On the promotion energy of an atom in a molecule
I. Mayer
⇑Chemical Research Center, Hungarian Academy of Sciences, H-1525 Budapest, P.O. Box 17, Hungary
a r t i c l e
i n f o
Article history: Received 26 July 2010 In final form 3 September 2010 Available online 7 September 2010
Dedicated with affection to my wife, Dr. Márta Révész.
a b s t r a c t
Using the concept of the effective minimal basis set introduced some time ago, a proper definition is pro-posed for the atomic promotion energy in the molecule, which the atom can be assigned after the orbital deformations are introduced but before any bonding, delocalization and charge transfer effects are taken into account. The first pivoting calculations indicate that these promotion energies can be quite substan-tial and are characteristic for the chemical nature of the atom.
Ó2010 Elsevier B.V. All rights reserved.
1. Introduction
Our qualitative understanding of molecular structure strongly relies on the notion of the minimal basis set. We think in terms that the atoms enter the molecules with their 1s;2s;2p;etc. orbi-tals (or their hybrids), while we do our calculations by using larger and larger basis sets. This contradiction can be resolved by intro-ducing an effective minimal basis set of an atom, which can be extracted from the results of a large scaleab initiocalculation using an extended basis set. That can be done either in the framework of a ‘Hilbert-space analysis’ performed in terms of the basis orbitals assigned to the different atoms [1,2] or in terms of the ‘three-dimensional’ (3 D) analysis[2,3]performed in the physical space. It has always been found for SCF wave functions1that such ana posteriori analysis results in as many effective atomic orbitals (AO-s) with non-negligible occupation numbers as is the number of functions in a classical minimal basis set. The delimitation between the strongly occupied and the essentially unoccupied effec-tive AO-s is usually very sharp—the occupation numbers fall by orders of magnitude.2
Originally[1]the orbitals of the effective minimal basis were found by looking for molecular orbitals for which Mulliken’s net atomic population on the given atom is maximal, or at least sta-tionary (Magnasco–Perico localization criterion[4]), and renormal-izing the intraatomic parts of the orbitals obtained—it was shown
that they form an orthonormalized atomic basis[1]. The further analysis[2]revealed that these effective atomic orbitals are in a close connection with McWeeny’s natural hybrid orbitals[5], and the stationary values obtained for the net orbital populations can be considered as occupation numbers—our free program[6]also uses an algorithm based on this analogy. At the same time these orbitals are only in a quite loose connection with the concepts of ‘natural hybrid orbitals’ and ‘natural atomic orbitals’ of Weinhold and coworkers[7–9]. In the 3 D case the effective AO-s can be ob-tained[3]by applying a localization functional representing the di-rect 3 D generalization of Mulliken’s net atomic populations.
Although it was obvious that the ‘effective minimal basis’ orbi-tals are essentially those by which the atom participates in the molecular wave function, and thus represent its promoted state, no actual atomic many-electron wave functions built up of these orbitals were constructed till date and, accordingly, no promotion energies were calculated by their use. Now they became of some importance in light of the ‘dilemma’ we have recently encountered
[10]. We considered several related energy decomposition schemes falling in two main categories depending on the treatment of the kinetic energy integrals. One of them gives numbers which are ‘on the chemical scale’ and have quite appealing values at the equi-librium molecular geometries, but exhibit a counter-intuitive dis-tance dependence, another results in numbers with too large absolute values (very large positive one-center ‘promotion ener-gies’ and very large negative bonding energy components) but ‘cor-rect’ distance behaviour. For that reason we started to consider in detail the individual effects contributing to the change of one-cen-ter energy contributions during the molecule formation—promo-tion, electron accumulation/depletion and the special effect of enhanced ionic terms in the Hartree–Fock wave function. The pres-ent Letter is devoted to the first of these—the promoted ‘reference state of an atom in a molecule’ and the respective promotion energy.
0009-2614/$ - see front matterÓ2010 Elsevier B.V. All rights reserved. doi:10.1016/j.cplett.2010.09.007
⇑Fax: +36 1 325 7554.
E-mail address:[email protected]
1 It was mentioned in[2]that these concepts can bemutatis mutandisapplied to correlated wave functions, too, but they are out of our present scope—here we deal with SCF wave functions only.
2 Exceptions are some hypervalent atoms which can exhibit also a tail of slightly populated orbitals of basicallyd-character[1]. But even in such cases it is always evident which orbitals are the true valence ones and which orbitals are only participating in the ‘back-donation’.
Chemical Physics Letters 498 (2010) 366–369
Contents lists available atScienceDirect
Chemical Physics Letters
2. The promoted reference state of an atom in a molecule
It is usual to speak about ‘valence state’ of an atom, for instance four-valent carbon has a valence stateð1sÞ2ð2sÞ1ð2pÞ3, as opposed to itsð1sÞ2ð2sÞ2ð2pÞ2ground state atomic configuration. When dis-cussing the valence state, one usually does not consider explicitly the distortions (and possible mixing) of the free atom’s orbitals, although the importance of ‘orbital contraction’ had been pointed out as early as in 1962[11]. For that reason we define here the pro-moted ‘reference state’ of an atom, in which the orbitals of the
effective minimal basis(the effective AO-s having significant occu-pation numbers) of the atom in the molecule are either doubly or singly occupied. It is usually trivial to determine by human inspection what orbitals should be doubly occupied, singly occu-pied or left empty, but it was desirable to have an automated pro-cedure. That was achieved by transforming the overlap and density matrices to the basis of effective AO-s, performing a Mulliken’s population analysis for them and rounding the gross orbital popu-lations obtained to the nearest integers. In some cases the refer-ence state obtained in this manner does not correspond to a neutral atom but to a positive or negative ion; this may carry important information about the molecule studied.3
After determining the orbitals and their occupation scheme, one can construct a wave function by their use. As different spin cou-plings within the atom are of no chemical interest, a high-spin sin-gle determinant wave function with all the singly occupied orbitals having the same (
a
) spin in accord with Hund’s rule seems to be the adequate choice. Thus the energy of such a determinant is to be considered the energy of the promoted reference state; the pro-motion energy of the atom is then given by the difference of this energy and that of the atomic ground state ROHF wave function. In cases like carbon, this promotion energy can be decomposed into two terms: one is determined by the difference of the ROHF energies obtained for the ground state and valence configurations, another is the energy of the further promotion (orbital distortion) taking place with respect to the valence state. Of course, the corre-spondence between the reference state and the valence state is usually only approximate, as the effective AO-s defining the former are usually not pures, petc. orbitals; often they have a dominatings or p character, but in some cases (e.g., for nitrogen atom in ammonia) they must be considered hybrids. For carbon, however, the fact that the effective AO-s represent hybrids in some cases
[2]does not have any importance, as the ones- and the threep -orbitals span the same subspace as the four hybrids, so the energy of the determinant in which all the four orbitals are filled with the same spin does not depend on the hybridization.
The promoted ‘reference state’ is obviously only a hypothetical intermediate entity which is devoted to facilitate the analysis. It is important to point out once again that the promotion energy ob-tained by its use reflects only the change in the orbital configura-tion scheme of the atom (if any) and the orbital deformaconfigura-tions occurring in the molecule, but neither delocalization and electron transfer effects nor the appearance of the ionic configurations con-nected with the bond formation are accounted for. Thus that pro-motion energy does not necessarily reflect the fact that the interactions between the bonds formed by the atom cause the resulting occupation of ans-orbital to be usually higher than that of the correspondingp-orbitals even if all of them are considered
singly occupied in the valence and reference states—although this delocalization effect should have a significant impact on the final one-center energies.
3. Method of calculations
The ‘natural hybrid orbitals’ were originally defined by McWe-eny[5]for the case of an orthonormalized basis. Similarly to the (spatial) natural orbitals which diagonalize the spinless first order density matrix, the natural hybrid orbitalsfwAigof atom A diago-nalize the intraatomic part of the electron density:
.
AAð~rÞ ¼If an overlapping basis is used, then one should not simply diago-nalize the intraatomic block of the matrix Dbecause that would lead to some spurious non-orthogonal atomic hybrids. Instead, in line with the known fact that in the overlapping case the natural orbitals are the eigenvectors of the matrix DS, one should solve the non-symmetric eigenvalue problem[2]
DAASAACA ¼CA
K
A; ð2Þ
whereDAAandSAAare the atomic blocks of the density and overlap
matrices, respectively,KAis the diagonal matrix of the atomic
nat-ural occupation numbers
m
Ai, and the columns of matrixC
Acontain
the expansion coefficients of the atomic natural hybrids in the ori-ginal basis. Instead of doing that explicitly, we turn to an auxiliary orthogonal basis on the atom in question by using Löwdin orthog-onalization, transform the density matrix to this basis as
Dk¼ ðSAA
Þ1=2DAAðSAAÞ1=2; ð3Þ
diagonalize the Hermitian matrixDkby the unitary matrix Uas
and then one obtains the atomic natural hybrid orbitals in the ori-ginal basis as
This provides a set oforthonormalizedeffective atomic hybrid orbi-talswAi, coinciding with those introduced in[1,2]; the eigenvalues obtained in the diagonalization are the respective occupation num-bers
m
Ai entering the expansion (1). As it is easy to see, the sum of
eigenvalues equals Mulliken’s net population of the atom in question.
There is a unique set of orthogonal localized orbitals, different for each atom, for which the intraatomic parts of the orbitals (up to the normalization) coincide with the atomic natural hybrids ob-tained in the above manner. These localized orbitals are right those which make stationary the Mulliken’s net atomic population for each orbital[1].
4. Sample calculations
The effective AO-s and their gross orbital populations have been computed by a modified version of the program[6], the energies of the promoted reference states have been calculated either by anad hocmodification of an older program of ours or by imputing the orbitals to a commercial program.
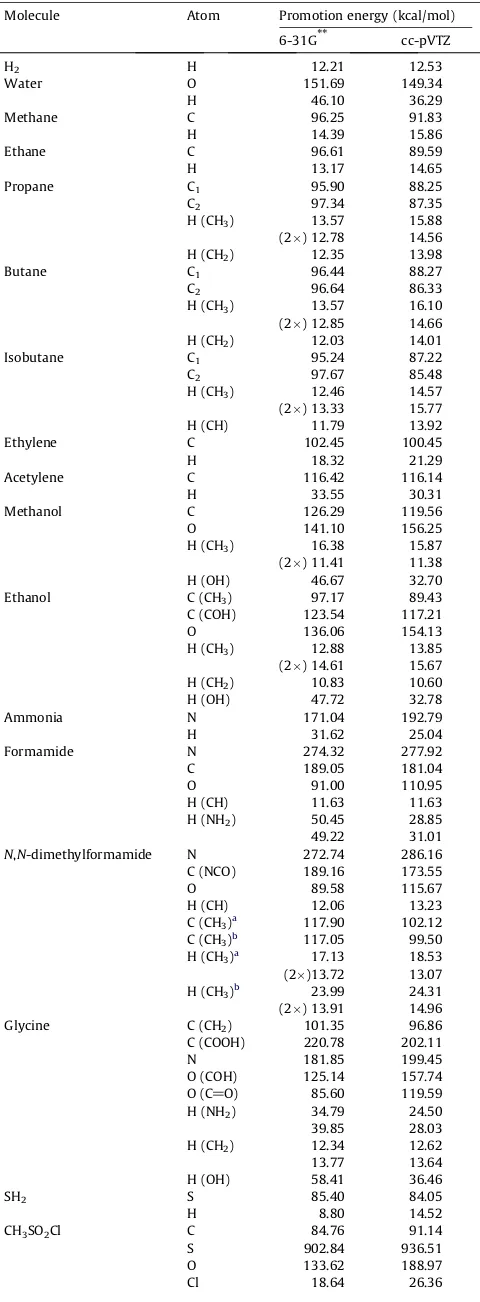
Table 1displays the energies of the promoted reference state of different atoms with respect to the free atom energies for a set of
3 For instance, in the CH
3SO2Cl molecule one obtains that the sulfur is a doubly positive ion and both oxygens carry a negative charge. It may happen that the rounding scheme described leads to violation of the overall neutrality of the system. Then minor corrections may be desirable—e.g., rounding a gross orbital population 1.52 to 1 instead than to 2, if the latter produces a disbalance. Alternatively—as noted by a Referee—it might be of meaning to consider in such a case some average of the energies of the two states in question.
selected molecules calculated by using the standard 6-31G** and cc-pVTZ basis sets. (According to our experience, both of them are of sufficiently adequate atomic character as to permit reason-able ‘Hilbert-space analyzes’ of chemically relevant quantities.)
The results are, of course, basis dependent, but some overall tendencies can be observed. First of all, the promotional energies are quite substantial in some cases and are well characteristic to the chemical nature of the atom in question. When looking at the numbers, one has to keep in mind that they are not variation-ally determined quantities but represent only some components of the optimized total SCF energy. This means that the atomic promo-tion energies may change significantly if that permits some—even rather small—overall energy improvement. This may be well illus-trated on the example of the enhanced promotion energy of one of the CH3hydrogens in theN,N-dimethylformamide molecule. This
molecule represents one of the simplest systems in which the exis-tence of a C–H O interaction has been postulated on the basis of experimental results[12]. The direct H O interaction energy had been estimated[10]around 24 kcal/mol for the 6-31G** basis. This is, however, almost compensated by the increase of other en-ergy components—among them the one-center enen-ergy of the hydrogen involved—so the resulting net energy effect is at most a few kcal/mol. In full accord with that, the promotion energy of the hydrogen involved in that interaction represents the highest value for all CH3hydrogens included inTable 1.
Disregarding that special case, the promotion energies of the hydrogens scatter around 12–15 kcal/mol in the ‘neutral’ cases (H2 molecule, saturated hydrocarbons and most of the CH3 and
CH2groups). For C–H bonds in the molecular planes the promotion
energy is somewhat larger than for out-of-the–plane ones, indicat-ing the presence of some additional interaction in these planes. The hydrogen promotion energies (as well as the carbon ones) are sig-nificantly larger in the unsaturated systems, indicating them to be more ‘stressed’.
Very characteristic are the promotion energies of the hydrogen atoms linked to a heteroatom: the more electronegative is that atom the higher is the hydrogen promotion energy. (Similar obser-vation holds for carbon, too—see below.) That can be explained by taking into account that the orbital in question will be depleted of electrons during the bond formation, so larger orbital deformations may become possible.
When considering the carbon promotion energies, one has to take into account that ca. 56 kcal/mol of these values (56.23 and 56.48 for the two basis sets considered) is the energy of promotion from the ð1sÞ2ð2sÞ2ð2pÞ2 ground state to the ð1sÞ2ð2sÞ1ð2pÞ3 va-lence state. That may be considered the structure of the carbon ‘reference state’, too, in all cases considered—as noted above, if all the four valence orbitals are occupied by spin
a
, it does not have any importance that some effective AO-s are obtained hybrids. (The situation is, however, different for nitrogen and oxygen atoms.) So orbital deformation is responsible for some further 30–50 kcal/mol energy increase—if the carbon in question is not linked to any electronegative atom. If that is case, however, then the more electronegative is the environment of the carbon atom, the higher is its promotion energy—that very pronounced effect seems analogous to the situation discussed for the hydrogen. TheTable 1
Atomic promotion energies calculated for selected molecules by using the 6-31G** and cc-pVTZ basis sets.
Molecule Atom Promotion energy (kcal/mol)
6-31G** cc-pVTZ
Propane C1 95.90 88.25
C2 97.34 87.35
H (CH3) 13.57 15.88
(2) 12.78 14.56
H (CH2) 12.35 13.98
Butane C1 96.44 88.27
C2 96.64 86.33
H (CH3) 13.57 16.10
(2) 12.85 14.66
H (CH2) 12.03 14.01
Isobutane C1 95.24 87.22
C2 97.67 85.48
N,N-dimethylformamide N 272.74 286.16
C (NCO) 189.16 173.55
Table 1(continued)
Molecule Atom Promotion energy (kcal/mol)
6-31G** cc-pVTZ
H 25.79 23.84
(2) 23.56 22.85
aCH
3group intransposition with respect to the oxygen atom. bCH
3group incisposition with respect to the oxygen atom.
results for ethanol molecule clearly show the difference of the car-bons of these two types: one of them is similar to those in hydro-carbons another to that in methanol. The comparison of the two carbon atoms in ethanol again indicates that orbitals which are going to loose a significant amount of their populations undergo larger deformations when the overall molecular wave function is optimized.
It is customary to speak about the valence state of carbon differ-ing from its ground state in order to explain why carbon is four-valent. However, nitrogen and oxygen atoms undergo more-or-less similar changes, even if this is not manifested by changes in their overall valences. Thus only the carboxylic oxygen atoms have a ref-erence state close to theð1sÞ2ð2sÞ2ð2pÞ4ground state, but even in these cases there is somep-admixture to the doubly filled 2sorbital, surely contributing to the relative high promotion energy. But the oxygen atoms in the COH moieties have ansp-type lone-pair, so their reference state may be symbolically denotedð1sÞ2ð2spÞ3ð2pÞ3, lead-ing to an even significantly higher promotion energy. Similarly, pyramidal nitrogen atoms are characterized by a reference state of type ð1sÞ2ð2spÞ3ð2pÞ2 while the planar ones entering a local
p
-system have in a first approximation a lone-pair ofp
-symmetry, so they may be characterized asð1sÞ2ð2sÞ1ð2pÞ4. That explains why the promotion energies of the later are significantly higher.The calculations for the CH3SO2Cl molecule exhibit the specific
behaviour of hypervalent systems: as opposed to the non-hyperva-lent sulfur in the SH2molecule, the sulfur in CH3SO2C is very
posi-tive and its reference state is the double posiposi-tive ion. Thus more than 730 kcal/mol, i.e., most of the enormous sulfur promotion energy, comes from the ionization. It may be noted in this connec-tion that the role of sulfur d-orbitals is very important for the chemistry of this system and their aggregated gross orbital popu-lation is significant (exceeds one in the case of the cc-pVTZ basis set). These orbitals describe the important ‘back donation’ effects, but neither of the five d-type effective AO-s has a gross orbital pop-ulation reaching 0.3, so one cannot consider them as true valence orbitals to be filled in the ‘reference state’.
5. Summary
Using the concept of the effective minimal basis set introduced some time ago, we discuss that the reference state of an atom in a molecule can be defined as a high-spin determinant built up by occupying the orbitals of the effective minimal basis by one or two electrons, depending on their gross orbital populations. The energy of that determinant gives the atomic promotion energy in the molecule, which the atom can be assigned after the orbital deformations are introduced but before any bonding, delocaliza-tion and charge transfer effects are taken into account. The first pivoting calculations indicate that these promotion energies can be quite substantial and are characteristic for the chemical nature of the atom.
Acknowledgments
The author is grateful to both Referees for their valuable com-ments and acknowledges the partial financial support of the Hun-garian Scientific Research Fund (Grant OTKA 71816).
References
[1] I. Mayer, Chem. Phys. Lett. 242 (1995) 499. [2] I. Mayer, J. Phys. Chem. 100 (1996) 6249.
[3] I. Mayer, P. Salvador, J. Chem. Phys. 130 (2009) 234106. [4] V. Magnasco, A. Perico, J. Chem. Phys. 47 (1967) 971. [5] R. McWeeny, Rev. Mod. Phys. 32 (1960) 335.
[6] I. Mayer, Program ‘‘EFF-AO”, Budapest 2008; may be downloaded from the web-site <http://occam.chemres.hu>.
[7] J.P. Foster, F. Weinhold, J. Am. Chem. Soc. 102 (1980) 7211. [8] A.E. Reed, R.B. Weinstock, F. Weinhold, J. Chem. Phys. 83 (1985) 735. [9] A.E. Reed, L.A. Curtiss, F. Weinhold, Chem. Rev. 88 (1988) 899. [10] I. Mayer, Faraday Discuss. 135 (2007) 439.
[11] K. Ruedenberg, Rev. Mod. Phys. 34 (1962) 326. [12] G. Schultz, I. Hargittai, J. Phys. Chem. 97 (1993) 4966.