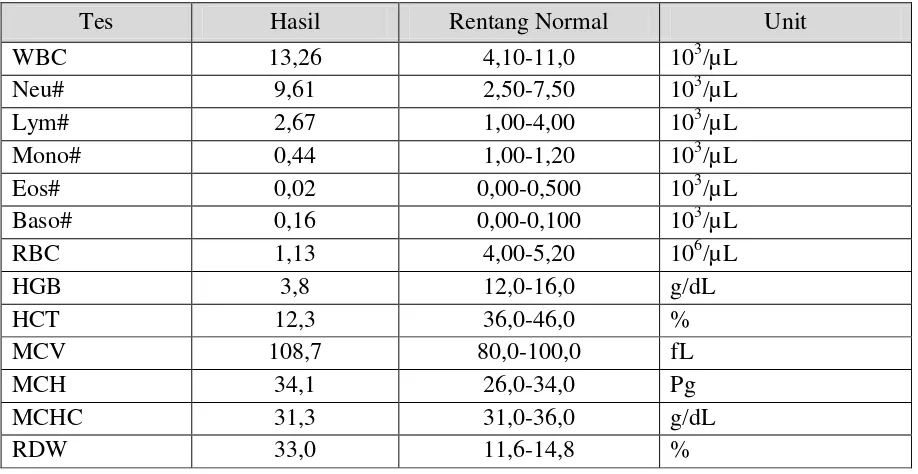
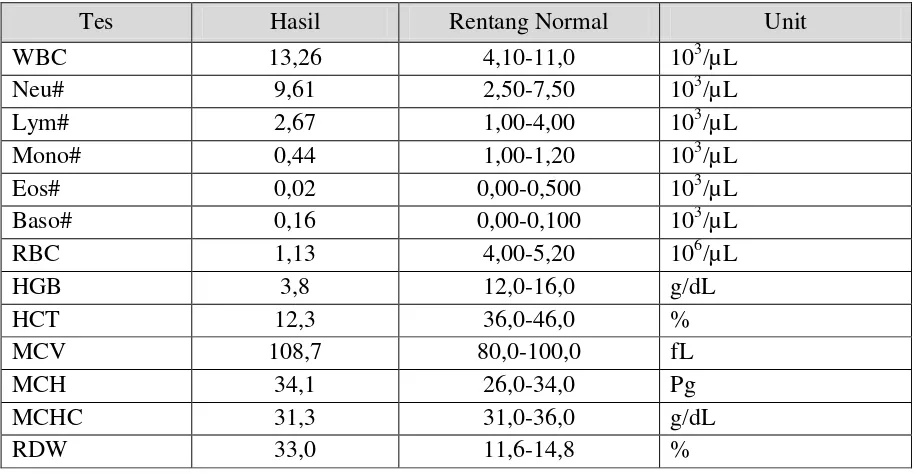
Hasil pemeriksaan darah dapat dilihat pada Tabel 1 dan Tabel 2. Dimana hasilnya yaitu terjadi peningkatan WBC (leukositosis), penurunan HGB disertai peningkatan MCV dan retikulosit (anemia hemolitik), dan ditemukan juga penurunan trombosit (trombositopenia). Dari hasil tes fungsi ginjal tidak didapatkan kelainan, sedangkan pada tes fungsi hati didapatkan kelainan berupa peningkatan bilirubin dan SGOT.
Tabel. 1 Hasil Tes Darah Lengkap
Tes Hasil Rentang Normal Unit
WBC 13,26 4,10-11,0 103/µL
Neu# 9,61 2,50-7,50 103/µL
Lym# 2,67 1,00-4,00 103/µL
Mono# 0,44 1,00-1,20 103/µL
Eos# 0,02 0,00-0,500 103/µL
Baso# 0,16 0,00-0,100 103/µL
RBC 1,13 4,00-5,20 106/µL
HGB 3,8 12,0-16,0 g/dL
HCT 12,3 36,0-46,0 %
MCV 108,7 80,0-100,0 fL
MCH 34,1 26,0-34,0 Pg
MCHC 31,3 31,0-36,0 g/dL
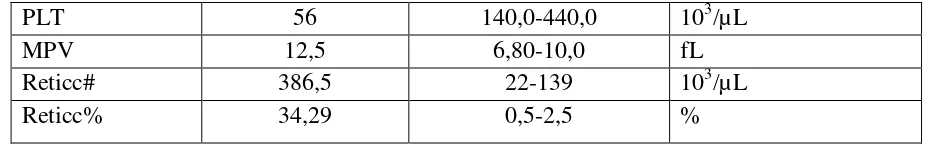
4
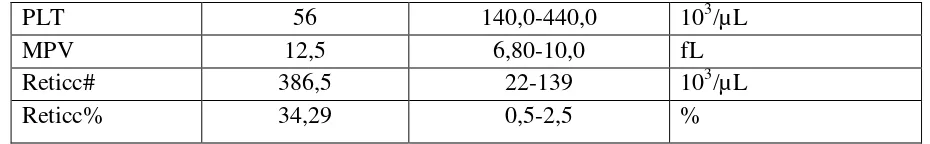
PLT 56 140,0-440,0 103/µL
MPV 12,5 6,80-10,0 fL
Reticc# 386,5 22-139 103/µL
Reticc% 34,29 0,5-2,5 %
Tabel. 2 Hasil tes Kimia Darah
Pemeriksaan Hasil Rentang
Normal Satuan
Faal Ginjal :
- Urea 8 6 - 20 mg/dl
- Creatinin 0,63 0,5 – 0,9 mg/dl
- Uric acid 5,1 2,4 – 5,7 mg/dl
Faal Hati
- Total Protein 6,7 6,4 – 8,3 g/dl
- Albumin 3,9 3,4 – 4,8 g/dl
- Globulin 2,8 1,8 – 3,5 g/dl
- Bilirubin total 4,2 0 – 1,1 mg/dl
- Bilirubin direk 0,7 0 – 0,30 mg/dl
- Bilirubin indirek 3,5 0 – 0,8 mg/dl
- SGPT 15 0 - 34 U/L
- SGOT 72 0 – 27 U/L
- Alkali phosphatase 45 42 – 98 U/L
- Gamma GT 14 < 39 U/L
Secara radiologis, kondisi jantung dan paru tidak tampak adanya kelainan. Dari hasil hapusan darah tepi didapatkan eritrosit normokromik normositik, polikromasia dan normoblast meningkat. Leukosit jumlahnya meningkat (leukositosis), differensial count normal, blast menurun. Trombosit jumlahnya menurun (trombositopenia) dan bergerombol (clamping). Direct Coombs Test (DCT) positif dimana ditemukan adanya Auto Immune Antibody juga ditemukan IgG dan C3 yang terikat pada sel darah merah penderita. Indirect Coombs Test (ICT) positif yaitu ditemukan adanya Ireguller Allo Antibody yang bebas di dalam
serum penderita pada suhu 200C dan suhu 370C. Dari hasil tes urinalisis didapatkan pH 6,5 (Normal: 7,35 – 7,45), Leukosit (+) 25 leuco/µL, Protein (+) 25 mg/dL, Urobilinogen (+) 1 mg/dl, Eritrosit (+) 50 ery/µL, dimana normalnya pada urin tidak ditemukan leukosit, protein, urobilinogen, dan eritrosit. Dari hasil anamnesis, pemeriksaan fisik dan hasil pemeriksaan penunjang yang telah dilakukan, maka pasien ini didiagnosis menderita sindrom Evans dimana terjadi AIHA dan ITP secara bersamaan.
5 terhadap metilprednisolon dan transfusi
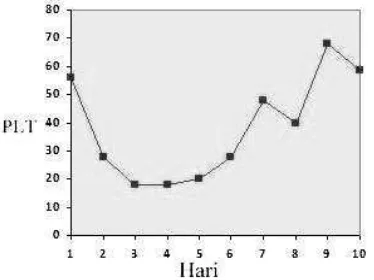
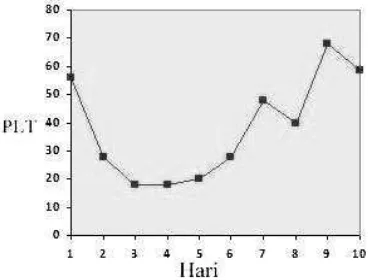
WRC dapat dilihat pada Gambar 1. Dimana hasilnya terjadi peningkatan hemoglobin setiap harinya selama 10 hari perawatan sehingga dapat dikatakan AIHA berespon baik terhadap pemberian terapi metilprednisolon dan transfusi WRC. Respon trombosit terhadap pemberian metilprednisolon dan transfusi TC dapat dilihat pada Gambar 2. Dimana hasilnya pada awal terapi trombosit mengalami penurunan akan tetapi pada hari ke enam trombosit mulai mengalami peningkatan, sehingga dapat dikatakan ITP berespon baik terhadap pemberian terapi metilprednisolon dan transfusi TC. Selain itu pasien juga diberikan terapi suportif berupa terapi cairan IVFD NaCl 0,9% dan terapi oksigen.
Gambar 1. Respon Hemoglobin terhadap pemberian metilprednisolon dan transfusi WRC selama 10 hari.
Gambar 2. Respon Trombosit terhadap pemberian metilprednisolon dan transfusi TC selama 10 hari.
DISKUSI
Pada tahun 1951, Evans dan rekan menggambarkan sindrom Evans dengan sekelompok pasien yang secara klinis ditandai dengan Autoimmune Hemolytic Anemias (AIHA) dan Idiopathic Trombocytopenias Purpura (ITP). Anemia dan trombositopenia bervariasi dalam hal onset dan durasi. Meskipun sindrom Evans tampaknya menjadi gangguan regulasi imun, patofisiologi yang tepat tidak diketahui.2 Sindrom Evans dapat diklasifikasikan menjadi dua yaitu primer (idiopatik) dan sekunder (berhubungan dengan beberapa penyakit). Ada laporan kasus Sindrom Evan berhubungan dengan SLE, incomplete lupus, sindrom antifosfolipid primer, sindrom Sjogren, defisiensi IgA, B dan T sel limfoma Hodgkins non ganas dan leukemia limfositik kronis.5
Sindrom Evans berisiko berkembang menjadi masalah autoimun lainnya, hipogammaglobulinemia, dan 58% dari anak-anak dengan sindrom Evans memiliki sel T CD4-/CD8- yang merupakan prediktor kuat untuk mengalami sindrom limfoproliferatif autoimun.4 Kebanyakan pasien dengan sindrom Evans mengalami penurunan tingkat serum IgG, IgM, IgA dan penurunan sintesis in vitro dari IgG dan atau IgM. Terjadi penurunan limfosit T -helper dan peningkatan limfosit T - supresor yang mirip dengan anemia hipoplasia kongenital dan trombositopenia amegakaryocytic. Temuan ini telah menimbulkan spekulasi bahwa sitopenia pada sindrom Evans mungkin berhubungan dengan kelainan sel T. Peran imunisasi anak dalam pengembangan sindrom Evans telah diteliti, namun asosiasi tertentu belum dilaporkan.2
6 67% dari pasien memiliki anemia.
Selain itu, 24% pasien memiliki neutropenia dan 14% memiliki pansitopenia.2
Kriteria untuk diagnosis sindrom Evans, yaitu: (1) anemia hemolitik dengan tes Coombs direk positif; (2) trombositopenia yang terjadi baik secara simultan maupun secara berurutan; dan (3) tidak adanya etiologi mendasari yang dikenal.6Presentasi klinis sindrom Evans mencakup fitur biasa anemia haemolitik seperti pucat, kelelahan, pusing, lesu, sakit kuning, gagal jantung. Selain itu ditemukan juga tanda-tanda trombositopenia termasuk purpura, peteki, ekimosis, memar, perdarahan mucocutaneous. Pemeriksaan fisik dapat mengungkapkan limfadenopati, hepatomegali dan atau splenomegali.1,2
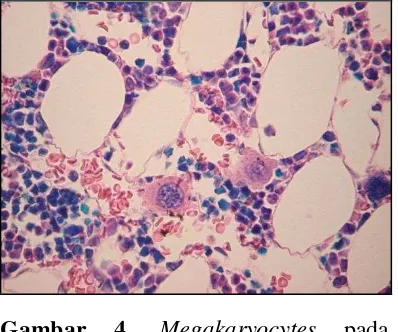
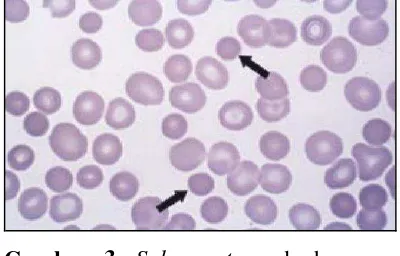
Pemeriksaan darah lengkap dan jumlah retikulosit mengungkapkan anemia, trombositopenia, neutropenia, atau sitopenia pada pasien dengan sindrom Evans. Hapusan darah tepi harus diperiksa untuk menyingkirkan diagnosis lain yang mendasarinya seperti keganasan, anemia hemolitik mikroangiopati, hemolitik kongenital dan kondisi-kondisi trombositopenia. Pasien dengan AIHA pada hapusan darah tepi ditemukan polychromasia dan spherocytes (Gambar 3). Normal atau peningkatan jumlah megakariosit mengkonfirmasi bahwa trombositopenia disebabkan oleh meningkatnya kehancuran dalam darah.1,2
Gambar 3. Spherocyte pada hapusan darah tepi.11
Tanda hemolisis harus dicari termasuk peningkatan jumlah retikulosit, hiperbilirubinemia tak terkonjugasi dan penurunan haptoglobin. Pada kasus anemia hemolitik sebaiknya pasien diperiksa uji antiglobulin langsung (DAT), juga dikenal sebagai tes Coombs direk, yang bertujuan untuk menunjukkan adanya antibodi atau komplemen pada permukaan sel darah merah dan merupakan ciri khas hemolisis terutama untuk anemia hemolitik autoimun.3 Uji antiglobulin langsung/Tes Coombs direk hampir selalu positif dan mungkin positif untuk IgG, komplemen (C3), atau keduanya. Hasil tes antiglobulin tidak langsung mungkin juga positif dalam 52-83 % pasien. Berbagai antibodi yang bereaksi terhadap sel darah merah dan trombosit (misalnya antieritrosit, antineutrofil, antibodi antitrombosit) terjadi dalam hubungan dengan sindrom Evans. Pengujian autoantibodi trombosit dan granulosit mungkin positif, tetapi hasil negatif tidak mengecualikan diagnosis.1,2
7
Gambar 4. Megakaryocytes pada sumsum tulang.12
Pemeriksaan imunologi juga diperlukan untuk melihat adanya antitrombosit IgG pada permukaan trombosit atau dalam serum. Sebanyak 91% dari pasien yang terkena sindrom Evans ditemukan antibodi antitrombosit sementara sekitar 81% menunjukkan antibodi anti-neutrofil.7 Yang lebih spesifik adalah antibodi terhadap gpIIb/IIIa atau gpIb untuk mendiagnosis ITP.8
Pada kasus dilakukan pemeriksaan LDH (lactic dehydrogenase) dimana terjadi peningkatan LDH yaitu 2048H U/L, normalnya 240 – 480H U/L. Hal tersebut dilakukan untuk mendeteksi adanya hemolisis, dimana hemolisis intravaskuler menyebabkan sumsum tulang merespon dengan mengaktifkan proses eritropoesis, yang mengakibatkan beredarnya sel darah merah yang imatur. Sel darah merah imatur ini mudah mengalami destruksi, dan mengeluarkan isoenzim eritrosit. Isoenzim ini akan terikat dengan plasma LDH. Peningkatan kadar LDH tanpa disertai peningkatan kadar SGOT dan SGPT menunjukkan terjadinya hemolisis.9
Selain peningkatan LDH, kerusakan dari sel darah merah intravaskuler menyebabkan hemoglobin keluar dari intravaskuler. Lepasnya hemoglobin ini akan terikat dengan haptoglobin, dimana kompleks
hemaglobin-haptoglobin akan dimetabolisme di hepar dengan cepat. Hemoglobin bebas pada sistim retikuloendotel akan berubah menjadi bilirubin. Peningkatan kadar bilirubin menunjukkan terjadinya hemolisis.9 Hal tersebut ditunjukkan dari hasil tes bilirubin pada kasus ini yaitu 4,2 mg/dl (normal 0 – 1,1 mg/dl).
Sindrom Evans adalah diagnosis eksklusi dan menurut definisi gangguan pengganggu lainnya tidak harus hadir. Sebelum menerima diagnosis sindrom Evans penyebab lain dari cytopenia acquired immune harus singkirkan, khususnya SLE (systemic lupus erythematosis), defisiensi IgA, CVID, acquired immunodeficiency syndrome dan ALPS karena semua memerlukan manajemen yang berbeda. Kondisi lain yang menyebabkan anemia hemolitik dan trombositopenia bersamaan dan dapat meniru sindrom Evans termasuk paroxysmal nocturnal hemoglobinuria (PNH).3 Oleh karena itu untuk menyingkirkan SLE harus dicari dengan mengukur antibodi antinuclear (ANA), double-stranded DNA (dsDNA) dan faktor rheumatoid. Yang paling penting adalah perbedaan dengan ALPS (Autoimmune Lymphoproliferative Syndrome), maka pengukuran darah perifer subset sel - T dengan sitometri sangat penting dalam semua kasus sindrom Evans.1,2
8 pasien terbebas dari prednison.4 Pada
kasus, pasien telah diberikan methiprednisolon 2 x125 mg IV. Hal ini bertujuan untuk menurunkan ikatan immunoglobulin ke reseptor permukaan sel dan menghambat sintesis dan atau pelepasan interleukin, sehingga T-limfosit blastogenesis menurun dan mengurangi perluasan respon immun primer.10
Imunoglobulin intravena (IVIG) dapat membantu pasien yang bergantung pada steroid. Terapi lain yang efektif dalam seri kecil termasuk danazol, siklosporin, azathioprine, cyclophosphamide, dan vincristine. Baru-baru ini, rituximab (antibodi monoklonal yang menargetkan CD20 pada limfosit B) telah digunakan dalam pengelolaan pasien refrakter. Tanggapan bervariasi, dalam suatu penelitian penggunaan rituximab pada 18 pasien (usia 0,3-65 tahun) hasilnya menggembirakan, dengan remisi lengkap selama 17 bulan. Pertimbangkan splenektomi dalam kasus-kasus refrakter. Splenektomi dapat mengurangi kebutuhan steroid, walaupun kekambuhan biasanya terjadi
dalam waktu 1-2 bulan
postsplenektomi.4
Dalam pengaturan akut, transfusi darah dan transfusi trombosit mungkin diperlukan untuk mengurangi gejala, walaupun penggunaannya harus diminimalkan. Pada kasus pasien diberikan transfusi WRC (washed red cell) yang diperoleh dengan mencuci eritrosit pekat 2-3 kali dengan NaCl fisiologis dalam jumlah yang sama. Keuntungan transfusi sel darah merah adalah tidak membebani sirkulasi, tidak memperberat fungsi ginjal, dan sedikit mengurangi reaksi alergi karena tidak disertai pemberian plasma yang tinggi protein. Sediaan ini aman bagi resipien yang alergi terhadap plasma manusia, anemia hemolitik yang didapat, dan transfusi pada transplantasi ginjal.10 Pada kasus, setelah pemberian WRC
mulai hari ke pertama, hemoglobin pasien mengalami peningkatan setiap harinya. Transfusi trombosit juga dapat digunakan dalam pendarahan parah dengan jumlah trombosit yang sangat rendah. Pada kasus, tidak ditemukan tanda-tanda pendarahan akan tetapi trombosit pasien 18x103/uL pada oleh karena itu pada hari keempat diberikan transfusi berupa TC (Trombosit consentrate). TC biasanya digunakan untuk mengatasi keadaan trombositopenia berat. 10
9 menunjukkan adanya anemia hemolitik
autoimun tipe hangat. Pemeriksaan darah lengkap juga mendapatkan hasil penurunan trombosit yaitu sebesar 56x103/μL hal ini menunjukkan adanya trombositopenia, dengan tidak adanya penyakit yang mendasari yang disebut juga Idiopathic Thrombocytopenic Purpura (ITP). Dari dua diagnosis di atas yaitu anemia hemolitik autoimun (AIHA) dan ITP maka pasien menderita sindrom Evans. Terapi yang diberikan yaitu transfusi WRC, tranfusi TC, methylprednisolone, NaCl 90%, dan terapi oksigen. Diberikan edukasi pasien dan keluarga tentang sifat kronis kondisi ini, yang dapat mencakup periode remisi dan eksaserbasi. Pasien dan keluarga juga dijelaskan efek potensial yang merugikan dari obat-obatan, khususnya penggunaan steroid jangka panjang
DAFTAR PUSTAKA
1. Alice Norton, Irene Robert. Management of Evans syndrome. British journal of Haematology. 2005. 132 : 125-37
2. Prasad Mathew, Gary R Jones, Mary L Windle, Gary D Crouch, Samuel Gross, Robert J Arceci, et al. Evans Syndrome. Diakses 21 November
2013. Diunduh
dari:http://emedicine.medscape.com /article/955266-overview.
3. Marc Michel, Valérie Chanet, Agnès Dechartres, Anne-Sophie Morin, Jean-Charles Piette, Lorenzo Cirasino, et al. The Spectrum of Evans Syndrome in adults : new insight into the disease based on analysis of 68 cases. The American Society of Hematology. 2009. Vol 114, number 15, p 3167-72.
4. Liliana Isacianu, Miron Ingrid, Tansanu Iroan. Evans Syndrome : A rare Cause of Hemolysis (And Thrombocytopenia) in Children.
Fascicula. 2012. Vol. 17, number 1, p 19-22.
5. Priti Dave, Kavita Krishna, AG Diwan. Evans Syndrome Revisited. JAPI. 2012. Vol. 60, p 60-1.
6. Ahmendul Kabir, Jayanta Banik, Ratan Dasgupta, Robed Amin, AM Wasiq Faisal, ASM Mafidul Islam, et. al. Evans Syndrome. J Medicine. 2010. Vol. 11, p 78-82.
7. Manuel Monti, Lucia Stefanecchia, Manolo Filippucci, Alessio Monti, Giovanni Maria Vincentelli, Francesco Borbognoni. A Strange case of Evans Syndrome. Italian Journal of Medicine. 2012. Vol. 7, p 305-9.
8. Reksodiputro, A.H, Madjid, A., Rachman, A.M. Buku Ajar Ilmu Penyakit Dalam. Edisi kedua. Jakarta. 2007.
9. Bakta, I Made. Hematologi Klinik Ringkas. Edisi pertama. Jakarta. 2007.
10.Betram G. Katzung. Farmakologi Dasar dan Kinik. Edisi keenam. Jakarta : 1997. P 897-98.
11.Gurpreet Dhaliwal, M.D., Patricia A. Cornett, M.D., and Lawrence M. Tierney, Jr., M.D. Hemolytic Anemia. American Family Physician. 2004. Vol 69, number 11. 12.Anonim. Megakaryocytes. diakses
1
CASE REPORT
EVANS SYNDROME IN 24 YEARS OLD WOMEN
Made Gita Ratnasari, Sianny Herawati, I Wayan Putu Sutirta Yasa, Anak Agung Wiradewi Lestari
Departement of Clinical Pathology, Medical Faculty of Udayana University Sanglah Hospital Denpasar
ABSTRACT
Evans syndrome is a rare case, a condition which occurs with simultaneously or sequentially Autoimmune Hemolytic Anemias (AIHA) and Idiopathic Trombocytopenias Purpura (ITP) and sometimes together with neutropenia, in the absence of etiology. This syndrome may be associated with other autoimmune or lymphoproliferative diseases. Course of the disease is very serious and, in rare cases, can even be life-threatening. First-line treatment consists of steroids and or immunoglobulin. We reported a woman, 24 years old came with complaints of weakness for 1 week, sometimes feel dizziness, jaundice, colour of urine is like tea. Of a complete blood count showed hemoglobin decreased, equal to 3,8 g/dL, and a decrease in platelets that is equal to 56x103/μl. Physical examination did not reveal any organ enlargement. The patient was given WRC (washed red cell) transfusion, TC (trombosit consentrate) transfusion, and methylprednisolone. In addition, patient were also given supportive theraphy such as fluid therapy IVFD NaCl 0,9% and oxygen therapy. Patient was hospitalized to establish diagnosis and was discharged after ten day of hospitalization. Prognosis is good, where no complication such as bleeding or sepsis.
Keywords : Evans syndrome, Autoimmune disease, AIHA, ITP
LAPORAN KASUS
EVANS SYNDROME PADA WANITA USIA 24 TAHUN
Made Gita Ratnasari, Sianny Herawati, I Wayan Putu Sutirta Yasa, Anak Agung Wiradewi Lestari
Bagian/SMF Patologi Klinik, Fakultas Kedokteran Universitas Udayana Rumah Sakit Umum Pusat Sanglah Denpasar
ABSTRAK
2 56x103/μl. Pada pemeriksaan fisik tidak ditemukan adanya pembesaran organ. Terapi yang diberikan yaitu transfusi WRC (washed red cell), tranfusi TC (trombosit consentrate), dan metilprednisolon. Selain itu pasien juga diberikan terapi suportif berupa terapi cairan IVFD NaCl 0,9% dan terapi oksigen. Pasien masuk rumah sakit untuk diagnosis dan dipulangkan setelah sepuluh hari perawatan. Prognosis pasien baik, dimana tidak terjadi komplikasi seperti pendarahan ataupun sepsis.
Kata kunci: Sindrom Evans, Penyakit Autoimun, AIHA, ITP
PENDAHULUAN
Sindrom Evans didefinisikan sebagai kombinasi baik secara bersamaan atau berurutan dari Autoimmune Hemolytic Anemias
(AIHA) dan Idiopathic
Trombocytopenias Purpura (ITP), dan terkadang bersama-sama dengan neutropenia, dengan tidak adanya etiologi yang mendasari.1,2 Sindrom Evans adalah penyakit langka dimana hanya 0,8% - 3,7% yang terdiagnosis dari pasien yang menderita ITP dan AIHA. Kejadian familial jarang ditemukan.3
Di Malaysia tahun 1992, sindrom Evans ditemukan pada 12 dari 220 pasien dewasa dengan ITP dan 102 dengan AIHA.2 Sebuah penelitian oleh Silverstein dan Heck di Amerika, sebanyak 399 kasus AIHA dan 367 kasus ITP pasien dewasa, hanya enam dari 766 pasien ini mengalami sindrom Evans.1 Sindrom ini dominan terjadi pada ras kulit putih, dari 42 pasien yang dilaporkan dalam survei nasional terdapat 29 berkulit putih, 7 hitam, dan 6 memiliki latar belakang ras lainnya. Tidak ada predileksi pada sindrom Evans, pada suatu studi yang dilakukan oleh Liliana di Rumania tahun 2012 menyebutkan sindrom ini mempengaruhi anak laki-laki lebih sering daripada perempuan dengan rasio 1,4:1.4 Dan dalam suatu penelitian oleh Genty di Prancis tahun 2002, sebanyak 67% kasus sindrom evans terjadi pada wanita. Sindrom Evans terjadi pada
individu dari segala usia. Dalam sebuah survei tahun 1997 di Amerika Utara, melaporkan rata-rata usia saat diagnosis adalah usia 7,7 tahun.2
Etiologi sindrom Evans masih belum diketahui. Autoantibodi bereaksi terhadap antigen spesifik untuk sel darah merah, trombosit, atau neutrofil, tetapi autoantibodi ini tidak bereaksi silang.4 Sindrom Evans adalah diagnosis eksklusi. Gangguan seperti infeksi, penyakit rematologi, dan keganasan harus dikesampingkan. Hasil pemeriksaan darah lengkap dapat dijumpai anemia, trombositopenia, neutropenia, atau sitopenia pada pasien dengan sindrom Evans.1,2
Terapi lini pertama yang paling umum digunakan adalah kortikosteroid dan atau imunoglobulin intravena (IVIG).1,2 Sindrom Evans kadang-kadang fatal dan memiliki tingkat kematian sebesar 7%. Penyebab kematian terutama disebabkan oleh perdarahan dan sepsis.4
ILUSTRASI KASUS
3 mendenging. Satu minggu sebelumnya
pasien berobat ke dokter swasta didiagnosis anemia dan sakit kuning lalu diberikan vitamin. Sehari sebelum pasien datang ke RS. Sanglah, pasien berobat ke RS. Swasta. Kadar hemoglobin saat itu adalah 3,8 g/dL, dan kadar trombosit 56x103/µl. Akhirnya RS. Swasta tersebut merujuk pasien ke RS. Sanglah. Dari hasil anamnesis didapatkan pasien mengeluh lemas, terdapat riwayat panas badan, tidak ada riwayat ke wilayah timur, BAK (buang air kecil) dikatakan seperti teh, serta tidak terdapat rambut rontok.
Dari riwayat penyakit dahulu, pasien mengatakan tidak pernah menderita hipertensi, kencing manis, penyakit jantung, asthma, stroke, liver, ginjal TBC Paru. Riwayat keluhan yang sama dalam keluarga disangkal. Tidak ada riwayat penyakit jantung, hipertensi, kencing manis dan asthma di keluarga pasien. Tidak ada kebiasaan merokok dan mengkonsumsi alkohol. Pada pemeriksaan fisik, keadaan umum baik, tekanan darah 110/80 mmHg, nadi 100 kali/menit, respirasi 22 kali/menit, suhu aksila 37,8C.
Pada pemeriksaan mata ditemukan tanda anemia serta ikterus,
refleks pupil normal, dan tidak ditemukan adanya edema palpebral. Pemeriksaan THT dan leher dalam batas normal, tidak ditemukan pembesaran kelenjar dan kaku kuduk. Torak terlihat simetris, paru serta jantung berada dalam batas normal, suara jantung S1S2 tunggal reguler, serta
tidak ditemukan murmur, rhonki, dan ‘wheezing’. Pada pemeriksaan abdomen tidak ditemukan adanya kelainan seperti distensi, meteorismus, ascites, nyeri tekan, dan tidak ada pembesaran organ. Pada keempat ekstremitas teraba hangat serta tidak ditemukan edema.
Hasil pemeriksaan darah dapat dilihat pada Tabel 1 dan Tabel 2. Dimana hasilnya yaitu terjadi peningkatan WBC (leukositosis), penurunan HGB disertai peningkatan MCV dan retikulosit (anemia hemolitik), dan ditemukan juga penurunan trombosit (trombositopenia). Dari hasil tes fungsi ginjal tidak didapatkan kelainan, sedangkan pada tes fungsi hati didapatkan kelainan berupa peningkatan bilirubin dan SGOT.
Tabel. 1 Hasil Tes Darah Lengkap
Tes Hasil Rentang Normal Unit
WBC 13,26 4,10-11,0 103/µL
Neu# 9,61 2,50-7,50 103/µL
Lym# 2,67 1,00-4,00 103/µL
Mono# 0,44 1,00-1,20 103/µL
Eos# 0,02 0,00-0,500 103/µL
Baso# 0,16 0,00-0,100 103/µL
RBC 1,13 4,00-5,20 106/µL
HGB 3,8 12,0-16,0 g/dL
HCT 12,3 36,0-46,0 %
MCV 108,7 80,0-100,0 fL
MCH 34,1 26,0-34,0 Pg
MCHC 31,3 31,0-36,0 g/dL
4
PLT 56 140,0-440,0 103/µL
MPV 12,5 6,80-10,0 fL
Reticc# 386,5 22-139 103/µL
Reticc% 34,29 0,5-2,5 %
Tabel. 2 Hasil tes Kimia Darah
Pemeriksaan Hasil Rentang
Normal Satuan
Faal Ginjal :
- Urea 8 6 - 20 mg/dl
- Creatinin 0,63 0,5 – 0,9 mg/dl
- Uric acid 5,1 2,4 – 5,7 mg/dl
Faal Hati
- Total Protein 6,7 6,4 – 8,3 g/dl
- Albumin 3,9 3,4 – 4,8 g/dl
- Globulin 2,8 1,8 – 3,5 g/dl
- Bilirubin total 4,2 0 – 1,1 mg/dl
- Bilirubin direk 0,7 0 – 0,30 mg/dl
- Bilirubin indirek 3,5 0 – 0,8 mg/dl
- SGPT 15 0 - 34 U/L
- SGOT 72 0 – 27 U/L
- Alkali phosphatase 45 42 – 98 U/L
- Gamma GT 14 < 39 U/L
Secara radiologis, kondisi jantung dan paru tidak tampak adanya kelainan. Dari hasil hapusan darah tepi didapatkan eritrosit normokromik normositik, polikromasia dan normoblast meningkat. Leukosit jumlahnya meningkat (leukositosis), differensial count normal, blast menurun. Trombosit jumlahnya menurun (trombositopenia) dan bergerombol (clamping). Direct Coombs Test (DCT) positif dimana ditemukan adanya Auto Immune Antibody juga ditemukan IgG dan C3 yang terikat pada sel darah merah penderita. Indirect Coombs Test (ICT) positif yaitu ditemukan adanya Ireguller Allo Antibody yang bebas di dalam
serum penderita pada suhu 200C dan suhu 370C. Dari hasil tes urinalisis didapatkan pH 6,5 (Normal: 7,35 – 7,45), Leukosit (+) 25 leuco/µL, Protein (+) 25 mg/dL, Urobilinogen (+) 1 mg/dl, Eritrosit (+) 50 ery/µL, dimana normalnya pada urin tidak ditemukan leukosit, protein, urobilinogen, dan eritrosit. Dari hasil anamnesis, pemeriksaan fisik dan hasil pemeriksaan penunjang yang telah dilakukan, maka pasien ini didiagnosis menderita sindrom Evans dimana terjadi AIHA dan ITP secara bersamaan.
5 terhadap metilprednisolon dan transfusi
WRC dapat dilihat pada Gambar 1. Dimana hasilnya terjadi peningkatan hemoglobin setiap harinya selama 10 hari perawatan sehingga dapat dikatakan AIHA berespon baik terhadap pemberian terapi metilprednisolon dan transfusi WRC. Respon trombosit terhadap pemberian metilprednisolon dan transfusi TC dapat dilihat pada Gambar 2. Dimana hasilnya pada awal terapi trombosit mengalami penurunan akan tetapi pada hari ke enam trombosit mulai mengalami peningkatan, sehingga dapat dikatakan ITP berespon baik terhadap pemberian terapi metilprednisolon dan transfusi TC. Selain itu pasien juga diberikan terapi suportif berupa terapi cairan IVFD NaCl 0,9% dan terapi oksigen.
Gambar 1. Respon Hemoglobin terhadap pemberian metilprednisolon dan transfusi WRC selama 10 hari.
Gambar 2. Respon Trombosit terhadap pemberian metilprednisolon dan transfusi TC selama 10 hari.
DISKUSI
Pada tahun 1951, Evans dan rekan menggambarkan sindrom Evans dengan sekelompok pasien yang secara klinis ditandai dengan Autoimmune Hemolytic Anemias (AIHA) dan Idiopathic Trombocytopenias Purpura (ITP). Anemia dan trombositopenia bervariasi dalam hal onset dan durasi. Meskipun sindrom Evans tampaknya menjadi gangguan regulasi imun, patofisiologi yang tepat tidak diketahui.2 Sindrom Evans dapat diklasifikasikan menjadi dua yaitu primer (idiopatik) dan sekunder (berhubungan dengan beberapa penyakit). Ada laporan kasus Sindrom Evan berhubungan dengan SLE, incomplete lupus, sindrom antifosfolipid primer, sindrom Sjogren, defisiensi IgA, B dan T sel limfoma Hodgkins non ganas dan leukemia limfositik kronis.5
Sindrom Evans berisiko berkembang menjadi masalah autoimun lainnya, hipogammaglobulinemia, dan 58% dari anak-anak dengan sindrom Evans memiliki sel T CD4-/CD8- yang merupakan prediktor kuat untuk mengalami sindrom limfoproliferatif autoimun.4 Kebanyakan pasien dengan sindrom Evans mengalami penurunan tingkat serum IgG, IgM, IgA dan penurunan sintesis in vitro dari IgG dan atau IgM. Terjadi penurunan limfosit T -helper dan peningkatan limfosit T - supresor yang mirip dengan anemia hipoplasia kongenital dan trombositopenia amegakaryocytic. Temuan ini telah menimbulkan spekulasi bahwa sitopenia pada sindrom Evans mungkin berhubungan dengan kelainan sel T. Peran imunisasi anak dalam pengembangan sindrom Evans telah diteliti, namun asosiasi tertentu belum dilaporkan.2
6 67% dari pasien memiliki anemia.
Selain itu, 24% pasien memiliki neutropenia dan 14% memiliki pansitopenia.2
Kriteria untuk diagnosis sindrom Evans, yaitu: (1) anemia hemolitik dengan tes Coombs direk positif; (2) trombositopenia yang terjadi baik secara simultan maupun secara berurutan; dan (3) tidak adanya etiologi mendasari yang dikenal.6Presentasi klinis sindrom Evans mencakup fitur biasa anemia haemolitik seperti pucat, kelelahan, pusing, lesu, sakit kuning, gagal jantung. Selain itu ditemukan juga tanda-tanda trombositopenia termasuk purpura, peteki, ekimosis, memar, perdarahan mucocutaneous. Pemeriksaan fisik dapat mengungkapkan limfadenopati, hepatomegali dan atau splenomegali.1,2
Pemeriksaan darah lengkap dan jumlah retikulosit mengungkapkan anemia, trombositopenia, neutropenia, atau sitopenia pada pasien dengan sindrom Evans. Hapusan darah tepi harus diperiksa untuk menyingkirkan diagnosis lain yang mendasarinya seperti keganasan, anemia hemolitik mikroangiopati, hemolitik kongenital dan kondisi-kondisi trombositopenia. Pasien dengan AIHA pada hapusan darah tepi ditemukan polychromasia dan spherocytes (Gambar 3). Normal atau peningkatan jumlah megakariosit mengkonfirmasi bahwa trombositopenia disebabkan oleh meningkatnya kehancuran dalam darah.1,2
Gambar 3. Spherocyte pada hapusan darah tepi.11
Tanda hemolisis harus dicari termasuk peningkatan jumlah retikulosit, hiperbilirubinemia tak terkonjugasi dan penurunan haptoglobin. Pada kasus anemia hemolitik sebaiknya pasien diperiksa uji antiglobulin langsung (DAT), juga dikenal sebagai tes Coombs direk, yang bertujuan untuk menunjukkan adanya antibodi atau komplemen pada permukaan sel darah merah dan merupakan ciri khas hemolisis terutama untuk anemia hemolitik autoimun.3 Uji antiglobulin langsung/Tes Coombs direk hampir selalu positif dan mungkin positif untuk IgG, komplemen (C3), atau keduanya. Hasil tes antiglobulin tidak langsung mungkin juga positif dalam 52-83 % pasien. Berbagai antibodi yang bereaksi terhadap sel darah merah dan trombosit (misalnya antieritrosit, antineutrofil, antibodi antitrombosit) terjadi dalam hubungan dengan sindrom Evans. Pengujian autoantibodi trombosit dan granulosit mungkin positif, tetapi hasil negatif tidak mengecualikan diagnosis.1,2
7
Gambar 4. Megakaryocytes pada sumsum tulang.12
Pemeriksaan imunologi juga diperlukan untuk melihat adanya antitrombosit IgG pada permukaan trombosit atau dalam serum. Sebanyak 91% dari pasien yang terkena sindrom Evans ditemukan antibodi antitrombosit sementara sekitar 81% menunjukkan antibodi anti-neutrofil.7 Yang lebih spesifik adalah antibodi terhadap gpIIb/IIIa atau gpIb untuk mendiagnosis ITP.8
Pada kasus dilakukan pemeriksaan LDH (lactic dehydrogenase) dimana terjadi peningkatan LDH yaitu 2048H U/L, normalnya 240 – 480H U/L. Hal tersebut dilakukan untuk mendeteksi adanya hemolisis, dimana hemolisis intravaskuler menyebabkan sumsum tulang merespon dengan mengaktifkan proses eritropoesis, yang mengakibatkan beredarnya sel darah merah yang imatur. Sel darah merah imatur ini mudah mengalami destruksi, dan mengeluarkan isoenzim eritrosit. Isoenzim ini akan terikat dengan plasma LDH. Peningkatan kadar LDH tanpa disertai peningkatan kadar SGOT dan SGPT menunjukkan terjadinya hemolisis.9
Selain peningkatan LDH, kerusakan dari sel darah merah intravaskuler menyebabkan hemoglobin keluar dari intravaskuler. Lepasnya hemoglobin ini akan terikat dengan haptoglobin, dimana kompleks
hemaglobin-haptoglobin akan dimetabolisme di hepar dengan cepat. Hemoglobin bebas pada sistim retikuloendotel akan berubah menjadi bilirubin. Peningkatan kadar bilirubin menunjukkan terjadinya hemolisis.9 Hal tersebut ditunjukkan dari hasil tes bilirubin pada kasus ini yaitu 4,2 mg/dl (normal 0 – 1,1 mg/dl).
Sindrom Evans adalah diagnosis eksklusi dan menurut definisi gangguan pengganggu lainnya tidak harus hadir. Sebelum menerima diagnosis sindrom Evans penyebab lain dari cytopenia acquired immune harus singkirkan, khususnya SLE (systemic lupus erythematosis), defisiensi IgA, CVID, acquired immunodeficiency syndrome dan ALPS karena semua memerlukan manajemen yang berbeda. Kondisi lain yang menyebabkan anemia hemolitik dan trombositopenia bersamaan dan dapat meniru sindrom Evans termasuk paroxysmal nocturnal hemoglobinuria (PNH).3 Oleh karena itu untuk menyingkirkan SLE harus dicari dengan mengukur antibodi antinuclear (ANA), double-stranded DNA (dsDNA) dan faktor rheumatoid. Yang paling penting adalah perbedaan dengan ALPS (Autoimmune Lymphoproliferative Syndrome), maka pengukuran darah perifer subset sel - T dengan sitometri sangat penting dalam semua kasus sindrom Evans.1,2
8 pasien terbebas dari prednison.4 Pada
kasus, pasien telah diberikan methiprednisolon 2 x125 mg IV. Hal ini bertujuan untuk menurunkan ikatan immunoglobulin ke reseptor permukaan sel dan menghambat sintesis dan atau pelepasan interleukin, sehingga T-limfosit blastogenesis menurun dan mengurangi perluasan respon immun primer.10
Imunoglobulin intravena (IVIG) dapat membantu pasien yang bergantung pada steroid. Terapi lain yang efektif dalam seri kecil termasuk danazol, siklosporin, azathioprine, cyclophosphamide, dan vincristine. Baru-baru ini, rituximab (antibodi monoklonal yang menargetkan CD20 pada limfosit B) telah digunakan dalam pengelolaan pasien refrakter. Tanggapan bervariasi, dalam suatu penelitian penggunaan rituximab pada 18 pasien (usia 0,3-65 tahun) hasilnya menggembirakan, dengan remisi lengkap selama 17 bulan. Pertimbangkan splenektomi dalam kasus-kasus refrakter. Splenektomi dapat mengurangi kebutuhan steroid, walaupun kekambuhan biasanya terjadi
dalam waktu 1-2 bulan
postsplenektomi.4
Dalam pengaturan akut, transfusi darah dan transfusi trombosit mungkin diperlukan untuk mengurangi gejala, walaupun penggunaannya harus diminimalkan. Pada kasus pasien diberikan transfusi WRC (washed red cell) yang diperoleh dengan mencuci eritrosit pekat 2-3 kali dengan NaCl fisiologis dalam jumlah yang sama. Keuntungan transfusi sel darah merah adalah tidak membebani sirkulasi, tidak memperberat fungsi ginjal, dan sedikit mengurangi reaksi alergi karena tidak disertai pemberian plasma yang tinggi protein. Sediaan ini aman bagi resipien yang alergi terhadap plasma manusia, anemia hemolitik yang didapat, dan transfusi pada transplantasi ginjal.10 Pada kasus, setelah pemberian WRC
mulai hari ke pertama, hemoglobin pasien mengalami peningkatan setiap harinya. Transfusi trombosit juga dapat digunakan dalam pendarahan parah dengan jumlah trombosit yang sangat rendah. Pada kasus, tidak ditemukan tanda-tanda pendarahan akan tetapi trombosit pasien 18x103/uL pada oleh karena itu pada hari keempat diberikan transfusi berupa TC (Trombosit consentrate). TC biasanya digunakan untuk mengatasi keadaan trombositopenia berat. 10
9 menunjukkan adanya anemia hemolitik
autoimun tipe hangat. Pemeriksaan darah lengkap juga mendapatkan hasil penurunan trombosit yaitu sebesar 56x103/μL hal ini menunjukkan adanya trombositopenia, dengan tidak adanya penyakit yang mendasari yang disebut juga Idiopathic Thrombocytopenic Purpura (ITP). Dari dua diagnosis di atas yaitu anemia hemolitik autoimun (AIHA) dan ITP maka pasien menderita sindrom Evans. Terapi yang diberikan yaitu transfusi WRC, tranfusi TC, methylprednisolone, NaCl 90%, dan terapi oksigen. Diberikan edukasi pasien dan keluarga tentang sifat kronis kondisi ini, yang dapat mencakup periode remisi dan eksaserbasi. Pasien dan keluarga juga dijelaskan efek potensial yang merugikan dari obat-obatan, khususnya penggunaan steroid jangka panjang
DAFTAR PUSTAKA
1. Alice Norton, Irene Robert. Management of Evans syndrome. British journal of Haematology. 2005. 132 : 125-37
2. Prasad Mathew, Gary R Jones, Mary L Windle, Gary D Crouch, Samuel Gross, Robert J Arceci, et al. Evans Syndrome. Diakses 21 November
2013. Diunduh
dari:http://emedicine.medscape.com /article/955266-overview.
3. Marc Michel, Valérie Chanet, Agnès Dechartres, Anne-Sophie Morin, Jean-Charles Piette, Lorenzo Cirasino, et al. The Spectrum of Evans Syndrome in adults : new insight into the disease based on analysis of 68 cases. The American Society of Hematology. 2009. Vol 114, number 15, p 3167-72.
4. Liliana Isacianu, Miron Ingrid, Tansanu Iroan. Evans Syndrome : A rare Cause of Hemolysis (And Thrombocytopenia) in Children.
Fascicula. 2012. Vol. 17, number 1, p 19-22.
5. Priti Dave, Kavita Krishna, AG Diwan. Evans Syndrome Revisited. JAPI. 2012. Vol. 60, p 60-1.
6. Ahmendul Kabir, Jayanta Banik, Ratan Dasgupta, Robed Amin, AM Wasiq Faisal, ASM Mafidul Islam, et. al. Evans Syndrome. J Medicine. 2010. Vol. 11, p 78-82.
7. Manuel Monti, Lucia Stefanecchia, Manolo Filippucci, Alessio Monti, Giovanni Maria Vincentelli, Francesco Borbognoni. A Strange case of Evans Syndrome. Italian Journal of Medicine. 2012. Vol. 7, p 305-9.
8. Reksodiputro, A.H, Madjid, A., Rachman, A.M. Buku Ajar Ilmu Penyakit Dalam. Edisi kedua. Jakarta. 2007.
9. Bakta, I Made. Hematologi Klinik Ringkas. Edisi pertama. Jakarta. 2007.
10.Betram G. Katzung. Farmakologi Dasar dan Kinik. Edisi keenam. Jakarta : 1997. P 897-98.
11.Gurpreet Dhaliwal, M.D., Patricia A. Cornett, M.D., and Lawrence M. Tierney, Jr., M.D. Hemolytic Anemia. American Family Physician. 2004. Vol 69, number 11. 12.Anonim. Megakaryocytes. diakses