JOURNAL TOPIK KHUSUS,SEM.2, NO.20914009;20914014, MEI 2015 1
Simulasi Dinamika Molekular Menggunakan
Potensial Lennard-Jones untuk Sistem Silikon
*Arnita Irianti, M. Yusuf Hakim Widianto
Sains Komputasi, Institut Teknologi Bandung, Jl. Ganesha 10. Bandung 40132 Indonesia
*
[email protected]
,
[email protected]
Abstract—Simulasi dilakukan pada fluida sistem Silikon.
Metode yang digunakan adalah dinamika molekular menggu-nakan potensial Lennard-Jones. Sistem menggumenggu-nakan 64-atom Silikon dengan jumlah atom (N) adalah 216. Meninjau perg-erakan fluida sistem silikon pada fasa cair dengan menghitung fungsi pasangan korelasi (PCF)
Index Terms—Dinamika molekular,Potensial Lennard-Jones Silikon, Fungsi Pasangan Korelasi.
I. PENDAHULUAN
Simulasi dinamika molekular merupakan alat bantu un-tuk mempelajari dinamika dari cairan, molekul mikroskopis dan makro molekul[1]. Simulasi dinamika molekular di-gunakan untuk menyelesaikan persamaan Newton tentang gerak[2], dimana gaya ditentukan oleh massa dan percepatan. Partikel-partikel pada molekular dinamik bergerak secara ran-dom dan berinteraksi satu dengan yang lainnya. Pemodelan dari interaksi partikel-partikel cairan memotivasi untuk mem-pelajari dinamika cairan. Dari data dinamika partikel-partikel tersebut dapat dipelajari struktur, potensial dan termodinamika molekul dengan simulasi.
Tulisan ini tentang simulasi dinamika molekular pada fluida sistem Silikon. Dinamika molekular digunakan sebagai metode penyelesaian gerak sistem partikel Silikon pada fasa cair. Menggunakan potensial Lennard-Jones sebagai interaksi antar molekul Silikon. Meninjau pergerakan sistem silikon pada fasa cair pada box dengan ukuran L untuk menghitung Fungsi Pasangan Korelasi/Pair Correlation Function (PCF) se-hingga diharapkan lebih lanjut dapat divisualisasikan tekanan, temperatur terhadap perubahan posisi dan waktu
II. DINAMIKAMOLEKULAR
Dinamika molekular pada dasarnya memecahkan persamaan gerak sengan menggunakan mekanika klasik (persamaan Newton). adadua persoalan utama yang harus diselesaikan dalam penggunaan motede simulasi dinamika molekular yaitu:
1) Menentukan model energi potensial yang mengatur hubungan antar atom-atom atau molekul-molekul yang saling berinteraksi dalam sistem. Dari energi potensial inilah gaya-gaya yang mempengaruhi dinamika sistem dapat di tentukan
2) Menentukan algoritma dan metode numerik
III. MODEL
Simulasi ini menggunakan algoritma dinamika moleku-lar. Interaksi antar partikel-partikel Silikon memenuhi kaidah potensial Lennard-Jones[3]. Potensial tersebut menolak bila jarak terlalu dekat dan menarik bila jarak menjauh. Titik se-imbang yang potensialnya minimum terletak pada posisirmin memenuhiF(rmin) = 0. Ketika jarak antar partikel terlampau jauh maka interaksi antara partikel-partikel diabaikan diberi nilai rcutof f. Potensial Lennard-Jones pada interaksi antar atom yang berjarak lebih besar dari nilai rcutof f dituliskan sebagai berikut: dimana σ merepresentasikan diameter partikel denganrin =
216σ. Ketika partikel-partikel berinteraksi pada jarak (rij ≤
rcutof f) maka terdapat gaya yang bekerja pada partikel-partikel tersebut dimana F(rij) = −∇U(rij) sehingga per-samaan 1 dituliskan sebagai berikut:
F(rij) = 24
Bersesuaian dengan Hukum Newton tentang gerak se-hingga dapat dihitung semua gaya yang bekerja pada interaksi partikel-partikel. Dilakukan integrasi untuk menyelesaikan persamaan 3 dengan algoritma Verlet[4].
r(t+δt) =r(t) +v(t)δt+1 2a(t)δt
2
(4)
untuk mengetahui posisi baru partikel-partikel tersebut algo-ritma 4 dapat diekspresikan sebagai berikut :
ri(t0+△t) = 2ri(t0)−ri(t0− △t) +ai(t0)△t2 (5) Suhu untuk setiap waktu t, dengan energi kinetik 32 maka didapatkan
JOURNAL TOPIK KHUSUS,SEM.2, NO.20914009;20914014, MEI 2015 2
merupakan probabilitas untuk menemukan pusat partikel jarak tertentu dari pusat partikel lain yang didefinisikan sebagai berikut:
g(r) = V
N
"
n(r) 4πr2△r
#
(7)
dimana n(r) adalah jumlah atom yang berada pada jarak antara r danr+△r, N adalah jumlah atom dan V adalah volume.
Simulasi ini menggunakan sistem 64-atom Silikon. Perg-erakan fluida sistem Silikon didalam kotak dengan ukuran
L×L×L. Jumlah atom (N) dalam simulasi adalah 216. Nilai cut off σ = 2.80 A˚. Simulasi dilakukan pada suhu konstan pada 1◦K dan 175◦K dengan perubahan waktu δt sebesar
0.001 detik.
IV. ALGORITMAMOLEKULARDINAMIKAPADA SISTEM
SILIKON
Algoritma yang digunakan sebagai berikut:
• inisiasi ukuran box • Input jumlah partikel • Input densitas
• Input posisi awal semua partikel • Input lama perhitungan partikel (tmax)
• kecepatan awal partikel • Input∆t
• Input massa partikel
• Hitung total gaya yang bekerja dan percepatannya pada
partikel ke i sebagai berikut:
¯
Fi(t) = Σ j=1
j6=iFij(t)ai(t) =
Fi
mi
• Hitung posisi baru partikel ke-i berdasarkan metode verlet
sebagai berikut:
ri(t+ ∆t) = 2ri−ri(t−∆t) +ait0(∆t)2
• ulangi langkah diatas hingga i=N
• Plot fungsi korelasi pasangan g(r) dengan posisi baru
V. HASIL DANPEMBAHASAN
Posisi awal untuk semua partikel adalah random, se-hingga diperoleh hasil sebagai berikut:
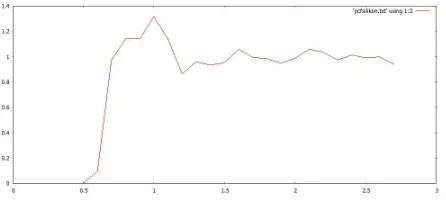
Fig. 1: Grafik PCF terhadap perubahan posisi pada T= 1 K
Simulasi menggunakan potensial Lennard-Jones pada sis-tem silikon pada fasa gas d sis-temperatur 1 K dan 175 K. Dari gambar di peroleh bahwah perubahan suhu mempengaruhi perubahan pair correlation (PCF) dari sistem silikon terse-but.Jurnal rujukan mempunyai nilai yang berbeda dengan hasil simulasi yang diperoleh karena parameter-parameter yang di-gunakan berbeda, dimana pada jurnal rujukan mengdi-gunakan parameterisasi ikatan kuat [5].
Fig. 2: Grafik PCF terhadap perubahan posisi pada T= 175 K
VI. KESIMPULAN
Simulasi menggunakan potensial Lennard-Jones pada sis-tem silikon pada fasa gas d sis-temperatur 1 K dan 175 K.Perubahan suhu mempengaruhi perubahan pair correlation (PCF) dari sistem silikon tersebut. Diperlukan penelitian lebih lanjut yang mendalam dalam memepelajari parameter-parameter yang digunakan pada Potensial Lennard-Jones pada sistem silikon pada fasa cair untuk menggambarkan PCF.
REFERENCES
[1] Berendsen, H. J. C. and Postma, J. P. M. and Gunsteren, W. F. van and DiNola, A. and Haak, J. R.,Molecular dynamics with coupling to an external bath,81,3684–3690,The Journal of Chemical Physics,1984,http: //dx.doi.org/10.1063/1.448118
[2] Plimpton, Steve,Fast Parallel Algorithms for Short-Range Molecular Dynamics,117,1–19,Journal of Computational Physics, 1995,http://dx.doi. org/10.1006/jcph.1995.1039
[3] Lennard-Jones, J. E. and Devonshire, A. F.,Critical Phenomena in Gases. I,16353–70,Proceedings of the Royal Society of London. Series A, Math-ematical and Physical Sciences,1937,http://www.jstor.org/stable/97067
[4] Verlet, Loup,Computer ”Experiments” on Classical Fluids. I. Thermodynamical Properties of Lennard-Jones,159,98–103,Phys. Rev.,1967,http://dx.doi.org/10.1103/PhysRev.159.98
[5] Dipojono, K, Herman,Simulasi Dinamika Molekul(Sebuah Pengantar),ISSN,1410-7686,Prosiding Seminar Nasional Hamburan Neutron dan Sinar X ke 4, 2001