The plastidic glutamine synthetase activity is directly modulated by
means of redox change at two unique cysteine residues
Yang Ae Choi, Sang Gu Kim, Young Myung Kwon *
Department of Biology,College of Natural Sciences,Seoul National Uni6ersity,Seoul,151-742,South Korea
Received 31 May 1999; received in revised form 20 July 1999; accepted 27 August 1999
Abstract
Two cDNA clones for glutamine synthetase (GS), Clgln1 and Clgln2, were isolated from a Cana6alia lineata cDNA library
constructed with poly(A)+ RNA from the mature plant leaves. From a comparison of the primary structures,Clgln1 was judged to encode a cytosolic isoform andClgln2 to encode a plastidic isoform. It was revealed by those sequence comparisons that the two cysteine residues (Cys-306 and Cys-371) ofClgln2 (chloroplastic GS encoding gene) were substituted by alanine and serine in theClgln1 clone (cytosolic GS encoding gene), respectively. These cysteine substitutions were found between chloroplastic GS (GS2) and cytosolic GS (GS1) sequences of all plants. These unique cysteine residues may account for the specific susceptibility of the plastidic GS by sulfhydryl reagents. To investigate this assumption the two additional cysteine residues of GS2 were mutated combinatorially and the resulting recombinant GS proteins as well as the two wild-type cytosolic GS and chloroplastic GS were examined in vitro. Only GS2, of the wild-type forms, was significantly activated by dithiotreitol. Moreover, the mutant form, mutated at both of the two additional cysteine residues, was not activated by the reductant, but the mutant forms mutated at only one of the two were also activated. © 1999 Elsevier Science Ireland Ltd. All rights reserved.
Keywords:Cana6alia lineata; Chloroplastic glutamine synthetase; Immunoscreening; cDNA cloning; Site-directed mutagenesis; Recombinant GS protein
www.elsevier.com/locate/plantsci
1. Introduction
Glutamine synthetase (GS: EC 6.3.1.2) catalyzes the ATP-dependent formation of glutamine from glutamate and ammonia. GS catalyzes the reas-similation of ammonia released from a variety of metabolic pathways such as photorespiration, catabolism of amino acids and metabolism of phenylpropanoids. It is also involved in nitrate (or nitrite) assimilation in leaves and roots and in dinitrogen fixation in root nodules of legumes [1]. In plants GS is encoded by a small multigene family that shows organ-specific patterns of ex-pression, with separate genes encoding leaf cytoso-lic and chloroplastic GS isoforms (GS1 and GS2, respectively), and root GS and nodule isoforms in
legumes [2 – 7]. Based on the site of localization, the different GS isoforms assimilate ammonia derived from different sources. Analysis of pho-torespiratory mutants of barley has established that reassimilation of photorespiratory ammonia is the function of the chloroplast-localized GS2 protein [8]. Furthermore, analysis of transgenic plants containing GS1 or GS2 promoters, has shown that GS1 genes are only expressed in the cells around the phloem, suggesting that the GS1 isoform functions in generating glutamine for ni-trogen transport [9].
The regulation of GS gene expression is not yet fully understood. The genes for GS1 and GS2 are differentially expressed during plant growth and development [6,10,11], reflecting the different roles and cellular compartmentalization of the two isozymes [3,12,13]. In addition, it has been shown that the pea GS2 promoter is light-induced [3,10]
* Corresponding author. Tel.: +82-2-880-6676; fax: + 82-2-872-6881.
E-mail address:[email protected] (Y.M. Kwon)
and the accumulation of GS2 mRNA may in part be due to a phytochrome-mediated response [14]. Photorespiratory production of ammonia also reg-ulates pea GS2 expression [13].
In the work on bacteria it has been shown that there is a single gene whose expression is con-trolled by two regulatory proteins in a manner which is sensitive to the nitrogen status of the cell. In addition the enzyme itself is regulated by a reversible activation/deactivation mechanism sen-sitive to the same cellular stimulus [15]. However, the work in higher plants has shown that the regulation of GS activity is quite different, involv-ing multiple GS genes which are individually con-trolled [6,7,10]. This complexity may reflect the greater compartmentalization and differentiation
of higher plant cells and/or organs. But, at
present, very little is known of the components that regulate GS activity unlike the situation in prokaryotes. In leaves there are the two forms of GS: GS1, cytosolic; GS2, chloroplastic. GS2 con-tains additional cysteine residues per subunit, which may account for the specific susceptibility of this isozyme to sulfhydryl reagents [16 – 18].
Cana6alia lineata, used in this study, contains
10% of canavanine in its seed dry weight [19].
Moreover, this compound, an analogue of
arginine, is known to have roles in chemical de-fense and nitrogen storage. It is catabolised to canaline and urea by arginase. The former com-pound is degraded to homoserine and ammonia by canaline reductase. The latter compound is de-graded to carbon dioxide and ammonia by urease.
Ammonia is then reassimilated by the GS/
GOGAT cycle to glutamate [20,21]. Thus, the understanding of the nitrogen metabolism in this plant, C. lineata, is important in the study of canavanine metabolism.
We previously reported the study of the
bio-chemical and immunological aspects of GS
isozymes in C. lineata [22]. The three organs each have a predominant form which is different from the others in their molecular sizes. In the present work we set out to clone cDNAs encoding cytoso-lic and chloroplastic GSs and to establish the molecular basis for the different regulation of the isozyme activities. We also confirmed that the two additional cysteine residues in GS2 are responsible for the specific susceptibility of this isozyme to sulfhydryl reagents.
2. Materials and methods
2.1. Plant material and chemicals
The leaves of 4 – 5-week-old plants ofC. lineata were used for RNA extraction. Restriction
en-zymes, DNA modifying enzymes and PCR
reagents were purchased from Boehringer
Mannheim (Germany). Medium components for the bacterial culture were obtained from DIFCO
(USA) and Sigma (USA). Radioisotope [a
-35S]dATP was obtained from Amersham (USA).
All other chemicals were purchased from Sigma-Aldrich.
2.2. Bacterial strains, media, and recombinant
DNA techniques
Bacterial strains, JM109, XLOLR and BL21 (DE3), were cultured in Luria – Bertani broth. The XL1Blue-MRF’ bacteria were grown in NZYP medium. When required, kanamycin was added at
50 mg/l. Plasmid DNA was purified with the
Mini-Prep kit (Promega, USA) and DNA frag-ments were isolated by restriction endonuclease digestion, electrophoresis and gel elution using a DNA purification system (Jet-Sorb, GENOMED, USA). Ligation and transformation were done as described by Sambrook et al. [23]. All sequences were determined on both strands. The nucleotide and deduced amino acid sequences were analyzed with the DNASIS and PROSIS programs. Multi-ple sequence alignments of GS1 (the cytosolic isoform) and GS2 (the chloroplastic isoform) copied from the GenBank database were per-formed using the Clustal W (1.7) multiple se-quence alignment program.
Leaves of C. lineata for nucleic acid isolation were harvested and ground to a fine powder in liquid nitrogen. Total RNA was isolated using the guanidium isothiocyanate methods [24]. Poly(A)+
RNA was affinity-purified on oligo(dT)-cellulose (Sigma-Aldrich, USA) according to Sambrook et al. [23] and used for cDNA library preparation.
2.3. Construction of a lZAP expression cDNA
library and isolation of chloroplast GS cDNA clones
The poly(A+) RNA from the leaves was used to
lZAP cDNA synthesis kit (Stratagene, USA). The recombinants were packaged using Gigapack III gold packaging extracts (Stratagene, USA). After infection of XL1-blue MRF’ cells and amplifica-tion, the recombinant phages were plated onto NZYP – agar plates. After incubation at 42°C for the small plaques to become visible, IPTG-treated nitrocellulose membranes (Amersham) were ap-plied onto the agar plates to express the cDNAs and to lift the expressed proteins. After another incubation of 3.5 h at 37°C, the membranes were hybridized with an antibody raised against the GS2 (1:8000 dilution) and visualized using horse radish peroxidase-conjugated antirabbit IgG. The immunopositive clones were selected and charac-terized further. Using properties of the ZAP Ex-press™ vector, the positive clones were excised with Exassist helper phage in XL1-blue MRF’ cells and the resulting phagemid-packaged phage particles were used to infect XLOLR cells. The transformed cells were plated on LB-kanamycin agar plates and colonies were selected for plasmid preparations and sequencing.
2.4. Cloning of cytosolic GS cDNA
After massive in vivo excision of thel bacterio-phage library, the resulting bacterio-phagemid-packaged phage particles were used to infect XLOLR cells. The transformed cells were cultured in LB-kanamycin media. The resulting plasmid library was used to transform an Escherichia coli DglnA mutant FDB213 [25,26]. FDB213 cells, generously provided by Professor Ausubel (Massachusetts General Hospital, Boston, USA), were
trans-formed by the CaCl2 procedure [23] and screened
by functional rescue[27].
2.5. Construction of expression 6ector
The open reading frame of the Clgln1 cDNA in
pBK-CMV vector at EcoRI and XhoI sites was
cloned into pET30+ (Novagen): An NcoI –XhoI
fragment containing the bases from 429 to 1345 of
Clgln1 cDNA was cloned into pET30+ to obtain
pETGS1nx. The remaining open reading frame (bases from 63 to 428) was prepared by PCR amplification using a 5% NcoI linker-primer (5%
-CCACATGTCTTTAGTCTCAGATCTC-3%) and
a 3% T7 primer (5%
-GTAATACGACTCAC-TATAGGGC-3%). The PCR product was digested
with NcoI and cloned into pETGS1nx to obtain
pETGS1.
The open reading frame ofClgln2cDNA, except for the transit peptide region, was cloned into
pET30+: an NcoI –SacI fragment containing
bases 241 – 414 was prepared by PCR amplification
using a 5% NcoI linker-primer (5%
-CCACATGC-CATGG CAACTAAGTCTGAAAA
TG-GCACC-3%) and a 3% T7 primer (5%-GTAATACG
ACTCACTATAGGGC-3%). The PCR product
was digested with NcoI and SacI and cloned into
the expression vector pET30a+ (Novagen, USA).
Subsequently, the remaining portion of the GS cDNA was inserted by using the internalSacI and
XhoI fragment to obtain pETGS2.
2.6. Site-directed mutagenesis
Site-directed mutagenesis was performed by PCR extension of the following complementary primers to the opposite site strands of pClgln2
[18]. Cys306 Ser (C306S), 5%-GG AAT GGT
GCG GTT AGC-3%(mismatched bases are shown
in and restriction enzyme sites underlined; muta-genic oligonucleotides were designed to generate new restriction sites to facilitate the confirmation of the mutation). The oligonucleotide primers were extended during thermal cycling (93°C for 30 s, 55°C for 60 s, 72°C for 12 min; 12 cycles) using Pow DNA polymerase (Boehringer Mannheim, Germany). The mutated plasmid containing stag-gered nicks was generated. Following the thermal cycling, the product was treated with DpnI (target sequence: 5%-Gm6ATC-3%) to digest parental DNA template and to select for mutation-containing synthesized DNA. The nicked DNA having the
desired mutations was transformed into E. coli
2.7. Expression and purification of recombinant proteins
E. coli strain BL21(DE3) (Novagen, USA) har-boring the recombinant plasmids were grown at 37°C in Luria – Bertani broth containing 50 mg/ml
kanamycin [23]. Expression of recombinant
proteins was induced with 1 mM IPTG when the cell density reached an A600 of 0.4 – 0.8. The cells were allowed to grow either for 3 h at 37°C or for 16 h at 25°C. The recombinant protein was
purified by the procedure recommended by
Novagen.
A typical enzyme assay was performed as fol-lows: to a 450 ml reaction mixture containing all components (80 mM glutamic acid, 6 mM
NH2OH, 2 mM DTT, 20 mM MgCl2, 150 mM,
Tris – HCl, pH7.8), variable concentrations of the sulfhydryl reagents and enzyme, incubated for 5
min at 35°C, was added with 50 ml 80 mM ATP.
Other details about the enzyme and protein assays performed are described by Choi and Kwon [22] and in Ref. [28].
3. Results and discussion
3.1. Isolation and sequence analysis of cytosolic
GS cDNA clone (Clgln1)
The E. coli strain FDB213, which contains a
deletion of the bacterial glutamine synthetase structural gene [25], was transformed with the
phagemid cDNA library of C. lineata leaves and
were plated on a selective medium lacking glu-tamine. The first clone to appear (more colonies appeared in 5 days) carried an insert of 1.4 kb and was analyzed with IPTG-induced expression in the mutant cell and by protein blotting (data not
shown). This clone was designated pClgln1 and
was chosen for further characterization. The
de-duced amino acid sequences from the Clgln1
nu-cleotide sequence (reported in the GenBank database with the accession number AF081566)
are shown in Fig. 1. The ORF of the Clgln1
translates into a 355 amino acid protein with a calculated molecular weight of 39 022 Da, which is similar to the GS1 sizes of other higher plants (35 000 – 38 000 Da) [29].
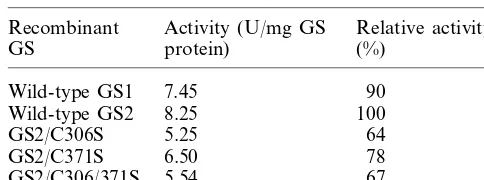
Table 1
Activities of the recombinant glutamine synthetasesa
Recombinant Activity (U/mg GS Relative activity (%)
aThe purified GS activity was measured without any thiol
reagent.
which would lead to a size of the mature plastidic GS of 41 835 Da which is appoximately the size of the native plastidic GS polypeptide (42 kDa) [22]. From these results we concluded that the pClgln2 insert represented a chloroplast GS cDNA.
3.3. Comparison of deduced amino acid sequences
of Clgln1 and Clgln2, with other plant GS
sequences
Comparison of deduced amino acid sequences
of the two C. lineata GS cDNA clones are shown
in Fig. 1 with the five conserved regions, identified by Eisenberg et al. [32]. Amino acid residues 62 – 414 of Clgln2 were found to be essentially colinear with the cytosolic GS coding sequence derived from the Clgln1 clone. While Clgln1 contains only three conserved cysteine residues (Fig. 1), Clgln2 has two more (Cys-306 and Cys-371). This com-parison revealed that the two cysteines (Cys-306 and Cys-371) of the Clgln2 clone are substituted by alanine and serine in the Clgln1 clone. These cysteine substitutions were found in all plant chloroplastic GS sequences [16,18]. These unique cysteine residues may account for the specific sus-ceptibility of the plastidic GS to sulfhydryl reagents reported by others [16,17].
3.4. Acti6ities of wild-type and mutant
recombinant GS enzymes
To investigate the susceptibility of the GS proteins to sulfhydryl reagents, a recombinant ex-pression system was used. Recombinant expres-sion plasmids, pETGS1 and pETGS2, were constructed by inserting the open reading frames
of Clgln1 and Clgln2 onto NcoI and XhoI of the
pET30a+ vector (Novagen, USA). The resulting
translational fusions have a His-tag (vector-en-coded residues) at the N-termini of the GS polypeptides. However, the N-termini of GS polypeptides are poorly conserved [6,18,32] and are not considered to be near the active site of the enzyme [32]. There was no significant difference in the specific activity between the tag-containing and tag-removed recombinant GS polypeptides (data not shown). Moreover, the recombinant GS2 has higher activity (8.25 U/mg) (Table 1) than the leaf-purified GS2 (6.68 U/mg protein) [22]. So, the His-tag fused recombinant proteins were used in all further experiments.
3.2. Isolation and sequence analysis of cDNA
clones encoding C. lineata chloroplast GS (Clgln2)
An expressed cDNA library from C. lineata
leaves was constructed using the lZAP Express™
system. About 3×105plaques were screened with the polyclonal antiserum raised against C. lineata chloroplast GS [18]. One of the positive clones which carried an insert of approximately 1.6 kb was designated pClgln2 and entirely sequenced. The deduced amino acid sequence from theClgln2 nucleotide sequence (reported in the GenBank database with the accession number AF031082) is presented in Fig. 1.
Clgln2 was believed to encode the leaf-enriched
42 kDa GS polypeptide. The ORF of Clgln2
en-codes a protein of 430 amino acids with a pre-dicted molecular weight of 47 411 Da. The
polypeptide encoded by the Clgln2 cDNA had
extensions at both N- and C-termini of 58 and 12 residues, respectively when compared to cytosolic GS polypeptides (Fig. 1). The N-terminal exten-sion had structural similarities to the presequence of nuclear-encoded chloroplast proteins [30,31]. Furthermore, the deduced amino acid sequence from the DNA sequence contains the N-terminal sequence obtained from the purified chloroplast GS polypeptide of the plant leaves and also showed very high homology with chloroplast GS cDNAs reported from other plant species [18]. The N-terminal sequence of the mature protein was also present after N-terminal transit peptide (50 amino acids).
In the study by Karlin-Neumann and Tobin [31]
the sequence Gly – Arg – Val was found 1 – 3
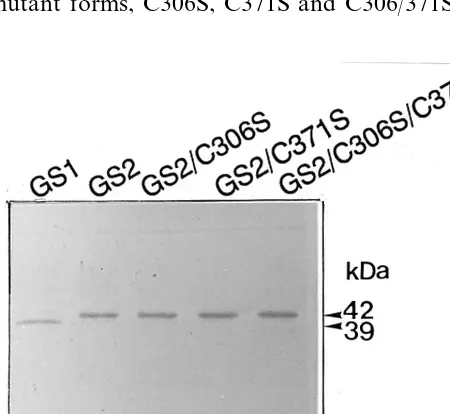
The wild-type GS1 (cytosolic GS) and GS2 (chloroplastic GS) and the three mutant forms of GS2 (i.e. GS2/C306S, GS2/C371S and GS2/C306/
371S) were purified to apparent homogeneity (Fig. 2). SDS-PAGE and Coomassie blue staining showed bands of the expected sizes for the recom-binant GS proteins. Thus the GS expression
plas-mids were being correctly transcribed and
translated.
Activities of the two forms of wild-type GS and the three forms of mutant GS2 were measured without any thiol reagent (Table 1) and the results
indicated that they were catalytically active
proteins. In addition, all of the recombinant GS proteins from C. lineata were completely
inacti-vated following treatment with 1 mM N
-ethyl-maleimide (NEM) for 10 min. These results are consistant with the activities of GS1 and GS2 being completely inhibited by the sulfhydryl block-ing agents and that they may also be stabilized by sulfhydryl reducing agents [16,17]. Mutations were introduced combinatorially at Cys 306 and Cys 371, two highly conserved and unique residues present in the same locations in all reported chloroplastic GS proteins [17,18]. When these Cys residues were altered to serine residues, the
mutant forms, C306S, C371S and C306/371S, has Fig. 3. Effects of thiol reagents on the activity of recombinant GS proteins. (A) The effect of three reducing agents, b-SH (2-mercaptoethanol), GSH, (reduced form of glutathione) and DTT (dithiothreitol), on the GS2 activity. (B) The effect of DTT on the activity of GS1 and GS2, and the three GS2 mutant forms (C306S, C371S, C306/371S).b-SH, GSH, and DTT were dissolved into 10 mM imidazole (pH 7.8) contain-ing 1 mM phenylmethylsulfonyl fluoride. The activity is rela-tive to that without any thiol reagent, taken to be 1. Results are the means of duplicate determinations that differed by less than 5%.
Fig. 2. Polyacrylamide gel electrophoresis of purified wild-type GS1 and GS2 and the three mutant GS2 forms. The recombinant proteins were purified as described in Section 2 and the N-terminal tag was removed by using S-protein agarose and enterokinase. The trimmed recombinant proteins (each 5 mg) were electrophoresed on a 12% (w/v) polyacry-lamide slab gel contained SDS and 2-mercaptoethanol fol-lowed by staining with Coomassie Brilliant Blue.
64, 78 and 67% relative activity, respectively (Table 1).
3.5. Effect of thiol reagents on GS acti6ity
of the cytosolic GS was insignificantly increased by DTT, compared to the chloroplastic GS, indi-cating that the requirement for DTT was specific to the plastidic GS. These results suggest that a redox-responsive site is present in the plastidic GS and that reduction of sulfhydryl group(s) is needed for its higher activation. In other words, the two cysteine residues (Cys-306 and Cys 371 atClgln2), unique to the chloroplast enzymes (Fig. 1), are required to be maintained in their reduced form for the activation of plastidic GS.
This assumption was confirmed by mutagenesis of the residues in this study: the resulting recombi-nant GS2 mutant proteins (C306S, C371S and C306/371S) were examined for the effects of the sulfhydryl-reducing agent DTT on their activities (Fig. 3B). The mutant forms mutated at only one of the two cysteine residues (C306S or C371S) were also activated by the reductant (Fig. 3B). However, the mutant mutated at both additional cysteine residues (C306/371S) was not activated by the reductants, like cytosolic GS (GS1). These results indicated that the susceptibility of the plas-tidic GS (GS2) to sulfhydryl reagents is caused by the two cysteines unique to GS2 and that this redox-response site is very important to the GS activity in the sense that the subcompartmental isoform has a role in assimilation of photorespira-tory ammonia [8,12,13]. The flux of ammonium through the photorespiratory nitrogen cycle is esti-mated to be 10-fold greater than that resulting from primary assimilation [29]. Therefore, a link between the light reaction of photosynthesis and
the GS/GOGAT cycle is needed for a cooperated
control during illumination. One possible link be-tween the two reactions is via a redox cascade using thioredoxin light-induced changes. The ac-tivity of many enzymes that function in chloro-plast is light-modulated by the regulatory proteins (ferredoxin/thioredoxin) which transduce the light signal in the form of a reduced disulfide bridge to the regulated enzyme [33]. Hence, in the light, the target enzyme is continuously part of an oxida-tion – reducoxida-tion cycle, but in the dark, cycling stops and the ratio shifts from the reduced to the oxidized enzyme so that the oxidized form is mainly present. The findings presented above sug-gest that GS, the first regulatory step in the cycle, can be regulated by these means analogous to the thioredoxin-linked enzymes of the reductive pen-tose phosphate cycle [33]. These regulation
mecha-nisms acting in the enzyme itself, which afford a more rapid regulation of GS activity in response to changing environmental conditions, may be needed in the chloroplastic form (GS2).
Acknowledgements
We are very grateful to Professor Ausubel of Massachusetts General Hospital, Boston, USA, for his generous gifts ofE. colistrain FDB213 The authors wish to acknowledge the financial support of the Korea Research Foundation in the program year 1998 (1998-15-D00251) and the grant from the Korea Science and Engineering Foundations through SRC for Cell Differentiation (98-5-1).
References
[1] B.J. Miflin, P.J. Lea, The pathway of nitrogen assimila-tion in plants, Phytochemistry 15 (1976) 873 – 885. [2] S.V. Tingey, E.L. Walker, G.M. Coruzzi, Glutamine
synthetase genes of pea encode distinct polypeptides which are differentially expressed in leaves, roots and nodules, EMBO J. 6 (1987) 1 – 9.
[3] S.V. Tingey, F.-Y. Tasi, J.W. Edwards, E.L. Walker, G.M. Coruzzi, Chloroplast and cytosolic glutamine syn-thetase are encoded by homologous nuclear genes which are differentially expressed in vitro, J. Biol. Chem. 263 (1988) 9651 – 9657.
[4] C. Gebhardt, J.E. Oliver, B.G. Forde, R. Saaelainen, B.J. Miflin, Primary structure and differential expression of glutamine synthetase genes in nodules, roots and leaves of Phaseolus 6ulgaris, EMBO J. 5 (1986) 1429 – 1435.
[5] J.V. Cullimore, C. Geghardt, R. Saarelainen, B.J. Miflin, K.B. Idler, R.F. Barker, Glutamine synthetase ofPhase
-olus6ulgarisL.: organ specific expression of a multigene family, J. Mol. Appl. Genet. 2 (1984) 589 – 599. [6] G. Forde, J. Woodall, Glutamine synthetase in higher
plants, in: R.M. Wallsgrove (Ed.), Amino Acids and their Derivatives in Higher Plants, Cambridge University Press, Cambridge, 1995, pp. 1 – 18.
[7] T.K. Peterman, H.M. Goodman, The glutamine syn-thetase gene family ofArabidopsis thaliana: light-regula-tion and differential expression in leaves, roots and seeds, Mol. Gen. Genet. 230 (1991) 145 – 154.
[8] R.D. Blackwell, A.J.S. Murray, P.J. Lea, Inhibition of photosynthesis in barley with decreased levels of chloro-plastidic glutamine synthetase, J. Exp. Bot. 38 (1987) 1799 – 1809.
[10] J.W. Edwards, E.L. Walker, G.M. Coruzzi, Cell specific expression in transgenic plant reveals non-overlapping roles for chloroplastic and cytosolic glutamine syn-thetase, Proc. Nat. Acad. Sci. 87 (1990) 3459 – 3463. [11] K. Kamachi, T. Yamaya, T. Mae, K. Ojima, A role of
glutamine synthetase in the remobilization of leaf nitro-gen during natural senescence in rice leaves, Plant Phys-iol. 96 (1991) 411 – 486.
[12] A.F. Mann, P.A. Fentem, G.R. Stewart, Tissue localiza-tion of barley glutamine synthetase isoenzymes, FEBS Lett. 110 (1980) 265 – 267.
[13] J.W. Edwards, G.M. Coruzzi, Photorespiration and light act in concert to regulate the expression of the nuclear gene for chloroplastic glutamine synthetase, Plant Cell 1 (1989) 241 – 248.
[14] A. Migge, E. Carrayol, B. Hirel, M. Lohmann, G. Meya, T.W. Becker, Regulation of the subunit composition of plastidic glutamine synthetase of the wild-type and of the phytochrome-deficient aurea mutant of toamto by blue/
UV-A or by UV-B-light, Plant Mol. Biol. 37 (1998) 689 – 700.
[15] B. Magasanik, Reversible inactivation of an enhancer binding protein regulates the transcription of bacterial nitrogen utilization genes, Trends Biochem. Sci. 13 (1988) 475 – 479.
[16] D.A. Rhodes, A.P. Sims, G.R. Stewart, Glutamine syn-thetase and the control of nitrogen assimilation inLemna minor, in: E.J. Hewitt, C.V. Cutting (Eds.), Nitrogen Assimilation of Plants, Academic Press, New York, 1979, pp. 509 – 516.
[17] J.F. Francisco, P. Gadal, B.B. Buchanan, The thiore-doxin-linked activation of the chloroplast and cytosolic forms of Chlamydomonas reinhardtii glutamine syn-thetase, Plant Physiol. Biochem. 31 (1993) 649 – 655. [18] Y.A. Choi, Molecular and biochemical characterization
of glutamine synthetases from Cana6alia lineata leaves, PhD Thesis, Seoul National University, 1998.
[19] G.S. Park, Y.M. Kwon, The analysis of canavanine content in leaves, roots, and xylem exudate ofCana6alia
lineata, Korean J. Bot. 33 (1990) 119 – 126.
[20] G.A. Rosenthal, Purification and characterization of the higher plant enzyme L-canaline reductase, Proc. Natl. Acad. Sci. USA 89 (1992) 1780 – 1784.
[21] G.A. Rosenthal, The preparation and colorimetric analy-sis of L-canaline, Anal Biochem. 51 (1973) 354 – 361.
[22] Y.A. Choi, Y.M. Kwon, Comparative characterization of glutamine synthetase iso forms fromCana6alia lineata
in biochemical and immunological aspects, Plant Sci. 134 (1998) 171 – 180.
[23] J. Sambrook, E.F. Fritsch, T. Maniatis, Molecular Cloning, a Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 1989. [24] J.J. Chirgwin, A.E. Prozbyla, R.J. MacDonald, W.J.
Rutter, Isolation of biologically active nucleic acid from source enriched in ribonuclease, Biochemistry 18 (1979) 5294 – 5299.
[25] F.J. De Brujin, F.M. Ausubel, The cloning and transpo-son Tn5 mutagenesis of the glnA region of Klesiella pneumoniae: Identification of glnR, a gene involved in the regulation of the nif and hut operons, Mol. Gen. Genet. 183 (1981) 289 – 297.
[26] S. Drassarma, E. Tischer, H.M. Goodman, Plant glu-tamine synthetase complements a glnA mutation in E.
coli, Science 232 (1986) 1242 – 1244.
[27] D.P. Snustad, J.P. Hunsperger, B.M. Chereskin, J. Mess-ing, Maize glutamine synthetase cDNAs: isolation by direct genetic selection in Escherichia coli, Genetics 120 (1988) 1111 – 1124.
[28] M.M. Bradford, A rapid and sensitive method for quan-titation of microgram quantities of protein utilizing the principle of protein-dye binding, Anal. Biochem 72 (1976) 248 – 254.
[29] H.A. Lam, K.T. Coschigano, J.C. Oliveira, R. Melo-Oliveira, G.M. Coruzzi, The molecular genetics of nitro-gen assimilation into amino acids in higher plants, Annu. Rev. Plant Physiol. Plant Mol. Biol. 47 (1996) 569 – 593. [30] S. Smeekens, J. van Oosten, M. de Groot, P. Weisbeek, Silence cDNA clones for a divergent chlorophyll-a/ b-binding protein and a small subunit of ribulose bisphos-phate carboxylase, Plant Mol. Biol. 7 (1986) 433 – 440. [31] G.A. Karlim-Neumann, E.M. Tobin, Transit peptide of
nuclear encoded chloroplasts share a common amino acid framework, EMBO J. 5 (1986) 6 – 9.
[32] D.R. Eisenberg, J. Almassy, C.A. Janson, et al., Some evolutionary relationships of the primary biological cata-lysts glutamine synthetase and Rubisco, Cold Spring Harb. Symp. Quant. Biol. 11 (1987) 483 – 490.
[33] B.B. Buchanan, Role of light in the regulation of chloro-plast enzymes, Annu. Rev. Plant Physiol. 31 (1980) 341 – 374.
.