© 2016 UBM. All rights reserved. No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical including by photocopy, recording, or information storage and retrieval without permission in writing from the publisher. Authorization to photocopy items for internal/educational or personal use, or the internal/educational or personal use of specific clients is granted by UBM for libraries and other users registered with the Copyright Clearance Center, 222 Rosewood Dr. Danvers, MA 01923, 978-750-8400 fax 978-646-8700 or visit http://www.copyright.com online. For uses beyond those listed above, please direct your written request to Permission Dept. fax 440-756-5255 or email: [email protected].
P h a r mTe c h . c o m CONTINUOUS IMPROVEMENT
4
Could Greater Transparency Improve
Pharmaceutical Quality and Compliance?
Agnes ShanleyEXCIPIENT QUALITY
9 Ensuring Quality in
Pharmaceutical Raw Materials
Agnes ShanleyBATCH RECORDKEEPING MANAGEMENT
14
Paperless Batch Records for the Masses
Agnes ShanleySTERILIZATION
21
Too Much by Half: Misapplication
of the Half-Cycle Approach to Sterilization
James AgallocoAGING FACILITIES
27 Is Global Regulatory
Gridlock Slowing Modernization?
Agnes ShanleyMICROBIAL TESTING
33
Microbiological Testing,
Time is of the Essence
Cynthia A. ChallenerINVESTIGATIONS
38
Investigation Effectiveness
Drives Human Performance Excellence
Clifford BerryQUALITY MANAGEMENT
48 Extending QMS to Contract Partners
Agnes Shanley51
Ad Index
Quality Througout the Supply Chain 2016
Issue Editor: Agnes Shanley
Animated Cover images – Shutterstock.com Images: AmerEagle/Rawpixel.com/Simon Mayer/ A_Lesik/Sarawut Aiemsinsuk/Sumroeng Chinnapan/Dmitry Kalinovsky
Static Cover Image – Olivier Le Moal/Shutterstock.com EDITORIAL
Editorial Director Rita Peters [email protected] Senior Editor Agnes Shanley [email protected] Managing Editor Susan Haigney [email protected] Science Editor Adeline Siew, PhD [email protected] Manufacturing Editor Jennifer Markarian [email protected]
Science Editor Randi Hernandez [email protected] Community Manager Caroline Hroncich [email protected]
Art Director Dan Ward
Contributing Editors Jill Wechsler [email protected]; Jim Miller [email protected]; Hallie Forcinio [email protected]; Susan J. Schniepp
[email protected]; Eric Langer [email protected]; and Cynthia A. Challener, PhD [email protected] Correspondent Sean Milmo (Europe, [email protected])
Address
485 Route One South, Building F, Second Floor, Iselin, NJ 08830, USA Tel. 732.596.0276, Fax 732.647.1235, PharmTech.com
SALES
Publisher Mike Tracey [email protected] Mid-West Sales Manager Irene Onesto [email protected]
East Coast Sales Manager Joel Kern [email protected] European Sales Manager Linda Hewitt [email protected] European Senior Sales Executive Stephen Cleland [email protected]
Executive Assistant Barbara Sefchick [email protected]
Address
485 Route One South, Building F, Second Floor, Iselin, NJ 08830, USA Tel. 732.596.0276, Fax 732.647.1235
PharmTech.com
Sr. Production Manager Karen Lenzen
International Licensing Maureen Cannon [email protected], tel. 440.891.2742 or toll-free 800.225.4569 ext 2742, fax. 440.756.5255 Audience Development Manager Rochelle Ballou [email protected]
UBM Americas provides certain customer contact data (such as customers name, addresses, phone numbers, and e-mail addresses) to third parties who wish to promote relevant prod-ucts, services, and other opportunities that may be of interest to you. If you do not want UBM America’s to make your contact information available to third parties for marketing purposes, simply call toll-free 866.529.2922 between the hours of 7:30 a.m. and 5 p.m. CST and a customer service representative will assist you in removing your name from UBM America’ lists. Outside
the US, please phone 218.740.6477.
Pharmaceutical Technology does not verify any claims or other information appearing in any of the advertisements contained in the publication, and cannot take responsibility for any losses or
other damages incurred by readers in reliance of such content.
Pharmaceutical Technology welcomes unsolicited articles, manuscripts, photographs, illustra-tions, and other materials but cannot be held responsible for their safekeeping or return.
Single issues, back issues: Call toll-free 800.598.6008. Outside the US call 218.740.6480. Reprints of all articles in this issue and past issues of this publication are available. Call 877-652-5295 ext. 121 or email [email protected]. Outside US, UK, direct dial: 281-419-5725. Ext. 121. Direct mail lists: Contact Tamara Phillips, Marketing Services, tel. 440.891.2773, [email protected]. Display, Web, Classified, and Recruitment
Advertising: Contact, tel. 732.346.3027.
Permissions: Contact Maureen Cannon, tel. 440.891.2742 or toll-free 800.225.4569 ext 2742, fax. 440.756.5255, [email protected].
Continuous Improvement
F
or years, leaders within the FDA, particularly Janet Wood-cock, director of the Center for Drug Evaluation and Re-search (CDER), (1) have noted the need for FDA to break with old “command-and-control” methods. A few years ago, Woodcock proposed an incentive program that would reward companies for strong compliance and product quality records, and penalize those with lax practices. Results of a 2015 survey (2) of in-dustry experts in California suggested that such a “scorecard” might increase industry investment in quality.Originally, mention of the scorecard brought immediate ques-tions of whether FDA should supervise the effort. A number of different ideas have been proposed, but a new approach was out-lined at the Parenteral Drug Association (PDA) Annual Meeting in September 2016 by Martin Van Trieste, former chief quality of-ficer at Amgen, who retired in June 2016. Van Trieste had founded Rx360, the industry supply-chain safety coalition that is working to combat pharmaceutical counterfeiting, substandard drugs, and illegal diversion.
He has been collaborating with veteran quality executive Richard Love, founder of HarborView LLC, on software development and new algorithms that are based on key pharmaceutical quality perfor-mance indicators. Their goal is a website, the first version of which went live in November 2016, and a smartphone app that will help consumers make informed buying decisions, and allow proactive pharmaceutical manufacturers to use product quality as a sales and marketing strategy. He discussed the concept with Pharmaceutical Technology.
Could Greater Transparency
Improve Pharmaceutical
Quality and Compliance?
Agnes ShanleyA new website and methodology grades manufacturers, and their products, based on quality and good business practices.
Founder Martin Van Trieste explains the approach and the thinking behind it.
A
ME
R
E
A
G
LE
/S
H
U
T
T
E
R
S
T
O
CK
.C
O
Pharmaceutical Technology QUALITY THROUGHOUT THE SUPPLY CHAIN NOVEMBER 2016 5
A new approach
PharmTech: What inspired you to start working on this project?
Van Trieste: The more I’ve thought about it, the more I’ve seen that the industry needs a transfor-mational approach to product quality and patient safety. This is the age of the Internet. More power must go to the pharma consumer, just as it has to consumers in other sectors with sites such as Angie’s List and Yelp. Transparency has a positive impact on business, and the best performing com-panies tend to prosper.
PharmTech: Why isn’t the old approach working?
Van Trieste: In the old scenario, the head of qual-ity defends what is needed to improve qualqual-ity to senior management, but the top manager generally considers it an expense, or a cost of doing business, rather than an investment in the future.
If the consumer of your products changes buying habits based on product quality and the robust-ness of your supply chain, then product quality be-comes a business issue. Sales and marketing teams will see benefit, and invest more in product quality. This would result in a real change in dynamics that could be revolutionary.
We live in a very litigious society. Some con-sumer lawsuits have already been seen based on product problems that involve purchasing agents, pharmacy benefit managers, and buyer groups. The basic questions behind some of these suits are simple: ‘Why was I given a grade-D generic phar-maceutical product, which resulted in this adverse reaction, rather than a grade-A generic? Was this done just to save money?’ In today’s environment, large companies will lose suits like this.
If market dynamics are introduced, it could spur a logarithmic change in the industry’s approach
to quality and compliance. I’m a realist and rec-ognize that this approach could take many years to catch on, but I figured we could start with a ‘JD Edwards’-type scorecard. Currently there is a lot of motivation for improvement.
PharmTech: Today, the consumer at the typical re-tail pharmacy is not informed of the source of the medication that is being used to fill his or her pre-scription. Shouldn’t that information be provided?
Van Trieste: We don’t have that level of transpar-ency now, but I believe that the country of origin for both actives and finished drugs should be re-vealed to the consumer. At the PDA conference, I asked the audience a hypothetical question: ‘If you could choose between three products, one made in western Europe, one in India, and one in China, which one would you choose?’
No hands went up for India or China. Whatever else that says, it proves that consumers want sourc-ing information and somethsourc-ing that tells you more about how a drug was made.
PharmTech: So you are taking a ‘report card’ ap-proach?
Van Trieste: Richard Love at HarborView and I are developing software and an app that would allow the consumer to take a photograph of a prescrip-tion bottle at the pharmacy. The system will be set up so that it would check that photo against a database, providing information on the company that makes the drug and the factory where it was made, to let people know whether it was manufac-tured at a well- or poorly-managed facility.
PharmTech: Is this a realistic idea, given how se-cretive pharmaceutical companies are?
buy a Tesla from a dealership, and you can rate your Uber experience instantly. Technology will enable better quality.
PharmTech: Why do you think it is that country and manufacturer of origin are not disclosed to the consumer today?
Van Trieste: I’ve asked FDA, and they said that such information could be considered a trade se-cret, and the agency cannot disclose trade secrets.
Back in the day when current good manufactur-ing practices (cGMPs) took effect, or even earlier, in the 1930s when the Food Drug and Cosmetic Safety Act was established, most drugs and APIs were made in the United States, so our laws and practices are based on the fact that most drugs were produced in the US. But times have changed and, today, consumers have a right to know.
PharmTech: What data can consumers realistically be expected to check?
Van Trieste: Well, some data is already publicly available. The tools that exist today for analyz-ing Big Data were not there, even three years ago. They allow users to go through 483s, warning let-ters, and drug shortages documentation in a very short amount of time.
So, we are developing a scorecard based mainly on information that is publicly available, adding on information from PDA’s quality-culture metrics. We’ve developed proprietary computer algorithms
to use that information.
PharmTech: What other data are you using for the algorithms?
Van Trieste: We also incorporate manufacturing data on process capability, and ask whether or not the company has redundant sourcing for raw ma-terials. Additional questions focus on inventory levels. From these data, we issue a quality grade
that also indicates the probability of the given manufacturer to have a drug shortage.
New tools for transparency
PharmTech: Tell us about the website.
Van Trieste: It is called medicine-i.com, and will allow users to sift through compliance data as well as data on the probability of shortages. I believe that companies with good quality programs will want to be in the system. For others, the program will start some difficult internal conversations.
PharmTech: Will APIs and their suppliers be in-cluded in the grading process?
Van Trieste: APIs will be part of the calculation but their manufacturers will not be graded. However, if a finished drug manufacturer buys API from a supplier, that supplier’s records will have been ana-lyzed too (e.g., whether or not the company and the facility were cited by FDA). The API manufac-turer’s customer will receive a higher grade if their API comes from a reputable company with a good
compliance history.
A website like this will improve transparency and give consumers more power. Over time, retail outlets will only want to stock products that have been made by good suppliers.
PharmTech: How do you think the industry will react to this program?
Van Trieste: I predict that one third of the industry will want to be involved, one third will be against the whole idea, and the other third will take their usual ‘let’s wait and see what the others do’ ap-proach to gage other companies’ response.
I do predict that pharma, as an industry, will resist the idea, which is rather controversial for this segment. But transparency is inevitable, and it has already hap-pened in other industries. Resistance is futile!
8 weeks.
;OH[»Z^OH[[OLOHUKVɈZ ramp-up and rework
from First in Human to Proof of Concept take from you.
^LLRZ`V\JHU»[HɈVYK[V^HZ[L ^LLRZWH[PLU[ZJHU»[HɈVYK[V^HP[
But when you partner with 7H[OLVU6UL:V\YJL™ you learn how
you can get your 8 weeks back.* And maybe even more.
We approach drug development HM\UKHTLU[HSS`KPɈLYLU[^H` Our way accelerates every step. (UKTVYLPTWVY[HU[S`LSPTPUH[LZ
the spaces between them. ;VNL[OLY^L»SSNL[`V\YTVSLJ\SL to Proof of Concept faster — and better prepared for what comes next.
We’ve got your weeks. Come get them.
A HEALTHIER WORLD. DELIVERED.
3LHYUHSS[OLILULÄ[ZVMZPUNSLZV\YJLV\[ZV\YJPUN
at 7H[OLVUJVT6UL:V\YJL
* 8-week time savings estimate based on applying Patheon OneSource™ optimization processes for typical multi-vendor Phase I – Phase IIb drug development program. ©2016 Patheon®. All rights reserved.
Continuous Improvement
PharmTech: How are you organizing the manage-ment of this program?
Van Trieste: I may not be the one to carry out the idea, ultimately, but I want to bring it to a certain level of development. Our algorithms will remain our trade secret and the structure of the enterprise, but the information within them will be made public (e.g., number of shortages, which products, length of time since last shortage, how the com-pany is handling inventories, and whether they have the drug on hand if need be).
Companies will also be penalized for failing to build redundancy into their supplier networks.So, for example, if Company X has had a drug shortage in the past, if they are not measuring process ca-pability, and if they rely on one supplier, the prob-ability of their having a shortage is higher than it is for the company with supply redundancy, 120 days of inventory, and process capability analysis. Drug counterfeiting will not be part of the analysis.
A focus on quality
PharmTech: Does the industry’s top management have a grasp of the cost of poor quality?
Van Trieste: In 33 years, I’ve worked for various companies and interacted with people at many different companies in different capacities. One group of companies is highly progressive. Qual-ity and manufacturing leaders at these compa-nies figured out a long time ago how to commu-nicate the importance of investing in quality to C-level executives. There are progressive phar-maceutical companies out there. But there is still a lot of work to do. Other companies are not progressive about quality and the industry is not at Six Sigma quality levels. There are some companies that are close (e.g., Lilly, BMS and
Amgen), but they still have a way to go. But at least they’ve realized that, if you measure some-thing, you can improve it.
PharmTech: What is the pharmaceutical industry’s Sigma level today? Ten years ago, people were put-ting it at 3 or 3.5.
Van Trieste: I’d say the industry is probably still at 3.5 Sigma level as a whole, and roughly 80% of the medicines sold today in the US are generic drugs. In discussions about quality management, the Ge-neric Pharmaceuticals Association has been most vocal about the thousands of products that their members manufacture, and how difficult it would be to measure CpK (process capability) for each product. Why is it that other industries have fig-ured out how to do this for even the least expensive products or components?
In the end, numbers tell the story. If a com-pany is performing at Three Sigma, the cost of poor quality accounts for 25% of manufacturing costs. For a company operating at the Six-Sigma level, it is only 2% of manufacturing costs. For a large company with cost of goods sold of roughly $1 billion per year, that can mean the difference between $20 million and $250 million per year. Pressure within the healthcare sector in general should drive greater interest in reducing the cost of poor quality through investment and best practices.
References
1. T. Sullivan, “FDA Drug Shortages: Fundamental Problem is the
Inability for the Market to Observe and Reward Quality,”
Poli-cymed.com, April 12, 2013,
www.policymed.com/2013/04/fda-
drug-shortages-fundamental-problem-is-the-inability-for-the-market-to-observe-and-reward-quality.html
2. C. Medina and F. Richmond, Therapeutic Innovation and
Pharmaceutical Technology QUALITY THROUGHOUT THE SUPPLY CHAIN NOVEMBER 2016 9
Excipient Quality
G
elatin is commonly used for drug encapsulation. Jean-Philippe Talmon, chief procurement officer at Cap-sugel spoke to Pharmaceutical Technology about the critical quality attributes of gelatin as an excipient in capsule production, as well as measures that can be taken to ensure an uninterrupted supply of quality raw materials.Critical to quality
PharmTech: What are the most important ‘critical-to-quality’ attri-butes for gelatin, for encapsulation?
Talmon (Capsugel): In today’s increasingly interconnected and highly regulated global pharmaceutical market, adherence to ‘critical-to-quality’ attributes is required to deliver best-in-class products.
First, sourced gelatin must comply with the different pharmaceuti-cal and food regulations in all the markets where the end product is sold. In global pharmacopeias, there are mandatory attributes to assure the quality, identity, and purity of the excipients used in the drug product. There are also non-mandatory attributes that pro-vide information on control parameters for specific excipient use and include functionality-related characteristics. Various European Pharmacopoeia monographs, for example, have listed relevant pa-rameters specific to the intended use of the excipient, which can form part of the regulatory filing process. Manufacturers are increasingly expected to understand the functionality of excipients and their po-tential impact on the final drug product.
Second, acquired gelatin must meet specific certification expec-tations and environment, health, and safety (EH&S) requirements. There are more than 15 regulatory references that form the basis of
global gelatin purchasing specifications, including those from FDA,
Ensuring Quality in
Pharmaceutical Raw Materials
Agnes Shanley
Gelatin is widely used for encapsulation, and requires close attention to “critical-to-quality” attributes. Jean-Philippe Talmon, chief procurement officer at Capsugel, discusses best practices.
R
A
W
P
IX
E
L
.CO
M
/S
HU
T
T
E
R
S
T
O
C
K
.CO
Excipient Quality
United States Pharmacopeia (USP), Japanese Phar-macopoeia (JP), Food Chemicals Codex (FCC), and World Organization for Animal Health (OIE).
For example, there are specific regulations on the sourcing of bovine raw materials for excipient use. While Bovine Spongiform Encephalopathy (BSE) is well under control as a disease, the OIE is leading a global movement to standardize BSE regulations and establish a common guidance on prevention, control, and eradication of the disease. Global regulators base their regulatory framework on OIE guidance. In Europe, manufacturers of bovine material must provide certificates of suit-ability (CEP) issued by the European Directorate for the Quality of Medicines (EDQM) to be in-corporated into marketing authorization dossiers for applicable medicinal products. This is just one example of the types of certifications required.
Third, sourced gelatin must maintain specified physical/chemical and microbiological require-ments to provide appropriate encapsulation of in-dividual APIs. These specifications cover a large number of criteria, including viscosity, pH level, loss on drying, and particle sizes, among others.
Through adherence to these three critical-to-quality attributes, companies with capsule and encapsulation capabilities can help to minimize the business continuation risks associated with gelatin raw materials and gelatin production. For
example, in 2013, when heavy levels of chromium were detected in gelatin capsules made in China, the USP–National Formulary (NF) monograph for pharmaceutical-grade gelatin was revised to include testing for heavy metals. Some companies scrambled to comply with the new requirements, which then had a negative impact on their custom-ers. However, companies whose gelatin purchase specifications included chromium restrictions could maintain product supply without delaying delivery of their customers’ products to market.
Animal-sourced materials
PharmTech: What issues are entailed with the use of animal-sourced materials, as compared with syn-thetic or non-animal-derived materials?
Talmon (Capsugel): Animal- and non-animal sourced materials are used as excipients in phar-maceutical products. Each of these forms offers its own unique characteristics, specifications, oppor-tunities, and challenges at various points in the development process. The positives and negatives associated with both forms must be well under-stood to ensure that any potential associated risks are identified in advance and controlled.
Gelatin, which is made from the hydrolysis of collagen of bovine, porcine, or fish origin, is well-suited for the encapsulation of pharmaceutical ingredients because of its excellent gelling and film-forming characteristics that support the man-ufacturing process. These characteristics are the result of the unique property of the gelatin poly-mer, which exhibits a reversible temperature-based gelification, turning into liquid when heating and hardening in the cooling process.
Hydroxypropyl methylcellulose (HPMC), which is made from plant/cellulose-based material, is an
Stringent raw material
©2016 Adare Pharmaceuticals, Inc.
Adare is a global specialty pharmaceutical company inspired to improve the lives of patients whose treatment needs are not fully addressed by current medications. We use our unique combination of experience, proprietary capabilities, and resources to create meaningful products for them.
Our entrepreneurial and performance-driven culture
encourages us to take risks, identify promising ideas, and see those opportunities through to completion. Our collaborative spirit and dedication to developing strong partnerships
provide Adare and our partners with signifi cant advantages in competitive markets.
Adare can help you overcome complex formulation
challenges and add valuable IP to commercialized products and products in development. Over 40 products incorporating Adare taste masking, customized drug release, and
bioavailability enhancement proprietary technologies have been commercialized around the world.
Adare also has an extensive patent portfolio, which includes more than 360 granted patents and 225 pending patent applications.
Experience a partnership focused on the needs of patients and your company’s goals. Schedule a meeting with us at
Discover how our taste masking, customized drug release, and bioavailability enhancement experts can help solve your complex formulation challenges. Email us at
Adare Pharmaceuticals:
Patients and our partners
are at the heart of
everything we do
alternative to gelatin for drug encapsulation. Recent
in-vivo and dissolution test results investigated the
clinical performance of second-generation HPMC capsules compared to that of gelatin capsules, and found that the second-generation HPMC capsule per-formance was equivalent to that of gelatin capsules (1). The choice between gelatin and HPMC materials is based on numerous factors including, for instance, the targeted product profile, characteristics of the fill, and consumer lifestyle considerations (i.e., dietary re-strictions based on religious practices, such as halal and kosher requirements, or on vegan or vegetarian diets). For example, while gelatin sets the benchmark for pH- and ionic strength-independent disintegra-tion and product dissoludisintegra-tion profiles, HPMC cap-sules maintain lower moisture absorption, more flexibility, resistance to chemical crosslinking, and a higher thermal stability than gelatin capsules, pro-viding greater benefit for hygroscopic and moisture-sensitive ingredients.
Ensuring supplier quality
PharmTech: How do you ensure supplier quality?
Talmon (Capsugel): First, stringent raw material
pur-chasing specifications and quality agreements are re-quired that cover a large number of criteria. Second, a robust supplier qualification program is needed to provide a sound framework to guarantee that hard capsules meet the highest standards for quality, trace-ability, and integrity. At Capsugel, our multi-phase program requires that our critical raw material suppli-ers undergo an intensive, year-long selection process that includes 150 critical parameters for evaluation to verify they meet the most stringent regulatory and industry standards, the most important being unique specifications related to dissolution profiles and impu-rities. The process includes the following:
• Step 1: Supplier screening and investigation. A review
of the quality system of the vendor, the state of their manufacturing technology, the scope of their product and service offerings, and a de-tailed understanding of their performance met-rics, regulatory compliance, and raw material traceability. For example, more than 15 regula-tory agency regulations inform the basis of our purchasing specs.
• Step 2: Quality evaluation. A review of supplier
ma-terial quality compliance with the company’s specifications.
• Step 3: Manufacturing suitability evaluation.An
anal-ysis of small-scale production trials and R&D pilot, when relevant. This analysis ensures com-patibility of materials with manufacturing pro-cesses and protocols and confirms both finished capsule product quality and manufacturing effi-ciency performance levels.
• Step 4: On-site supplier audit.A thorough
traceabil-ity analysis, from raw material delivery to fin-ished goods release, including a detailed review of quality systems. This audit also confirms compliance of production equipment and com-pliance to current good manufacturing practice (cGMP) requirements.
• Step 5:Scale-up and final approval. This step
in-volves a full-scale manufacturing run of cap-sules to confirm that material consistently meets specifications and is continuously suitable to our full-scale production. This also requires an eval-uation of the supplier’s ability to control product integrity and proper documentation across the entire supply chain.
Third, it is important that long-term supplier part-nerships are supported by a continuous supplier performance management program. This program
Pharmaceutical Technology QUALITY THROUGHOUT THE SUPPLY CHAIN NOVEMBER 2016 13
includes an ongoing evaluation process designed to ensure supply-chain traceability and finished capsules that comply with the highest integrity standards. This program helps to maintain a global network of qualified suppliers with whom there are established long-term partnerships. Key components of this program are constant testing to the highest standards, updated with the most advanced instrumentation for contaminants tests, for example; regular in-depth onsite audits of sup-pliers; continuous monitoring at manufacturing facilities, including performing incoming quality control on each lot of raw material prior to its use; and follow up on supplier performance. Finally, a global sourcing strategy is needed to ensure con-sistent quality and security of supply.
PharmTech: How do you prepare for potential
dis-ruptions in the supply chain?
Talmon (Capsugel): The key to preventing
supply-chain disruptions is to develop long-term partner-ships with suppliers across multiple production sites to ensure that alternative resources can be ramped quickly and effectively in the event of an unexpected issue. Companies must always look one step ahead of their current needs and the requirements of the regu-latory environment and business market. This process includes continuously screening potential new suppli-ers, monitoring industry and governmental standards and trends, building and maintaining an extensive knowledge base of international regulatory require-ments, and participating in membership associations and working groups. All of these efforts are aimed at anticipating and rapidly implementing changes to safeguard the integrity of supply chain and products.
PharmTech: Which supplier criteria are crucial?
Talmon (Capsugel): Supplier selection is guided by
several core principles, including the quality of the
suppliers’ products and services as well as their product traceability and integrity framework. Ad-ditionally, suppliers’ quality systems, certifications and cGMP, regulatory compliance, and EH&S compliance are important. Finally, it is critical to look for suppliers to maintain responsible sourc-ing initiatives and have viable business continuity plans. All of these attributes are central to ensuring that suppliers can consistently and reliably deliver.
PharmTech: What are best practices for working
with offshore suppliers?
Talmon (Capsugel): A robust framework is required
to ensure that offshore suppliers meet the global expectations of companies and their customers. At Capsugel, we have a global production footprint, and our raw material sourcing efforts are globally coordinated and managed.
We maintain supplier evaluation programs to secure identification of potential sourcing risks and development of appropriate mitigation plans in close collaboration with our suppliers. Supplier audit results, for example, are used to dictate the frequency, as well as the depth and breadth of the follow-up supplier audits we conduct.
In parallel, we also maintain a thorough busi-ness continuity risk assessment program across our entire global supplier base. These assessments are conducted along six main dimensions, including supplier production capacity constraints; compli-ance to Capsugel quality expectations; complicompli-ance to regulatory requirements; single-sourcing risks; supplier financial health; and supplier intellectual property risk and third-party misappropriation.
Reference
1. A.Shanley, “Establishing a New Performance Standard for HPMC Capsules,” PharmTech.com, May 20, 2016,
Batch Recordkeeping Management
J
ust as the paperless office, a utopian vision of the 1970s, has never quite taken shape, neither has the paperless pharmaceu-tical plant. Most large pharma companies have installed the enterprise research planning (ERP) backbone and manufac-turing execution systems (MES) software that is required for electronic data management. Most small to mid-sized pharmaceutical firms, however, continue to handle batch documentation the old-fashioned way, which does not make data management any easier. In this reli-ance on paper, much potential power is lost: potentially reduced cycle times and document management times are just the tip of the iceberg.High costs a factor
Smaller companies have not had too many choices for modernizing data management. Either they could invest hundreds of thousands of dollars on MES and ERP systems, and the same amount again on their implementation, or they could use and try to customize “job shop” software systems that were designed for other industries and do not come out of the box meeting FDA’s 21 Code of Federal Regu-lations (CFR) Part 11 requirements. This lack of FDA compliance would require users to generate thousands of standard operating procedures (SOPs).
Within the past few years, cloud computing has brought the po-tential power of paperless batch recordkeeping to a growing num-ber of smaller companies. Rick Soltero, an alumnus of AAI Pharma and former director of R&D at Smith-Kline Beecham, turned an interest in process control and programming into InstantGMP, a company that offers cloud-based batch documentation software designed to streamline quality control time to allow for manage-ment by exception.
Paperless Batch
Records for the Masses
Agnes Shanley
A cloud-based program offers a less expensive alternative to MES, bringing the power of paperless batch record management to small and mid-sized companies.
Rick Soltero, founder and CEO of InstantGMP, discusses the need for paperless data management that facilitates cGMP compliance.
S
IM
ON
M
A
Y
E
R
/SH
U
T
TE
RS
T
O
C
K
.C
O
ENGINEERING
MEDICINES TO LIFE
WWW.CAPSUGEL.COM
RISING TO THE CHALLENGE
Tomorrow’s complex medicines face challenges to overcome low bioavailability and optimize drug delivery. This calls for a partner with the credibility, ingenuity and flexibility to deliver both the product and process design required to make your compound a commercial reality. With a unique range of technology and integrated product development from design to commercial manufacturing, Capsugel is that partner.
Batch Recordkeeping Management
Over the past two to three years, Soltero says, more than 100 pharma companies have bought the software. Soltero talked about paperless data management issues and the company with Phar-maceutical Technology.
Still a paper industry?
PharmTech: How much of the pharmaceutical indus-try is still handling batch and other key manufac-turing and quality records on paper?
Soltero: Even in this age of smartphones and advanced communication, 50% of the industry is still using paper. I believe this is because the op-tions available to smaller manufacturers are not affordable.
PharmTech: Does the continued use of paper also partly ref lect a mindset issue? When FDA’s re-quirements for digital signatures, as set by 21 CFR Part 11, came out about 10 years ago, they were a source of misunderstanding and debate within the industry. Has that changed?
Soltero: The environment has definitely changed, and the industry’s understanding of digital signa-tures and records has crystallized. Now, the regu-lations for electronic signatures are very clearly written. They are easy to understand and comply
with. Today, when software packages tout the fact that they are Part 11 compliant, they are.
PharmTech: How did you decide to start the com-pany?
Soltero: InstantGMP started back in 2004. I saw a need in pharmaceutical manufacturing for soft-ware that could be used at multiple sites, and by different people, simultaneously. At the time, there weren’t any cloud-based software packages avail-able that could be used by multiple sites and people, at least not in a way that would allow one software platform to reinforce GMPs.
My consulting company had to build a manu-facturing plant for a non-profit research organiza-tion that was conducting Phase III clinical trials in developing nations. Instead of taking the tradi-tional manufacturing plant approach, which would have cost $6 million, we opted for an approach that would depend on outsourcing and handing off the project, after clinical trials were completed, to the World Health Organization (WHO).
At that point, technology had advanced to a point where remote data access via the Internet was possible. I wanted to create tools that would allow operations and quality staffers to create sys-tems that did not rely on the presence of perma-nent workers, and that could be used at more than one company facility.
The facility needed quality systems that would allow manufacturing data to be assessed remotely so that the client could minimize its dependence on permanent staff.
Those quality systems, in turn, had to work with a 21 CFR Part 11-compliant database and docu-ment managedocu-ment application, which included controls for specifications, purchases, materials receiving, inventory, and electronic batch records.
Most small- to mid-sized
Pharmaceutical Technology QUALITY THROUGHOUT THE SUPPLY CHAIN NOVEMBER 2016 17 Quality systems were written while the business
rules were programmed into the application. PharmTech: What are some best practices that can be employed with the software system?
Soltero: The system will only record data from materials that come from approved suppliers and that have met all the required materials checks. It also checks that specs have been approved and that the status is acceptable. As a result, one cannot use materials if they haven’t been approved. The process stops when a gap is reached.
The software also performs quality review of batch product records to ensure that requirements have been met, signatures are in place, and docu-mentation has been closed out, before reviews are done by the quality assurance person. The reviewer in the quality assurance department can then re-view these documents by exception, which saves a significant amount of time.
We’ve designed every module with current good manufacturing practice (cGMP) compliance in mind. New features are judged not only based on functionality but on how they comply with Part 11.
Data integrity
PharmTech: Pharmaceutical and ingredients manu-facturers continue to have problems ensuring data integrity. Does the software address this issue or help facilitate compliance?
Soltero: We’ve designed the software to eliminate the need to sign by hand or type in information. In fact, all signatures are electronic. The company scans barcodes on badges or materials, so one needn’t try to figure out signatures, for instance, to find out when materials were signed and by whom.
In addition, the software can be integrated with balances and other equipment, and data can be
transferred electronically. Every item in inventory has a bar code on it and can be brought up on a computer screen.
PharmTech: Why did you feel that this approach was needed?
Soltero: Not every pharma company is up to speed in batch documentation. Many are still using paper only. Big Pharma companies have electronic systems, but not middle-sized companies, because those systems are not affordable. An ERP system from a company like SAP, for example, can cost millions of dollars to buy and install.
We decided to use a cloud-enabled approach that is easy and intuitive. Users can access the system from anywhere by inputting their user name and password. The result is more fluid operations than possible in older systems.
We are now extending the software by embed-ding equipment control features, allowing manu-facturing and process equipment to be controlled automatically through the cloud.
PharmTech: How do you ensure data security? Soltero: Skeptics continue to question the secu-rity of cloud-based systems, but we use 128-bit encrypted data, just like the military does. Never-theless, Big Pharma companies are often reluctant to rely on cloud software, so we have developed a version of the software that they can run internally, behind their own firewalls.
PharmTech: Can the software enable in-process con-trol and use of process analytical technology (PAT)?
Soltero: We are currently building the capability to facilitate PAT and allow users to monitor data in real time and to handle multivariate data. The software offers users a dynamic view so that they
P h a r mTe c h . c o m 18 Pharmaceutical Technology
T
INDUSTRY OUTLOOK
MPI RESEARCH
Robert H. DeWit, PhD, DABT President MPI Research
INDUSTRY COLLABORATION IS KEY
The drug development industry has always been fastpaced, by necessity. With new discoveries come new regulatory considerations, new processes. When the goal of an industry is to improve quality of life, advancing quickly is not trendy—it’s a necessity.
The tools and resources now available to us have allowed the industry to advance at an unprecedented pace. In today’s land-scape of increased consolidation, a sharpened focus on biologics, and the emergence of new technologies, companies often strug-gle to effectively define themselves.
Consolidation, whether among contract research organizations, large pharma companies, or other entities, seems to be today’s normal. However, it’s important our industry doesn’t get ahead of it-self. Service expansion and consolidation should be done thoughtfully. Companies need to make sure they truly understand the needs of a market before, for instance, buying out small shops. We shouldn’t attempt to be all things to everyone. Niche operations with a complete dedication to certain specialized areas should always have a role.
Rather than worrying so much about deal flow and valuations, we could all stand to focus more on collaboration—among developers, CROs, government agencies, universities, etc. A collective deci-sion to work just a bit more in concert could have a significant impact on quality of life. Plus, it is also in everyone’s long-term business interests when compared to the innovation-stifling cloak-and-dagger attitude that often exists around data or technologies that prove useless when kept in a vacuum.
If organizations throughout the industry were more open to working together to achieve the best outcomes, drug and device development could advance at a more efficient and effective pace. To truly prioritize our collective goal of improving quality of life, the industry also needs to prioritize col-laboration.
One final charge for drug and device developers: work with organizations that employ a consulta-tive model. In an industry that evolves as quickly as ours, it pays (literally) to work with people who understand the practical details of new technologies and regulations. For example, the SEND initiative is top of mind with many today. With the initial SEND requirement date quickly approaching in Decem-ber, the industry is buzzing about SEND capabilities and ensuring readiness. Take the time to make sure your development team not only knows “letter of the law” regarding requirements, but also has a practical understanding of how to navigate the real-world process. A little experience can and will have a significant impact on your timeline, budget, and chances for success.
ABOUT ROBERT H. DEWIT, PHD, DABT
Robert H. DeWit, PhD, DABT, is the president of MPI Research. Dr. DeWit has more than 25 years of global preclinical drug development and toxicology experience. Rob joins MPI Research after serv-ing in leadership positions at the Southwest Michigan Innovation Center—first as president and CEO, then as Assistant Dean of Western Michigan University’s School of Medicine. Throughout his career Dr. DeWit has held scientific and leadership roles at Parke-Davis, The Upjohn Company, Pharmacia, and Pfizer, bringing robust industry experience to MPI Research. Rob is a graduate of Calvin College, and received his PhD from The University of Michigan, and is an active member of the American As-sociation for the Advancement of Science, and both the national and regional chapters of the Society of Toxicology.
Company Name : MPI Research
Phone : 269.668.3336
Email : [email protected] Website : www.mpiresearch.com
Batch Recordkeeping Management
can define the most important fields and set it up to capture any kind of information, operation, or data related to these priorities. These fields become the source of data for control.
A lot of pharmaceutical manufacturing equip-ment is already computer controlled (e.g., tablet presses, blenders, compressors), so operators dial in a setting when they use the equipment. What we’re doing is building an interface to connect that with the control panel.
Speeding up batch documentation review
PharmTech: What problems did you hope to address
with this approach?
Soltero: I wanted to reduce the time and effort
re-quired for quality review. I’ve been involved in man-ufacturing, from the clinical level through full-scale commercial manufacturing, and in my experience, reviewing batch documentation can take up to six weeks. The reviewer must look through every piece of documentation associated with a batch. We made all that part of the software so that it can be accom-plished in 20 seconds. This set-up allows the quality department to review by exception.
PharmTech: How do you handle training with this
approach?
Soltero: Generally, using the traditional approach, it
takes a year to train an operator on cGMPs. For us, training can be done quickly, especially since much of the required training material is already embedded in the software. If an operator needs to access a SOP, it’s right there.
PharmTech: You mentioned the fact that you set
com-panies up so that all badges and components are bar-coded. That would assume a higher level of sophistica-tion than can be found at many facilities today. How
do you get companies to the point where they can take advantage of the benefits of this software?
Soltero: Part of the package is process consulting,
which helps clients take paper-based reports and map them into the electronic systems to take full advan-tage of digital technology and streamline operations. Training can be done in one day.
Some paper-bound companies came to us after a regulatory inspection didn’t go well, and FDA issued a 483 or warning letter. After they’d installed our soft-ware, some of these same companies reported that FDA was satisfied during reinspection.
PharmTech: What are your plans for the platform?
Soltero: In the short term, we plan to make the use of
manufacturing and batch records more flexible. We recognize the fact that every company has its own unique way of doing things, so we are changing the design to be user friendly enough to allow people to retain some of their own originality when they move from paper to digital practices.
We also want to make the software more intuitive and to feature more responsive designs, so that the software will be able to respond to any kind of device being used, whether laptop, tablet, or smartphone. We already have the designs for this capability in place.
The final step will be providing more artificial intel-ligence. We’re also building wizards to help people by walking through key tasks.
One important plan is to incorporate more mod-ern manufacturing tools into the software. People talk about the teachings of Deming, statistical pro-cess control, and Six Sigma, but many people work-ing in the pharmaceutical industry today haven’t been trained in those approaches. We plan to have those methodologies be part of the underpinnings of the software. PT
Pharmaceutical Technology QUALITY THROUGHOUT THE SUPPLY CHAIN NOVEMBER 2016 21
Sterilization
T
he expectations for validation of sterilization were first es-tablished in the 1970s, when at first large-volume parenteral manufacturers and then the remainder of the pharmaceu-tical industry were obliged to scientifically support their processes (1). In the past 40 years, numerous training sessions and conferences have been given and many regulatory and compendial guidance documents, protocols, and reports have been published on this subject. Despite this effort, a substantial number of firms use the half-cycle approach, which is the crudest and least appropriate method for the development and validation of a sterilization process. This article reviews the half-cycle method and its limitations, and outline more appropriate means to develop and validate sterilization processes.Half-cycle approach
The half-cycle approach is believed to have originated in the late 1970s when validation was first expected for ethylene oxide (EO) sterilization of materials. The half-cycle approach relies on the fol-lowing conservative assumptions regarding the sterilization process:
• The population and resistance of the bioburden present during
routine usage of the sterilization process must be less than or equal to that of the biological indicator (BI) used in the devel-opment and validation effort.
• Complete destruction of a 106 population BI in the half cycle
as-sures at least a 12-log reduction of the BI (and thus the bioburden) population in the full cycle. (This assumption of a bioburden of one million colony forming units [CFU] is extremely conservative and highly unlikely to ever be encountered in actual practice.)
• A minimum 12-log BI destruction is required for sterilization.
Too Much by Half:
Misapplication of the Half-Cycle
Approach to Sterilization
James Agalloco
The half-cycle method for validating sterilization can have adverse effects on materials if used for steam sterilization. The author reviews historical and current thinking on validation of sterilization processes.
James Agalloco is president of Agalloco & Associates, [email protected]; he is also a member of Pharmaceutical Technology’s editorial advisory board.
A
_
L
E
S
IK
/SH
U
T
TE
RS
T
O
C
K
.C
O
Sterilization
Implicit in the use of the half-cycle is that varia-tions in EO concentration, relative humidity, and process temperature can be ignored. In its original context, this was considered necessary because of the key process parameters in EO sterilization (hu-midity, temperature, time, and gas concentration), only temperature and time could reliably be mea-sured. As real-time instrumentation for EO concen-tration and humidity within the sterilizing chamber simply did not exist, use of the half-cycle approach for EO sterilization was sufficiently robust to assure the desired outcome. To some extent, that situation remains largely unchanged today. EO sterilization has become the province of contract sterilization organizations, and the predominant means of estab-lishing the amount of EO introduced to the cham-ber is by weight or volume of the gas introduced. As 100% EO without a diluent gas in the cylinder is
the prevalent sterilant used, this method assures the minimum required quantity has been introduced into the sterilization chamber. While inefficient in the use of both sterilizing gas and chamber capacity, its basic simplicity cannot be denied.
Sterilization impact on materials
An element of the cycle development for any ster-ilization process is confirmation that the materials being sterilized are safe for their intended use after exposure. With respect to EO sterilization, material properties must be evaluated following exposure to the routine cycle. The comparatively low tempera-ture conditions of EO sterilization are generally be-nign to the materials sterilized by it; in most cases, minimal differences are observed in materials ex-posed to the half cycle as compared to the full cycle. The half-cycle method is also used for other ster-ilizing gases, liquids, and vapors. Application of
the half-cycle method for decontamination pro-cesses is unnecessary as these propro-cesses are not intended to sterilize. The absence of resistance data for common bioburden microorganisms exposed to these processes makes the half-cycle method more suitable than other sterilization development and validation approaches for these less widely used sterilization methods.
Unfortunately, the author has observed the half-cycle validation method being increasingly used for steam sterilization processes, where the conse-quences of its use are not benign. Steam steriliza-tion for many items is customarily performed at temperatures at or above 121 °C. In steam cycles,
complete kill of a 106 population of the thermophilic
spore former G. stearothermophilus is understood to
overkill any bioburden present with a high degree of reliability and safety (2). The overkill approach is accepted because there is a substantial body of information on the moist heat thermal resistance of pathogenic and environmental microorganisms (3).
Figures 1 and 2 depict the half-cycle and full-cycle
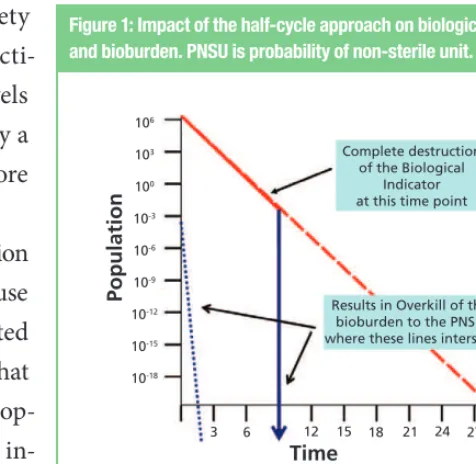
Pharmaceutical Technology QUALITY THROUGHOUT THE SUPPLY CHAIN NOVEMBER 2016 23 the bioburden provides a large measure of safety
that when the biological indicator is fully inacti-vated, bioburden destruction is assured to levels exceeding the minimum requirement (typically a probability of a non-sterility [PNSU] of not more than 1 chance in 1,000,000) (2).
Applying the half-cycle approach to a sterilization process that is already sufficiently lethal for safe use of the materials must consider an often neglected aspect of all sterilization processes—which is that the sterilized materials retain their essential prop-erties post-process. In the zeal to kill biological in-dicators, many sterilization scientists have failed to consider the implications of over-processing. This is most egregious when the half-cycle method is used for steam sterilization, in which doubling the process dwell stresses the materials unnecessarily. Consider the potential adverse impact on mate-rials that are exposed to moist heat excessively as listed in Table I. The only materials unaffected by
excessive heat might be parts made of stainless steel and other metals. The need to consider material ef-fects more fully was added to the most recent USP sterilization chapter (2); it was acknowledged that the numerous negative effects of over-processing on materials adversely affected their fitness for use. Simply stated, the half-cycle should not be used for any steam sterilization process. The half-cycle pro-vides overkill of the bioburden and is more than sufficient to render the bioburden fully inactivated with a substantial margin of safety. A consideration in this is the available body of knowledge on the moist heat resistance of microorganisms, especially G. stearothermophilus (3, 4). This sporeformer is universally used in the validation of moist heat ster-ilization where aqueous filled containers are not present. In those instances, its destruction,
espe-cially when a population of 1x106 spores per
chal-lenge unit is used, assures that the expected biobur-den will be reproducibly destroyed well below the minimum PNSU expectation of one in a million.
Alternative methods
The use of G. stearothermophilus as an indicator in the sterilization of aqueous filled containers is
Figure 1: Impact of the half-cycle approach on biological indicator and bioburden. PNSU is probability of non-sterile unit.
Figure 2: Impact of the full-cycle approach on biological indicator (BI) and bioburden. PNSU is probability of non-sterile unit.
Complete destruction of the Biological
Indicator at this time point
Results in Overkill of the bioburden to the PNSU where these lines intersect
Time
Contin. on page 26
T
INDUSTRY OUTLOOK
IMPACT ANALYTICAL
Katherine Robertson Business Technical Manager
Impact Analytical
NAVIGATING THE CHANGES IN USP <661>
If you are considering registering plastic material with FDA in a Drug Master File (DMF), evaluating the safety of that plastic is key. As of May 2016, a revision in the US Pharmacopeia’s monograph for <661> changes the requirements for the testing of polymericmate-rials used for pharmaceutical packaging. The updated monograph has been divided into <661.1>, “Plastic Materials of Construction,” and <661.2> “Plastic Packaging Systems for Pharmaceutical Use.” In order that a plastic material be considered suitable for pharma-ceutical use, it should be thoroughly characterized. While not all
polymers are cited specifically in the monograph, those that are, polyethylene, polypropylene, poly-olefin, poly (vinyl chloride), and polyethylene terephthalate, are most commonly used in the pharma-ceutical industry for packaging systems. Plastic materials of construction not included in this list must be characterized with the same intent. The evaluation of a polymer material of construction according to USP <661.1> is extensive, and includes identification, physico-chemical testing, biological reactivity, additives analysis, and extractable metals.
Plastic packaging systems include any container, whether it is bag, bottle, syringe, or inhaler, which comes into direct or indirect contact with the drug product. Therefore, the packaging system also includes secondary components such as labels, closures, and printing. Evaluation of a plastic pack-aging system according to USP <661.2> includes biological reactivity, and physicochemical testing. A chemical safety assessment of the plastic packaging system should also be considered, and this could be accomplished through extractables and leachables testing.
Impact Analytical is ready to help meet these changes and challenges. With a solid background and expertise in polymers and polymer additives, we are uniquely qualified to support our customers. We are able to perform traditional extraction for raw material as required in USP <661.1>, as well as autoclave extraction of containers required USP <661.2>. And, with our wide range of capabilities, we can support the analysis of polymer materials from physical property testing such as DSC, to addi-tives analysis by liquid and gas separations and mass spectrometry.
ABOUT KATHERINE ROBERTSON
Katherine currently serves as the business technical manger at Impact Analytical. She has been with Impact Analytical for 24 years.
ABOUT IMPACT ANALYTICAL
Impact Analytical is a contract analytical laboratory serving the pharmaceutical, medical device, and consumer products industries. ISO 9001:2008 certified, DEA licensed, FDA registered, and cGMP & GLP compliant.
Company Name : Impact Analytical
Phone : 855.427.6583
Email : [email protected] Website : www.impactanalytical.com
No matter what your industry
or market, product quality and
performance can make or break
your success. That’s why over
300 companies trust Impact
Analytical for comprehensive
testing and analysis.
Unparalleled Quality and Service
We are a registered FDA facility, our work processes adhere to the toughest regulatory standards, including:
• ISO 9001:2008 quality system standards
• Good Laboratory Practice (GLP) regulations required by the EPA and FDA
• Current Good Manufacturing Practices (cGMP)
• Drug Enforcement Administration (DEA) under schedules 1, 2, 2H, 3, 3N, 4, & 5
• Product Quality Research Institute (PQRI) best practices for extractables and leachables
855.427.6583
impact
analytical
.com
Sterilization
non-universal. It is generally avoided in the termi-nal sterilization of large volume parenterals and other aqueous liquids as its extreme resistance re-quires such lengthy exposure periods that product quality is essentially fully compromised (4). As a consequence, the use of a half-cycle approach for terminal sterilization is never recommended. The half-cycle approach for an aqueous liquid should also never be used to justify the use of an aseptic process rather than a terminal sterilization pro-cess. In fact, current thinking on the application of terminal sterilization is headed in the opposite direction, with increased usage of lower time-tem-peratures and the use of less resistant biological indicators (5).
The half-cycle will remain useful in selected settings, notably gas, liquid, and perhaps other sterilization processes as an expedient means to establish process lethality. There are opportunities in each of those processes to use alternative means
such as a bracketing approach that incorporates the potential variations in process parameters and results in a generally shorter, but more scientifi-cally defensible sterilization process (6–8).
References
1. Code of Federal Regulations, Title 21, Food and Drugs, Current Good Manufacturing Practices in Manufacturing, Processing, Packing or Holding of Large Volume Parenterals and Requests for Comments Regarding Small Volume Parenterals, Part 212, Federal Register 22202-22219 (Proposed June 1, 1976, withdrawn 1991). 2. USP, USP General Chapter <1229>, “Sterilization of compendial
articles,” USP–NF (US Pharmacopeial Convention, 2013). 3. I. Pflug, Microbiology & Engineering of Sterilization Processes
(Environmental Sterilization Laboratory, Minneapolis, MN, 14th edition, 2010).
4. S. Block, Ed., Disinfection, Sterilization, and Preservation (Lip-pincott Williams & Wilkins, Philadelphia, 5th ed., 2001). 5. J. Agalloco et al., PDA Letter, 47 (4) 14,16 (2011).
6. USP, USP General Chapter <1229.6>, “Liquid Phase Sterilization,” USP–NF (US Pharmacopeial Convention 2014).
7. USP, USP General Chapter <1229.7>, “Gas Phase Sterilization,” USP–NF (US Pharmacopeial Convention, 2014).
8. USP, USP General Chapter <1229.11>, “Vapor Phase Sterilization,”
USP–NF (US Pharmacopeial Convention, 2015). PT Table I: Potential changes from excessive sterilization treatment.
Change Possible result
Changes in elastomeric materials (e.g., bags, closures, gaskets, tubing)
• Hardening, embrittlement, loss of flexibility • Loss of strength
• Difficulty in automated feeding; machinability • Twinning and clumping (sticky or gummy) • Increases in extractables/leachables • Increases in visible and subvisible particles • Changes in appearance
Changes in glass • Delamination • Increases in particles
• Changes in extractables/leachables/pH
Changes in filters • Loss of integrity
• Increase in extractables/leachables • Changes in wetting properties
Changes in packaged materials (including those used in sterilization)
• Loss/weakening of package integrity • Appearance changes
Changes in APIs and excipients • Loss of potency
• Increased level of degradation • Changes in physical properties • Changes in appearance
Pharmaceutical Technology QUALITY THROUGHOUT THE SUPPLY CHAIN NOVEMBER 2016 27
Aging Facilities
I
n 2014, the Parenteral Drug Association (PDA) organized a task-force to study the impact of aging pharmaceutical manufactur-ing facilities on product quality, manufacturmanufactur-ing efficiency, and regulatory compliance. In 2016, the organization released results of its first survey, which summarized 2015 data (see Sidebar).Among other things, the survey results suggest that regulatory post-approval changes (PACs), and the time and investment that they require, are stifling conversion to new manufacturing technologies or methods. PDA has now established a PAC innovation for access to medicines (PAC iAM) working group to evaluate the issue and work with regulatory agencies for change.
As group members note in a op-ed published in the April 2016 edition of PDA Letter (1), global harmonization has not led to any improvement in the timeframes required for PACs. The op-ed states:
“Today, most companies operate globally; therefore, PACs are in-tended to apply globally. Many require approval, however, by the national regulatory authority of each country before the company can deliver a product manufactured using improved processes. In practice, this can result in submitting change filings for assessment to more than a hundred individual regulatory bodies ... it is not uncommon for a simple change to take more than five years to re-ceive approval. All of these complexities create a disincentive—albeit unintentional—for manufacturers to integrate growing product and process knowledge, continually improve, or innovate technologies. “In order to avoid the burden of implementing changes in such a complex process, many find it easier to postpone improvements to facilities, processes, and analytics, or simply to refrain from planning advancements at all.”
Is Global Regulatory Gridlock
Slowing Modernization?
Agnes Shanley
PDA’s first aging facilities survey suggests that post-approval changes are slowing investments in new technology. Maik Jornitz, who chaired the aging facilities taskforce for its first year, discusses challenges.
SA
R
A
W
U
T
A
IE
M
SI
N
S
UK
/S
HU
T
T
E
R
S
T
O
C
K
.CO
Aging Facilities
PAC iAM co-chair Anders Vinther sees a need to change the industry’s focus from “locking in what has already been approved” to continuously improving processes and reducing risk, quoting
Bill Paulson, editor of International
Pharmaceuti-cal Quality, “We are changing the way we think about GMPs to allow for continuous improvement. But on the chemistry, manufacturing, and control (CMC) side, we haven’t really seen that same push to change the basic paradigm. Rather than focus-ing on committfocus-ing to ‘where you are now,’ the ap-plication could include ‘a commitment to change and improve and indicate how you are going to do that’” (2), he said in a 2014 presentation at PDA’ s annual conference.
Among the solutions Vinther proposed are: hav-ing regulators ask companies for enhanced product and process control plans through continual
im-provement efforts rather than corrective actions, and allowing companies to report post-approval changes, even for critical changes, on the Annual Report, as long as comparability protocol require-ments were met.
Maik Jornitz, CEO of G-CON Manufacturing, former head of the aging facilities taskforce, and member of PDA’s PAC iAM taskforce, discussed
with Pharmaceutical Technology some of the issues
that the pharmaceutical industry is facing.
Stimulating modernization
PharmTech: Why does it seem that so little progress has been made in the area of stimulating modern-ization and the use of more modern technologies, especially since Janet Woodcock and others at FDA have been encouraging industry to make these changes for the past 10 years?
Earlier this year, the Parenteral Drug Association (PDA) published results of its first aging facilities survey (1). Most of those (64%) who responded work in the biopharmaceutical manufacturing sector, and the rest in small molecules. Nearly half said their facility was more than 20 years old. Of the 80% who responded to the survey’s question about the operation and condition of their facilities, 73% described their facilities’ operation as excellent or good, with the remainder saying it was fair or poor.
Two thirds responded to questions about batch rejection rates, with 80% citing rejection rates of up to 4%, with another 15% seeing a range from 5–10%; 88% of respondents said that up to 30% of their processes receive improvements.
When asked about regulatory requirements and whether they encourage or discourage investments in improvements, 56% said they encourage investments in modernization, while 24% said they discourage such investments, and 10% were undecided.
Respondents agreed that the time required for post-approval changes can be considerable, with 32% describing timeframes
of up to two years, and 62% saying that they can take from two to 10 years, and 3% relating that they take more than 10 years. The majority of respondents (92%) estimated the
regulatory costs for such changes to be below $110,000. Budget allocation for improvements went mainly to facilities, where 68% of respondents saw the focus, while 51% said they went to process improvements and 40% to better analytics. However, 13% of respondents said that their companies did not have an annual improvement budget, while another 18% did not know whether such a budget existed.
Forty-two percent said their companies did not plan to modernize their facilities, while 11% did not know.
Twenty-eight percent of respondents reported having an active technology scouting program, but 54% said their companies did not actively look for new technology, while 18% said that they did not know.
Reference