In the first part of the thesis, the synthesis, characterization and reactivity of “reverse pyridine” bis(phosphinite) pincer complexes are described. In the first case, more donating substituents decreased the potential required for Ni(II) to Ni(III) oxidation.

Homogeneous alkane transfer dehydrogenation by iridium PCP and POCOP pincer complexes. 16
X Bond Cleavage
This chapter describes the synthesis of a "reversed pyridine" POCOP pincer ligand from 3,5-dihydroxypyridine and the formation of a subsequent adduct between the pyridyl nitrogen and the potent Lewis acid tris(pentafluorophenyl)borane (BCF). Given the synthetic utility of late-transition metal bis(phosphinite) tang complexes with a central resorcinol core, further elaborations of this ligand class are warranted.1-2 Thus, the development of an "inverted pyridine" POCOP variant was pursued, starting with the synthesis of the bis(phosphinite) ligand itself.
Synthesis of a reverse pyridine POCOP pincer ligand 2.1
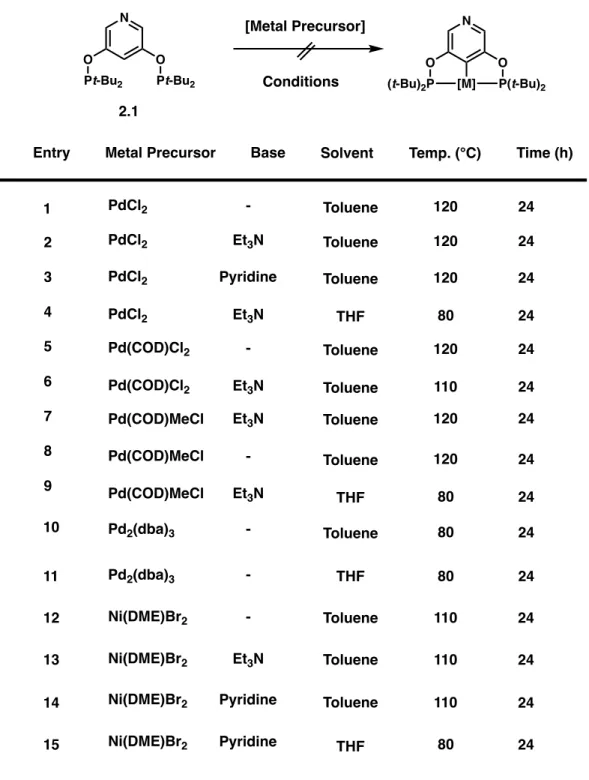
Brookhart and Jensen first reported the synthesis of POCOP ligands from substituted resorcinols, base, and dialkylchloro or diarylchlorophosphines.3-5 Modern methods have varied little from this approach except for the identity of the base and reaction solvent and temperature.6 Starting from commercially available 3,5-dihydroxypyridine, we screened reaction conditions using di-tert-butylchlorophosphine as a coupling partner. The importance of the organic base DBU probably stems from the increased solubility of the in situ generated aryl oxide anion as opposed to more typical alkali metal cations associated with metal hydride bases.
Attempted alkylation of 2.1 and successful BCF N-coordination
Bergman and Tilley used B(C6F5)3 for remote tuning of the electronics in platinum bipyrazine complexes.7 They found that biaryl-reductive elimination of square platinum "20ydroxy" complexes can be accelerated by a factor of 64,000 via BCF coordination to the backbone of their bidentate ligands (Scheme 2.3). Further work on 2,2'-bipyrimidyl platinum complexes with coordinated Lewis acids revealed a more complex mechanistic picture involving nitrogen ligand dissociation of platinum before reductive elimination.8.
Effect of remote Lewis acid binding on platinum biaryl complex reductive elimination. 8
Fortunately, coordination of BCF to ligand 2.1 proved straightforward, as stirring the Lewis acid and pyridine ligand in benzene at ambient temperature afforded the desired N-adduct 2.2 in high yield (Scheme 2.2). Metalation of the free pyridine ligand 2.1 was attempted with a variety of Group 9 and 10 metal precursors. In the case of ligand 2.1. is it also possible to imagine oligomeric or polymeric species that do not contain a cyclometalated seaweed complex at all.
When metalation of ligand 2.1 was attempted with Vaska's complex [IrCl(CO)(PPh3)2], some soluble products attributable to the desired cyclometalation were observed by 31P NMR spectroscopy in C6D6 after heating to 80°C. Fortunately, reaction of ligand 2.1 with [Rh(CO)2Cl]2 in the presence of triethylamine afforded the desired cyclometalated seaweed rhodium carbonyl complex 2.4 (Scheme 2.4).

Metalation of POCOP ligands with rhodium carbonyl chloride dimer
Reaction of the BCF POCOP adduct 2.2 with [Rh(CO)2Cl]2 afforded the desired seaweed carbonyl complex 2.5 in high yield in a remarkably short reaction time of ca. ten minutes. In the case of ligand 2.1 , added base enabled formation of reverse pyridine pincer complex 2.4 with an uncoordinated nitrogen backbone. Comparison of the spectroscopic data from both new rhodium compounds revealed the significant effect that Lewis acid coordination to an inverse pyridine pincer complex can exert.
Interestingly, the gap between the free base reverse pyridine complex and the BCF-coordinated analog was many times greater than In the latter case, heating a toluene solution of 2,2 and [Ir(COD)Cl]2 for one day gave the desired Ir(III) hydrochloride complex 2,6 cleanly in good yield (Scheme 2.5).
Metalation of BCF-bound ligand 2.2 with iridium and nickel
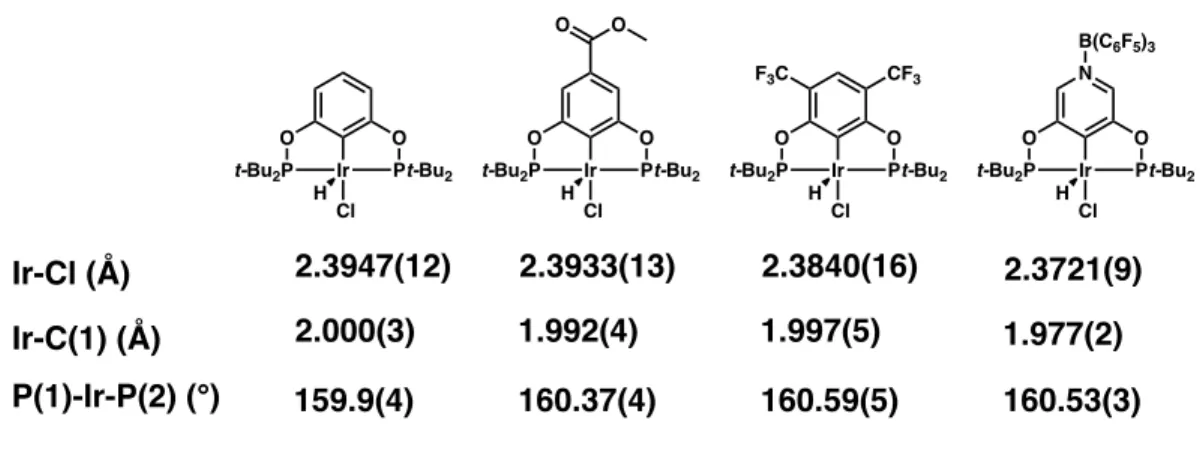
In the nickel bromide complex 2.7, the Ni-Br distance of 2.3163(6) Å is approximately 0.022 Å shorter than that observed for the parent complex (Table 2.7).17 This again may reflect an interaction that is more polarized from Ni to Cipso and subsequent greater Coulombic attraction between Ni+ and Br-. With both borane-attached complex 2.7 and borane-free complex 2.8 in hand, we decided to compare the two by their UV-visible absorption spectra. A potential rationalization for this shift to lower energy absorption is that electron-withdrawing substituents on the ligand backbone help to delocalize and thereby stabilize charge in the complex's excited state.
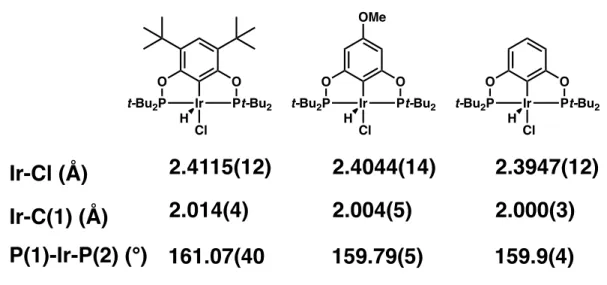
Finally, the borane-bound complex 2.7 was investigated and found to exhibit a non-reversible oxidation with Eox = 1.79 V vs Fc*+/Fc*. Although Guan and co-workers were able to obtain POCOP nickel hydrides using the same synthetic method in very high yields, it is possible that interactions between the borane in 2.7 and excess lithium and aluminum salts enabled degradation pathways that lowered the yield obtained. 27 Complex 2.9 is a bright yellow solid with two notable spectroscopic features that distinguish the metal hydride: a triplet in the 1H NMR spectrum (-7.92 ppm, 2JP-H = 52.0 Hz) and a stretch in the IR spectrum at 1804 cm-1.
Synthesis of pincer nickel hydride complexes
Empirically, this correlation between ligand basicity and CO2 insertion rates has been well known for kelp metal hydrides (Figure 2.9).31. We sought to investigate the rate of carbon dioxide insertion for our reversed pyridine-nickel hydride complex. Exposure of 2.9 to 1 atmosphere of CO2 in C6D6 led to slow and clean conversion to a nickel formate complex 2.11 over 16 h at ambient temperature (Scheme 2.8).
This rate of insertion is similar to that observed for a PNP-pyrrole-based nickel pincer complex.40 Therefore, coordination of a borane Lewis acid to the inverted pyridine POCOP pincer framework has a shift in the rate of a insertion reaction enabled after that seen for a tong with a completely different central donor atom.
Fast CO 2 Insertion at 25 °CSlow CO2 Insertion at 25 °C
Carbon dioxide insertion by complex 2.9
36 This microscopic reverse of the insertion reaction is clean for the parent complex under vacuum at 60 °C. Heating the formate complex to 60 °C under an atmosphere of CO2 also led to decomposition, especially to oxidized phosphine species. Such species may result from activation of the phosphinite donors to P-O and C-O cleavage through the inverted pyridine framework.
Unfortunately, when using catalytic 2.6 with added alkoxide base, none of the desired olefin product was observed over a temperature range. Attempts to remove the N-coordinated borane from 2.6 with DMAP or solid-supported DMAP were unsuccessful, most likely due to preferential binding of DMAP to the iridium metal center rather than to the coordinated borane.
Unsuccessful alkane transfer dehydrogenation with 2.6 and 2.12
- Ray Structure Determination
The crude reaction mixture was filtered through a thin plug of Celite into a Schlenk filter and the toluene was removed under vacuum. The flask was removed from the glovebox and vented to argon on a Schlenk line while the solution was stirred at ambient temperature. The crude reaction mixture was filtered through a thin plug of Celite and the toluene was removed under vacuum.
The flask was sealed, removed from the glove box and placed in an oil bath heated to 100 °C. After this time, the flask was cooled to ambient temperature, removed from the glovebox and then filtered through a Teflon syringe filter (0.45 µm), the toluene removed under vacuum.
BCF-pyrPOCOP)IrHCl
BCF-POCOP)NiBr
An unusual cleavage of C-O and C-S bonds with silanes in the presence of alkoxide bases is presented. Cleavage of both C(sp2)-O and C(sp3)-O bonds was achieved at high temperatures using excesses of both alkoxide bases and tertiary silanes. In the last few decades, the growing demand for energy combined with declining fossil fuel reserves has created a tremendous increase in interest in the efficient production of fuels and bulk chemicals from renewable bioresources.1 The natural heterobiopolymer lignin has emerged as a major target for cost-effective biomass conversion , because the repeating aromatic ether structural units could offer products with high energy content and potential access to useful derivatives for fine chemical applications.2 At present, however, the utilization of lignin is clearly limited, as current technology does not allow efficient degradation into its building blocks with the desired selectivity.1 One of the major challenges associated with such a process is the need to reductively cleave the various types of strong aromatic C-O bonds found in lignin,3 which is also a relevant problem for coal densification (Scheme 3.1). 4.
Structure of hardwood lignin
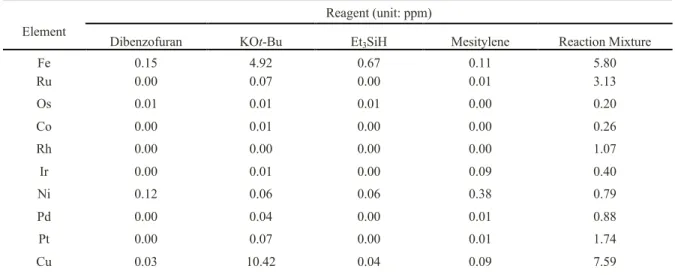
ICP-MS analysis of reaction mixtures, reactants and products showed that traces of transition metals did not exceed 10 ppm.16 Thus, heating 1 with 5 equiv. This result suggests that dihydrogen may be important to prevent the decomposition of active reducing species.26 In searching. These results demonstrate our ability to tune the selectivity of the reaction by optimizing the reaction conditions.
This substrate provided benzene and phenol in moderate yields (Table 2, Entry 1) with the remainder of the mass balance attributed primarily to Many of the diaryl ethers evaluated were more reactive compared to diphenyl ether and allowed the use of milder reaction conditions.
Reductive cleavage of lignin model substrates
This conclusion is supported by the high selectivity of the reductive ring opening of the dibenzofuran derivative 3.8, which mostly leads to 3.10. Similarly, we observe the preferential formation of phenol and anisole with similar selectivity to phenols 3.12 and 3.13 in the cleavage of lignin model 3.11. We speculate that such an effect can be rationalized by the resonance stabilization of the positive charge of the oxygen atom that occurs during the electrophilic activation of the CO bond that breaks.
To test this hypothesis, we subjected 3 to our reaction conditions and isolated the ring-opened phenols 3.5 and 3.6 along with the desilylated products 3.1 and 3.2 (Scheme 3.3, entry C). As an extension of the current work, we reacted dibenzothiophene under the conditions used for dibenzofuran separation (Scheme 3.4).
Dibenzothiophene reduction with alkoxide and silane
In a glove box, a 4 ml screw cap vial was loaded with the corresponding substrate (0.1 mmol, 1 equivalent), base (0.5-5 equivalent) and a magnetic stir bar, followed by addition of the solvent (1 ml) in a syringe. and triethylsilane (1–5 equiv.). After cooling to room temperature, the dark red to black reaction mixture was diluted with diethyl ether (3 mL) and carefully quenched with 1 mL of 1 N aqueous HCl. The mixture was allowed to reach ambient temperature and stirring was continued for 4 hours before chlorotriethylsilane (10.1 mL, 60 mmol, 5 equiv.) was added.
The crude reaction mixture was purified by chromatography on silica (hexanes) and the resulting product was recrystallized from a mixture of methanol and isopropanol (1:1) to give 4,6-bis(triethylsilyl)dibenzofuran (1.28 g, 2.45 mmol , 28%). like colorless needles. After acidic aqueous work-up, the crude reaction mixture was purified by chromatography on silica using hexanes and hexane-ether (10:1) to give, among other isolated products, 20 mg (0.05 mmol, 1%) 7. 9) Mechanism details of Ni-catalyzed aryl-oxygen cleavage were reported: p. 10).