OPTIMASI PENGEKSTRAKSI FITUR
SPACED
K-MERS
FREKUENSI MENGGUNAKAN ALGORITME GENETIKA
PADA
PENGKLASIFIKASIAN FRAGMEN
METAGENOME
ARINI AHA PEKUWALI
SEKOLAH PASCASARJANA INSTITUT PERTANIAN BOGOR
PERNYATAAN MENGENAI TESIS DAN
SUMBER INFORMASI SERTA PELIMPAHAN HAK CIPTA
Dengan ini saya menyatakan bahwa tesis berjudul Optimasi Pengekstraksi Fitur Spaced K-Mers frekuensi menggunakan Algoritme Genetika pada Pengklasifikasian Fragmen Metagenome adalah benar karya saya dengan arahan dari komisi pembimbing dan belum diajukan dalam bentuk apapun kepada perguruan tinggi mana pun. Sumber informasi yang berasal atau dikutip dari karya yang diterbitkan maupun tidak diterbitkan dari penulis lain telah disebutkan dalam teks dan dicantumkan dalam Daftar Pustaka di bagian akhir tesis ini.
Dengan ini saya melimpahkan hak cipta dari karya tulis saya kepada Institut Pertanian Bogor.
RINGKASAN
ARINI AHA PEKUWALI. Optimasi Pengekstraksi Fitur Spaced K-Mers Frekuensi menggunakan Algoritme Genetika pada Pengklasifikasian Fragmen Metagenome. Dibimbing oleh WISNU ANANTA KUSUMA dan AGUS BUONO.
Metagenomika adalah studi yang mempelajari keseluruhan informasi genetik tentang organisme-organisme dari sampel yang diambil langsung dari lingkungan. Lingkungan yang dimaksud seperti tanah, air, isi perut manusia, bangunan, limbah dan lain-lain yang merupakan tempat mikroba berkembang biak. Tahapan dalam metagenomika diawali dengan deoxyribonucleic acid (DNA) sequencing terhadap metagenome sampel. Karena diambil langsung dari lingkungan, fragmen yang dihasilkan mengandung berbagai mikroorganisme. Kondisi seperti ini memungkin terjadinya kesalahan perakitan terhadap fragmen metagenome, yaitu disambungkannya fragmen antara spesies yang satu dengan fragmen dari spesies yang lain. Permasalahan ini dapat diselesaikan menggunakan metode binning.
Ada 2 pendekatan binning, yaitu homologi dan komposisi. Penelitian ini berfokus pada pendekatan komposisi. Binning berbasis komposisi mengelompokan fragmen-fragmen dari berbagai organisme berdasarkan tingkat taksonominya menggunakan teknik-teknik dalam machine learning. Ada 2 komponen penting dalam machine learning, yaitu ekstraksi fitur dan pengklasifikasian atau pengklusteran.
Teknik machine learning yang digunakan dalam penelitian ini adalah spaced k-mers frekuensi sebagai metode pengekstraksi fitur dan Naïve Bayesian Classifier (NBC). Penggunaan metode spaced k-mers frekuensi menghasilkan banyak model posisi match (1) dan don’t care (0). Oleh karena itu, Algoritme Genetika digunakan untuk mengoptimasi model posisi yang menghasilkan akurasi pengklasifikasian tertinggi.
Penelitian ini bertujuan untuk mengetahui model posisi yang menghasilkan akurasi pengklasifikasian tertinggi. Tujuan kedua penelitian ini adalah mengetahui pengaruh penggunaan don’t care pada pengekstraksi fitur k-mers frekuensi.
Kromosom 111111110001 yang berarti [111 1111 10001] dengan nilai fitness 85,42 terpilih menjadi kromosom terbaik. Don’t care juga berperan dalam mereduksi jumlah fitur sehingga mempercepat waktu eksekusi 6 kali lebih cepat dibandingkan dengan k-mers frekuensi.
SUMMARY
ARINI AHA PEKUWALI. Optimization of Spaced K-mers Frequency Feature Extraction using Genetic Algorithms for Metagenome Fragments Classification. Supervised by WISNU ANANTA KUSUMA and AGUS BUONO.
Metagenomics is a study that learns the entire genetic information concerning organisms from the samples directly taken from the environment, such as from soil, water, building, waste and so on which are places of microbes to breed. The phases in metagenomics were started with sequencing on the metagenome sample. The resulting fragments contain various microorganisms. Such condition might occur errors in the assembling process of metagenome fragments, called as misassembly contigs. The misassembly process yields interspecies chimeras. To minimize the number of interspecies chimeras, we could perform binning process and assembly process simultenously. Binning is a process for grouping the various organism fragments based on the taxonomic level. There are two binning approaches including the homology and the composition approach. In the homology approach, sequence alignment is performed between metagenome fragments and sequence reference that exist in the database of National Center for Biotechnology Information (NCBI). On the other hand, in the composition approach, binning is conducted by classification using machine learning methods.
This research concerns with the problem that occur while conducting k-mers frequency extraction in metagenome fragment binning. K-k-mers frequency feature extractions produce larger features dimensions when the value of k is increased. In this research, the concept of spaced seed in PatternHunter was adapted to modify the k-mers frequency feature, known as spaced k-mers which consists of match positions (1) and don‟t care positions (0). The use of spaced k-mers frequency can reduce the size of dimension of k-k-mers frequency feature. The Genetic Algorithms was employed to find an optimum spaced k-mers frequency model to obtain high accuracy of Naïve Bayesian Classifier (NBC). The result show that the chromosome of 111111110001 representing the spaced k-mers frequency model of [111 1111 10001] was chosen as the best chromosome, with fitness of 85.42 higher than that of using only k-mers frequency features. Moreover, this approach also could reduce execution time almost six times. Keywords: don’t care, genetic algorithms, k-mers, metagenome, naïve bayesian
© Hak Cipta Milik IPB, Tahun 2015
Hak Cipta Dilindungi Undang-Undang
Dilarang mengutip sebagian atau seluruh karya tulis ini tanpa mencantumkan atau menyebutkan sumbernya. Pengutipan hanya untuk kepentingan pendidikan, penelitian, penulisan karya ilmiah, penyusunan laporan, penulisan kritik, atau tinjauan suatu masalah; dan pengutipan tersebut tidak merugikan kepentingan IPB
Tesis
sebagai salah satu syarat untuk memperoleh gelar Magister Ilmu Komputer
pada
Program Studi Ilmu Komputer
OPTIMASI PENGEKSTRAKSI FITUR
SPACED
K-MERS
FREKUENSI MENGGUNAKAN ALGORITME GENETIKA
PADA PENGKLASIFIKASIAN FRAGMEN
METAGENOME
SEKOLAH PASCASARJANA INSTITUT PERTANIAN BOGOR
BOGOR 2015
PRAKATA
Puji dan syukur penulis panjatkan kepada Allah yang Maha Kuasa atas segala kasih karunia, hikmat dan perkenanan-Nya sehingga karya ilmiah ini berhasil diselesaikan. Tema yang dipilih dalam penelitian yang dilaksanakan sejak bulan September 2014 ini ialah pengklasifikasian fragmen metagenome, dengan judul Optimasi Pengekstraksi Fitur Spaced K-mers Frekuensi menggunakan Algoritme Genetikapada Pengklasifikasian Fragmen Metagenome.
Terima kasih penulis ucapkan kepada seluruh pihak yang telah berperan dalam penelitian ini, yaitu:
1 Bapak Dr Wisnu Ananta Kusuma, ST MT dan Bapak Dr Ir Agus Buono, MSi MKom selaku pembimbing, yang telah memberikan banyak ide, masukan, dan dukungan kepada penulis.
2 Bapak Irman Hermadi, SKom MS PhD yang telah bersedia menjadi penguji, dan memberikan saran yang berharga sehingga tulisan ini menjadi lebih baik dari sebelumnya.
3 Ayahanda Marthen Yunus, Ibunda Adrifina Ndjurumbaha, dan adik Marvin Daniel Pekuwali, Ambu Ina, Yuni, serta Kakak Jonathan dan Frida Reese, atas kasih sayang, doa, semangat, dan dorongan kepada penulis sehingga dapat menyelesaikan penelitian ini.
4 Mamu Dorkas, Oma Ester Djera Naha, Kakak Amos dan keluarga, Kakak Ester, Kakak Anang dan Marlin Sinaga, Kakak Emanuel dan Okta Haramburu, Kakak Salmon dan Lina, Kakak Ria dan Aten, Mama Isto, Kakak Maung, Ishak, Tante Metty, Tante Nelce, atas kasih sayang, doa, semangat, dan dorongan kepada penulis sehingga dapat menyelesaikan penelitian ini.
5 Rekan-rekan satu bimbingan Bapak Wisnu: Om Yampi, Pak Syaif, Kakak Mulyati, Rosi, Kana yang telah memberikan dukungan dan bantuan.
6 Kakak Ichad, Rake, Basyith, Sodik dan Farino yang telah membantu mengatasi kesulitan pemrograman yang penulis hadapi.
7 Rekan-rekan llmu Komputer angkatan 15 yang saling menyemangati selama pengerjaan penelitian.
8 Teman-teman Wisma Fahmeda, Kakak Restu, Mia, Ria, Ad, Netty, Adik Kiki, Arista, Eka, Dini, dan Tini, yang telah memberikan dukungan dan bantuan. 9 Papi Peter, Mami Sarah dan rekan-rekan di UBF Depok, serta teman-teman PA
Oikumene Bogor yang telah mendukung dalam doa dan memberikan bantuan. Semoga karya ilmiah ini bermanfaat.
DAFTAR ISI
DAFTAR TABEL vi
DAFTAR GAMBAR vi
DAFTAR LAMPIRAN vi
1 PENDAHULUAN 1
Latar Belakang 1
Perumusan Masalah 2
Tujuan Penelitian 2
Manfaat Penelitian 2
Ruang Lingkup Penelitian 3
2 TINJAUAN PUSTAKA 3
Metagenomika 3
Pendekatan berbasis Homologi dan Komposisi 4
Algoritme Genetika 5
3 METODE 7
Tahapan Penelitian 7
Pengumpulan Data Penelitian 9
Praproses Data 9
Inisialisasi 10
Evaluasi Fitness 10
Seleksi 13
Crossover 13
Mutasi 14
Lingkungan Implementasi 14
4 HASIL DAN PEMBAHASAN 14
Implementasi 14
Optimasi menggunakan GA 15
Evaluasi 15
Fitur Potensial 17
Keterbatasan Penelitian 17
5 SIMPULAN DAN SARAN 18
Simpulan 18
Saran 18
Ucapan Terima Kasih 18
DAFTAR PUSTAKA 19
LAMPIRAN 21
DAFTAR TABEL
1 Data latih 9
2 Data uji 10
3 Matriks komposisi yang terbentuk dengan kromosom 111111110001 12
4 Parameter GA yang digunakan dalam penelitian 14
5 Matriks Confusion dari kromosom 111111110001 16
DAFTAR GAMBAR
1 Binning sampel metagenome 3
2 Mencari nilai similarity antar sequence dengan seed 4 3 Mencari nilai similarity antar sequence dengan spacedseed 4
4 Cara kerja k-mers frekuensi 5
5 Cara kerja spacedk-mers frekuensi 5
6 Siklus Algoritme Genetika sederhana 6
7 Ilustrasi Crossover One Cut Point 7
8 Tahapan penelitian 8
9 Proses evaluasi fitness 9
10 Kromosom k-mers yang terbentuk jika k=12 10
11 Ilustrasi inisialisasi kromosommenggunakan GA 11
12 Ilustrasi kemungkinan yang terbentuk dari E1, E2, dan E3 11
13 Ilustrasi kromosom yang terpilih 11
14 Fitur 22 sampai 27 dari kromosom 111111110001 12
15 Fitur 38 sampai 43 dari kromosom 111111110001 12
16 Percobaan GA menggunakan 9 parameter yang berbeda 15 17 Perbandingan nilai fitness dari k-mers dan spaced k-mers frekuensi 16
18 Perbandingan waktu eksekusi program 17
DAFTAR LAMPIRAN
1 Pseudocode program optimasi pengekstraksi fitur spacedk-mers 21
2 Pseudocode prosedur Ekstraksi Fitur 25
3 Pseudocode prosedur Naïve Bayesian Classifier (NBC) 30
4 Pseudocode Fungsi pembentuk fitur 35
1
PENDAHULUAN
Latar Belakang
Metagenomika adalah studi yang mempelajari keseluruhan informasi genetik tentang organisme-organisme dari sampel yang diambil langsung dari lingkungan. Lingkungan yang dimaksud seperti tanah, air, isi perut manusia, bangunan, limbah dan lain-lain yang merupakan tempat mikroba berkembang biak (Bouchot et al. 2013). Thomas et al. (2012) menjelaskan bahwa metagenomika bertujuan untuk mempelajari variasi spesies, memberikan kontribusi dalam penemuan keanekaragaman gen-gen baru, dan mempelajari interaksi antara mikroba dengan inangnya.
Tahapan dalam metagenomika diawali dengan proses sequencing terhadap metagenome sampel menghasilkan ururtan deoxyribonucleic acid (DNA) dari masing-masing organisme. Karena diambil langsung dari lingkungan, fragmen yang dihasilkan mengandung berbagai macam mikroorganisme (Bouchot et al. 2013). Kondisi seperti ini memungkinkan terjadinya kesalahan perakitan (assembly) terhadap fragmen metagenome, yaitu disambungkannya fragmen antara spesies yang satu dengan fragmen dari spesies yang lain, sehingga menghasilkan interspecies chimeras (Meyerdierks & Glockner 2010). Permasalahan ini dapat diselesaikan dengan melakukan proses assembly dan binning secara simultan. Binning, yaitu mengelompokan fragmen-fragmen dari berbagai organisme berdasarkan tingkat taksonomi. Ada dua pendekatan binning, yaitu homologi dan komposisi. Pada pendekatan homologi, dilakukan sequence alignment antara fragmen metagenome dengan sequence reference yang ada di basis data National Center for Biotechnology Information (NCBI). Pendekatan kedua adalah binning berbasis komposisi dengan melakukan klasifikasi atau pengelompokan terhadap fragmen metagenome menggunakan teknik-teknik dalam machine learning.
BLAST atau Basic Local Alignment Search Tool (Altschul et al. 1990) dan MEGAN (Huson et al. 2007) merupakan aplikasi yang menggunakan pendekatan homologi dalam mengidentifikasi spesies. Sementara itu penelitian yang telah dilakukan menggunakan pendekatan komposisi seperti supervised learning maupun unsupervised learning antara lain adalah PhyloPythia (McHardy et al. 2007), klasifikasi menggunakan Naïve Bayesian Classifier (NBC) oleh Rosen et al. (2008), klasifikasi menggunakan Support Vector Machine (SVM) oleh Kusuma (2012), dan pengelompokan fragmen metagenome menggunakan Growing Self Organizing Map (GSOM) oleh Overbeek et al. (2013).
2
pengekstraksi fitur k-mers frekuensi. Kesimpulan dari penelitian tersebut adalah GSOM dapat digunakan untuk pemetaan fragmen yang memiliki komunitas besar dan memiliki panjang fragmen yang pendek, yaitu 1 Kbp. Penelitian tersebut menghasilkan akurasi lebih dari sama dengan 80%.
Ukuran dimensi fitur yang besar merupakan tantangan yang dihadapi apabila menggunakan metode pengekstraksi fitur k-mers frekuensi. Penelitian yang dilakukan oleh Kusuma (2012) menggunakan pengklasifikasi SVM dan pengekstraksi fitur k-mers frekuensi yang melibatkan don’t care (0) atau yang dikenal dengan spaced k-mers frekuensi. Spaced k-mers frekuensi menghasilkan 192 fitur. Secara keseluruhan penelitian ini sudah menghasilkan akurasi yang baik bahkan untuk panjang fragmen kecil 400 bp yaitu 65,3% untuk takson genus, 72% untuk takson orde, 78,2% untuk takson kelas, dan 82,1% untuk takson filum. Pada panjang fragmen besar (10 Kbp) akurasi mencapai lebih dari 95% untuk semua level takson. Penggunaan berbagai panjang fragmen ini disimpulkan bahwa semakin panjang fragmen yang digunakan maka akan semakin besar hasil akurasi yang diperoleh dan sebaliknya.
Ekstraksi fitur menggunakan spaced k-mers frekuensi menghasilkan banyak variasi model posisi dari match (1) dan don’t care (0), sehingga perlu dicari model posisi yang dapat menghasilkan akurasi yang tinggi. Kusuma (2012) menggunakan metode exhaustive search untuk mencari model posisi dari match dan don’t care yang dapat menghasilkan akurasi yang tinggi.
Algoritme Genetika (GA) dapat digunakan untuk mengoptimasi pencarian model posisi yang dapat menghasilkan akurasi tinggi. Oleh karena itu, penelitian ini menggunakan GA untuk mengoptimasi model posisi match dan don’t care pengekstraksi fitur k-mers frekuensi dalam pengklasifikasian metagenome.
Perumusan Masalah
Berdasarkan latar belakang penelitian yang telah diuraikan sebelumnya, permasalahan yang menjadi bahan analisis dalam penelitian ini adalah bagaimana hasil optimasi terhadap model posisi match (1) dan don’t care (0) pada spaced seed menggunakan Algoritme Genetika yang menghasilkan akurasi yang tinggi?
Tujuan Penelitian
Tujuan penelitian ini adalah melakukan optimasi model spaced k-mers terhadap posisi match (1) dan don’t care (0) pada spaced seed menggunakan Algoritme Genetika yang menghasilkan akurasi yang tinggi.
Manfaat Penelitian
3 Ruang Lingkup Penelitian
Ruang lingkup dari penelitian ini, yaitu:
1. Data metagenome yang digunakan diambil dari basis data NCBI.
2. Fragmen yang digunakan dihasilkan dari perangkat lunak MetaSim yang mensimulasikan Illumina sequencer.
3. Fragmen yang dihasilkan memiliki panjang yang tetap dan tidak mengandung sequencing error.
4. Data latih berjumlah 10 000 fragmen dan 4 500 data uji.
5. Tingkat taksonomi yang digunakan dalam pengklasifikasian adalah genus. 6. Nilai k yang digunakan dalam proses ekstraksi fitur menggunakan spaced
k-mers adalah k=12.
2
TINJAUAN PUSTAKA
Metagenomika
Riesenfeld et al. (2004) menyatakan bahwa metagenomika merupakan suatu teknik yang secara khusus ditujukan untuk mengumpulkan gen-gen secara langsung dari suatu lingkungan, diikuti dengan menganalisis informasi genetika yang terkandung di dalamnya. Berbeda dengan teknik analisis genom bakteri pada umumnya, teknik metagenome dilakukan dengan langsung mengekstraksi DNA genom dari lingkungan dan tidak memerlukan proses pengkulturan bakteri pada media buatan (Handelsman 2007). Hal ini dilakukan untuk mempelajari bakteri yang tidak dapat dikultur yang diperkirakan terdapat lebih dari 99% dari populasi bakteri di seluruh lingkungan (Riesenfeld et al. 2004).
Tahapan pada metagenomika dimulai dengan pengambilan sampel dari lingkungan. Lingkungan dalam hal ini bisa berupa air laut, tanah, isi perut manusia, dan lain-lain. Sampel yang telah diambil selanjutnya diekstrak, kemudian hasil ekstrak dimasukkan ke dalam mesin sequencer. Mesin sequencer menghasilkan fragmen-fragmen DNA. Fragmen-fragmen DNA dirakit (assembly), namun bisa saja terjadi kesalahan pada tahap ini, seperti tersambungnya fragmen organisme A dengan fragmen dari organisme B. Kesalahan perakitan ini menghasilkan interspecies chimeras. Untuk menghindari terbentuknya interspecies chimeras, maka pada tahapan ini harus dilakukan perakitan (assembly) dan binning secara simultan. Binning adalah pengelompokan fragmen-fragmen metagenome sesuai tingkat taksonominya (Meyerdierks & Glockner 2010).
Gambar 1 Binning sampel metagenome (Kusuma 2012)
DNA Assembly
Binning
Finishing
4
Fragmen-fragmen yang sudah tersambung dan membentuk urutan (sequence) DNA disebut contigs (continuous sequence), adapun sequence DNA yang sudah lebih panjang contigs tapi masih mengandung gap yang disebut scaffolds. Kemudian scaffolds dianotasi, yaitu pengidentifikasian sequence DNA.
Pendekatan berbasis Homologi dan Komposisi
Pendekatan berbasis homologi bekerja dengan cara membandingkan data metagenome dengan data yang ada pada basis data menggunakan BLAST (Altschul et al. 1990) atau MEGAN (Huson et al. 2007). Pada dasarnya pendekatan ini digunakan untuk mencari kemiripan antar sequence DNA melalui proses pensejajaran sequence. Cara kerja BLAST adalah melakukan pensejajaran sequence secara local alignment dengan mencari daerah pendek yang berdekatan atau yang bagian nukleotida yang match antara metagenome sequence dengan sequence di basis data (Gambar 2).
Pendekatan berbasis homologi terus berkembang, sehingga oleh Ma et al. (2002) mengusulkan PatternHunter. Metode ini diusulkan untuk memodifikasi aplikasi BLAST. Ide dasar dari metode ini adalah seed yang dipakai tidak perlu semua match. Kombinasi dari match dan don’t care (spaced seed) dapat mempercepat proses pencarian nilai kesamaan dari 2 sequence DNA. Pada spaced seed, match disimbolkan dengan angka 1, sedangkan don’t care disimbolkan dengan angka 0. Keadaan yang disebut match adalah nukleotida yang ada pada metagenome sequence wajib sama dengan nukleotida yang ada pada sequence refrence di basis data. Sedangkan keadaan don’t careadalah nukleotida yang ada pada metagenome sequence tidak wajib sama dengan nukleotida yang ada pada sequencerefrence di basis data (Gambar 3).
* 1 1 1 1 1 * * 1 1 1 1 1 * * 1 1 1 1 1
A G C G A T G A C A G T A T C T A G A C
| | | | | | | | | | | | | | |
T G C G A T A G C A G T A A G C A G A T
Seed: 11111 Gambar 2 Mencari nilai similarity antara sequence metagenome dengan referensi sequence menggunakan pesejajaran sequence dengan seed (Kusuma 2012)
* 1 1 0 1 1 * * 1 1 0 1 1 * * 1 1 0 1 1
A G C T A T G A C A A T A T C T A G A C
| | | | | | | | | | | | | | |
T G C C A T A G C A T T A A G C A G A T
Spaced Seed: 11011 Gambar 3 Mencari nilai similarity antara sequencemetagenome dengan referensi
5 Berbeda dengan pendekatan homologi, pendekatan komposisi tidak perlu mensejajarkan metagenome sequence, tetapi dengan menghitung frekuensi kemunculan fitur dari metagenome sequence. K-mers frekuensi adalah salah satu metode pengekstraksi fitur yang dapat digunakan untuk menghitung frekuensi kemunculan fitur. Nilai k pada k-mers frekuensi adalah panjang fragmen metagenome (Choi & Cho 2002).
Pola kemunculan k dalam sequence dihitung menggunakan 4 basa utama, yaitu Adenine (A), Cytosine (C), Guanine (G) dan Thymine (T) yang dipangkatkan dengan rangkaian pasang basa yang ingin digunakan (pola kemunculan: 4k, dengan k ≥ 1). Gambar 4 menampilkan cara kerja k-mers frekuensi.
A A A G A A C A G A
Gambar 4 Cara kerja k-mers frekuensi (Kusuma 2012)
Metode yang berbasis pendekatan komposisi adalah spaced k-mers frekuensi (Kusuma 2012). Spaced k-mers mengadopsi ide dasar PatternHunter ke dalam metode pengekstraksi fitur k-mers frekuensi. Gambar 5 menampilkan cara kerja spacedk-mers frekuensi.
A A A G A A C A G A
Gambar 5 Cara kerja spacedk-mers frekuensi (Kusuma 2012)
Algoritme Genetika
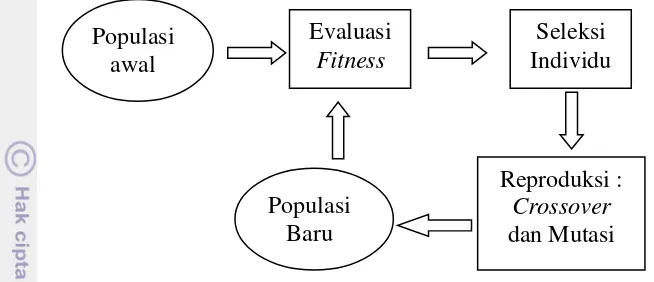
Goldberg (1989) adalah orang yang pertama kali memperkenalkan siklus algoritme genetika yang digambarkan seperti pada Gambar 6. Siklus dimulai dari membuat populasi awal secara acak, kemudian menghitung nilai fitness dari setiap kromosom. Proses berikutnya adalah menyeleksi kromosom terbaik, kemudian dilanjutkan oleh proses crossover dan mutasi sehingga terbentuk populasi baru.
Generate all position k = 3
AAA … AAT … CAA … CAT … TTT
1 0 0 0 0
Generate all position k = 3
spaced k-mers = 101
A*A … A*T … C*A … C*T … T*T
6
Selanjutnya populasi baru ini mengalami siklus yang sama dengan populasi sebelumnya. Proses ini berlangsung terus hingga maksimal generasi.
Gambar 6 Siklus Algoritme Genetika sederhana (Goldberg 1989)
Inisialisasi Populasi awal
Proses inisialisasi kromosom diawali dengan proses encoding. Encoding (pengkodean) berguna untuk mengodekan atau menyandikan nilai gen-gen pembentuk kromosom (Gen & Cheng 1997). Dalam penelitian ini kromosom yang dibentuk berupa kromosom biner yaitu terdiri dari bit 1 dan 0. Bit 1 mewakili match, sedangkan bit 0 mewakilidon’t care.
Seleksi Individu
Proses pemilihan dua kromosom sebagai orang tua biasanya dilakukan secara proporsional berdasarkan nilai fitness. Metode roulette wheel merupakan metode seleksi yang digunakan dalam penelitian ini. Sesuai dengan namanya, metode ini menirukan permainan roulette wheel. Masing-masing kromosom menempati potongan lingkaran pada roda roulette secara proporsional sesuai dengan nilai fitness (Gen & Cheng 1997).
Adapun formula yang digunakan dalam proses seleksi, yaitu: a. Total fitness untuk populasi
F = , k = 1, 2,..., pop_size b. Hitung probabilitas seleksi pkuntuk setiap kromosom vk
pk = , k = 1, 2,..., pop_size
c. Hitung probabilitas kumulatif qkuntuk setiap kromosom vk qk= , k = 1, 2,..., pop_size
Crossover
7 nilainya lebih kecil dari probabilitas pc tertentu, dengan kata lain r < pc. Crossover
dilakukan atas 2 kromosom untuk menghasilkan kromosom anak (offspring). Kromosom anak yang terbentuk akan mewarisi sebagian sifat kromosom induknya (Gen & Cheng 1997). Teknik crossover yang dipakai adalah crossover one cut point.
Orang tua 1 Orang tua 2
Gambar 7 Ilustrasi Crossover One Cut Point (Gen & Cheng 1997) Mutasi
Mutasi dilakukan untuk semua bit yang terdapat pada kromosom, jika bilangan acak yang dibangkitkan lebih kecil dari probabilitas mutasi pm yang
ditentukan. Untuk kode biner, mutasi dilakukan dengan cara menukar nilai bit 0 menjadi bit 1, sebaliknya bit 1 diubah menjadi bit 0 (Gen & Cheng 1997).
Elitisme
Elitisme adalah prosedur untuk menggandakan kromosom yang mempunyai nilai fitness tertinggi sebanyak satu (bila kromosom dalam suatu populasi adalah ganjil) atau dua (bila jumlah kromosom dalam suatu populasi adalah genap). Hal ini dilakukan agar kromosom ini tidak mengalami kerusakan (nilai fitness menurun) selama proses pindah silang maupun mutasi (Gen & Cheng 1997).
3
METODE
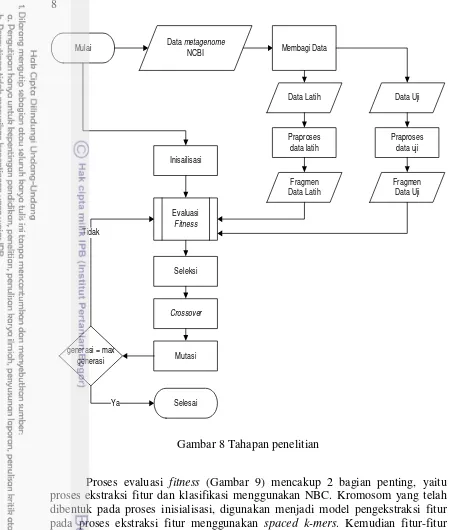
Tahapan Penelitian
Tahapan-tahapan dalam penelitian ini dibagi dalam 4 bagian, yaitu: 1. Pengumpulan, pembagian dan praproses data
2. Optimasi pengekstraksi fitur spaced k-mers mengggunakan GA 3. Ekstraksi fitur fragmen-fragmen metagenome
4. Klasifikasi fragmen metagenome menggunakan NBC Tahapan penelitian ditampilkan pada Gambar 8.
1 1 0 0 1 1 1 1 1 1 1 0
0 0 1 1 1 0 0 0 0 0 1 1
Anak 1
8
Mulai Data metagenome
NCBI Membagi Data
Praproses data latih
Data Latih Data Uji
Praproses data uji
Fragmen Data Latih
Fragmen Data Uji Inisailisasi
Evaluasi Fitness
Seleksi
Crossover
Mutasi generasi = max
generasi Tidak
Selesai Ya
Gambar 8 Tahapan penelitian
9
Menghitung rata-rata dan standar deviasi dari setiap kelas
Gambar 9 Proses evaluasi fitness Pengumpulan Data Penelitian
Penelitian ini menggunakan data mikroba yang diperoleh dari situs NCBI yang dapat diakses melalui (http://www.ncbi.nlm.nih.gov/). Format data yang digunakan adalah FASTA (*.fna). Ada 19 spesies yang termasuk ke dalam 3 genus (Kusuma 2012) yang digunakan di dalam penelitian ini. Data penelitian dibagi dalam 2 bagian yaitu data latih (Tabel 1) yang terdiri atas 10 spesies dan data uji (Tabel 2) yang terdiri atas 9 spesies.
Praproses Data
Proses praproses pada data latih dan data uji dilakukan menggunakan MetaSim (Richter et al. 2008) yaitu perangkat lunak yang dapat mensimulasikan DNA sequencer. Hasil simulasi sequencing menggunakan MetaSim diperoleh 10 000 fragmen dengan panjang fragmen 500 bp untuk data latih dan 4 500 fragmen dengan panjang fragmen 500 bp untuk data uji.
Tabel 1 Data latih
No Species Genus Jumlah
Fragmen
Panjang Fragmen
1 Agrobacterium radiobacter K84 chromosome 2 1000 500 bp
2 Agrobacterium tumefaciens str. C58 chromosome circular
Agrobacterium 1000 500 bp
3 Agrobacterium vitis S4 chromosome 1 1000 500 bp
4 Bacillus amyloliquefaciens FZB42 1000 500 bp
5 Bacillus anthracis str. Ames Ancestor Bacillus 1000 500 bp
6 Bacillus cereus 03BB102 1000 500 bp
10
No Species Genus Jumlah
Fragmen
Panjang Fragmen
8 Staphylococcus aureus subsp. Aureus JH 1000 500 bp
9 Staphylococcus epidermis ATCC 12228 Staphylococcus 1000 500 bp
10 Staphylococcus haemolyticus JCSC1435 1000 500 bp
Tabel 2 Data uji
No Species Genus Jumlah
Fragmen
Panjang Fragmen
1 Agrobacterium radiobacter K84 chromosome 1 500 500 bp
2 Agrobacterium tumefaciens str. C58 chromosome linear
Agrobacterium 500 500 bp
3 Agrobacterium vitis S4 chromosome 2 500 500 bp
4 Bacillus thuringiensis str Al Hakam 500 500 bp
5 Bacillus subtilis subsp. Subtilis str 168 Bacillus 500 500 bp
6 Bacillus pumilus SAFR-032 500 500 bp
7 Staphylococcus carnosus 500 500 bp
8 Staphylococcus saprophyticus subsp. Saprophyticus ATCC 1530S
Staphylococcus 500 500 bp
9 Staphylococcus Lugdunensis HKU09-01 500 500 bp
Inisialisasi
Inisialisasi kromosom menggunakan GA dapat dijelaskan sebagai berikut. Misalnya k yang digunakan 12, maka terbentuk kromosom yang terdiri atas 12 bit (Gambar 10).
1 1 1 1 1 1 1 1 1 1 1 1 x1 x2 x3 x4 x5 x6 x7 x8 x9 x10 x11 x12
Gambar 10 Kromosom k-mers yang terbentuk jika k=12
Saat menggunakan k-mers dengan k = 12 maka menghasilkan 412 fitur. Oleh karena itu, dalam penelitian ini menggunakan pengekstraksi fitur spaced k-mers dengan tujuan mereduksi jumlah fitur. Kromosom yang dibentuk terdiri dari bit yang bernilai 1 (mewakili match) dan bit yang bernilai 0 (mewakili don’t care).
Penelitian ini menggunakan k = 12. Pemilihan nilai k = 12 berpedoman pada penelitian yang telah dilakukan oleh Rosen et al. (2008), yang menyatakan bahwa semakin besar nilai k maka akurasi pengklasifikasian semakin meningkat. Dengan demikian, ruang pencarian GA berjumlah 4 096 kemungkinan.
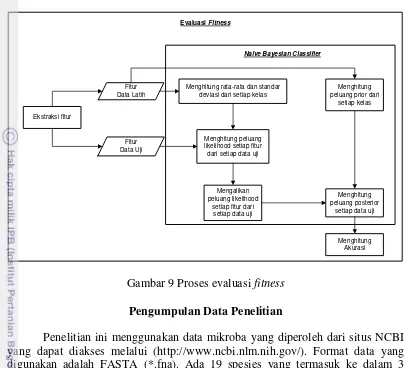
Evaluasi Fitness
11 digunakan dalam ekstraksi fitur dengan cara menghitung frekuensi kemunculan spaced k-mers. Model match (1) dan don’t care (0) dibentuk dari hasil inisialisasi GA. Model posisi selanjutnya disebut kromosom. Don’t care (0) berarti membolehkan pasangan basa apapun mengisi bit tersebut (Ma et al. 2002).
1 * 1 1 * * 1 1 * * * 1
x1 x3 x4 x7 x8 x12
Gambar 11 Ilustrasi inisialisasi kromosommenggunakan GA
Gambar 11 menunjukkan bahwa kromosom pengekstraksi fitur yang diinisilisasi terdiri atas 3 bagian. Bagian pertama E1 (Gambar 12a) terdiri atas 3 bit yang kemungkinan membentuk variasi sebanyak 21. Gambar 12b menunjukkan bagian kedua E2 terdiri dari 4 bit dengan kemungkinan variasi sebanyak 22.
Gambar 12 (a) Ilustrasi kemungkinan yang terbentuk dari E1 (b) Ilustrasi
kemungkinan yang terbentuk dari E2 (c) Ilustrasi kemungkinan yang
terbentuk dari E3
111 1111 10001
Gambar 13 Ilustrasi kromosom yang terpilih
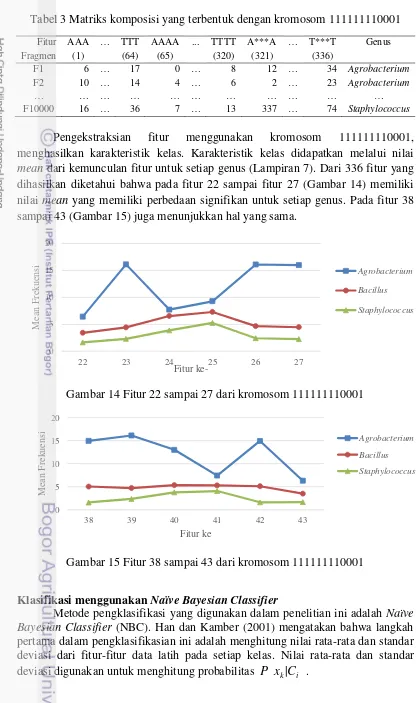
Kromosom pada Gambar 13 digunakan untuk mengekstraksi fitur, maka terbentuk matriks komposisi seperti pada Tabel 3. Matriks komposisi adalah matriks yang berisi frekuensi kemunculan fitur.
12
Tabel 3 Matriks komposisi yang terbentuk dengan kromosom 111111110001 Fitur
Pengekstraksian fitur menggunakan kromosom 111111110001, menghasilkan karakteristik kelas. Karakteristik kelas didapatkan melalui nilai mean dari kemunculan fitur untuk setiap genus (Lampiran 7). Dari 336 fitur yang dihasilkan diketahui bahwa pada fitur 22 sampai fitur 27 (Gambar 14) memiliki nilai mean yang memiliki perbedaan signifikan untuk setiap genus. Pada fitur 38 sampai 43 (Gambar 15) juga menunjukkan hal yang sama.
Gambar 14 Fitur 22 sampai 27 dari kromosom 111111110001
Gambar 15 Fitur 38 sampai 43 dari kromosom 111111110001
Klasifikasi menggunakan Naïve Bayesian Classifier
Metode pengklasifikasi yang digunakan dalam penelitian ini adalah Naïve Bayesian Classifier (NBC). Han dan Kamber (2001) mengatakan bahwa langkah pertama dalam pengklasifikasian ini adalah menghitung nilai rata-rata dan standar deviasi dari fitur-fitur data latih pada setiap kelas. Nilai rata-rata dan standar deviasi digunakan untuk menghitung probabilitas P x Ck| i .
13
C dan Ci adalah rata-rata dan standar deviasi, masing-masing memberikan nilai
atribut Ak untuk data latih dari kelas Ci.
sehingga didapatkan probabilitas P(X|Ci) untuk setiap kelasnya. Probabilitas P(X|Ci) dikalikan dengan probabilitas prior setiap kelas maka akan dihasilkan
Akurasi untuk hasil klasifikasi dapat dicari dengan menggunakan rumus :
Akurasi data uji benar x 100% data uji
Nilai akurasi yang didapat akan menjadi nilai fitness kromosom. Seleksi
Penelitian ini menggunakan operator seleksi Roulette Wheel. Gen dan Cheng (1997) mengatakan bahwa metode Roulette Wheel atau roda roulette ini merupakan metode yang paling sederhana, dan sering dikenal juga dengan nama stochastic sampling with replacement. Pada metode ini, kromosom-kromosom dipetakan dalam suatu segmen garis secara berurutan sedemikian hingga tiap-tiap segmen kromosom memiliki ukuran yang sama dengan ukuran fitness-nya. Sebuah bilangan random dibangkitkan dan kromosom yang memiliki segmen dalam kawasan bilangan random tersebut terseleksi.
Crossover
Operator crossover yang digunakan adalah penyilangan satu titik, posisi penyilangan k (k = 1, 2, …, N-1) dengan N adalah panjang kromosom (Gen & Cheng 1997). Kromosom yang terpilih menjadi parent adalah kromosom yang memiliki nilai acak kurang dari probabilitas crossover (Pc). Bit-bit ditukar antar kromosom pada titik tersebut untuk menghasilkan anak (offspring).
(3.4) (3.1)
(3.2)
14
Mutasi
Proses mutasi akan mengganti bit 1 dengan 0, atau mengganti bit 0 dengan 1. Pada proses mutasi ada parameter yang sangat penting yaitu probabilitas mutasi (Pm). Peluang mutasi menunjukkan persentasi jumlah total bit pada populasi yang akan mengalami mutasi. Untuk melakukan mutasi, terlebih dahulu kita harus menghitung jumlah total bit pada populasi tersebut. Kemudian bangkitkan bilangan random yang menentukan posisi yang bit dimutasi (Gen & Cheng 1997). Bit yang termutasi adalah bit yang mempunyai nilai random kurang dari nilai peluang mutasi.
Lingkungan Implementasi
Sistem perangkat lunak yang digunakan adalah: a. Sistem operasi: Windows 7 Ultimate
b. CodeBlocks 13.12 (sebagai compiler modul pengekstraksi fitur, optimasi GA dan pengklasifikasi NBC)
c. Notepad ++
Spesifikasi hardware yang digunakan adalah:
a. Processor Intel (R) Core (TM) i3-3217U CPU @ 1.80GHz 1.8GHz b. Memori 4,00 GB RAM
c. Hardisk 500 GB
4
HASIL DAN PEMBAHASAN
Implementasi
Program optimasi pengekstraksi fitur spaced k-mers frekuensi dikembangkan menggunakan bahasa C++. Implementasi Algoritme Genetika, sebagai metode pengoptimasi, dimulai dengan membuat proses inisialisasi populasi yang terdiri dari 20 kromosom dengan panjang kromosom 12 bit. Parameter GA yang digunakan dalam penelitian disajikan pada Tabel 4. Menurut Angelova dan Pencheva (2011) probabilitas mutasi lebih baik berkisar pada 0,002 sampai 0,1. Nilai probabilitas mutasi tidak boleh terlalu besar agar generasi baru yang didapatkan tidak mengalami perubahan yang signifikan.
Tabel 4 Parameter GA yang digunakan dalam penelitian
Paramater Nilai
Probabilitas Crossover (Pc) 0,650 0,700 0,750 Probabilitas Mutasi(Pm) 0,050 0,075 0,100
Populasi 20 kromosom
Jumlah Bit setiap kromosom 12 bit
15 Tahapan berikutnya adalah evaluasi fitness dengan cara mengekstraksi fitur fragmen-fragmen data latih dan data uji (Lampiran 2 dan 4). Fitur data latih dan data uji diklasifikasikan menggunakan metode NBC (Lampiran 3). Nilai akurasi merupakan keluaran dari proses pengklasifikasian. Nilai akurasi tersebut dijadikan nilai fitness untuk kromosom-kromosom yang telah diinisialisasi.
Seleksi kromosom menggunakan metode roulette wheel, nilai fitness dari setiap kromosom dijumlahkan. Total fitness tersebut digunakan untuk mendapatkan area pemilihan kromosom yang menempati posisi di generasi selanjutnya. Proses selanjutnya adalah kawin silang atau crossover, kromosom-kromosom yang mempunyai nilai acak kurang dari probabilitas crossover (Pc), merupakan kandidat kromosom-kromosom yang dikawin-silangkan. Metode crossover yang dipakai adalah one-cut-point, yaitu membangkitkan satu nilai acak yang dipakai sebagai titik potong dari kromosom yang dikawin-silangkan (Lampiran 5). Metode mutasi yang dipakai dalam penelitian ini adalah menukarkan bit 0 dengan 1 atau sebaliknya apabila bit tersebut memiliki nilai acak kurang dari probabilitas mutasi (Pm). Pseudocode algoritme genetika disajikan dalam Lampiran 1.
Optimasi menggunakan GA
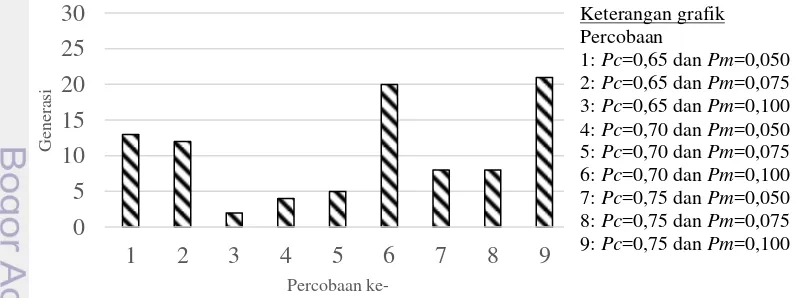
Ada 9 percobaan yang telah dilakukan dalam penelitian ini. Setiap percobaan menggunakan parameter yang berbeda (Gambar 16). 9 percobaan ini konvergen pada satu titik optimum yang sama (Lampiran 7). Namun, percobaan ke 3 lebih cepat menemukan titik optimum, karena berhasil menemukan titik optimum pada generasi ke 2 atau saat ruang pencarian sebesar 62,5%.
Titik optimum yang ditemukan oleh 9 percobaan tersebut adalah kromosom 111111110001 dengan fitness 85,42. Kromosom 111111110001 berarti [111 1111 10001] menghasilkan 366 fitur.
Gambar 16 Percobaan GA menggunakan 9 parameter yang berbeda
Evaluasi
Evaluasi dilakukan terhadap hasil percobaan yang telah dilakukan. Evaluasi yang dilakukan, yaitu confusion matrix, menunjukkan perbandingan
16
fitness dan waktu eksekusi program antara kromosom yang tidak melibatkan don’t care (k-mers frekuensi) dengan kromosom yang melibatkan don’t care (spaced k-mers frekuensi).
Confusion Matrix
Evaluasi menggunakan confusion matrix. Tabel 5 menampilkan hasil pengklasifikasian fragmen data uji menggunakan kromosom 111111110001.
Tabel 5 Confusion Matix dari kromosom 111111110001 Prediksi
Aktual
Agrobacterium Bacillus Staphylococcus
Agrobacterium 1462 38 0
Bacillus 40 1116 344
Staphylococcus 0 234 1266
Pada Tabel 5 dapat dilihat bahwa fragmen data uji dari genus Bacillus sebanyak 344 fragmen data uji diklasifikasikan salah ke genus Staphylococcus. Hal ini disebabkan genus Bacillus dan genus Staphylococcus berasal dari order yang sama yaitu order Bacillales.
Perbandingan k-mers dengan spaced k-mers
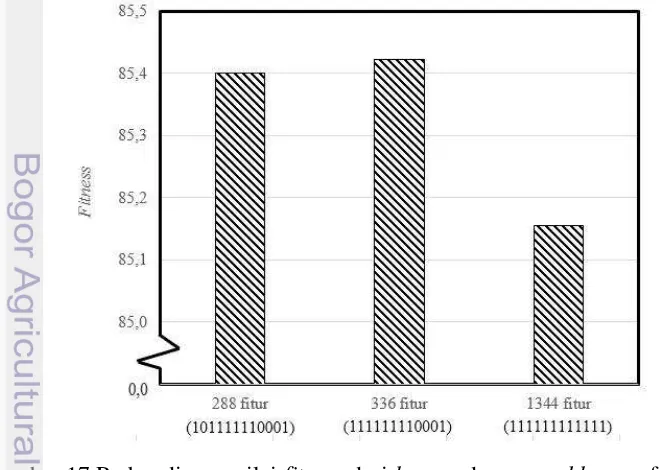
Kromosom 111111111111 mewakili pengekstraksi fitur k-mers frekuensi. Kromosom 111111110001 dan 101111110001 mewakili pengekstraksi fitur spaced k-mers. Kromosom 111111111111 menghasilkan 1344 fitur, sedangkan kromosom 101111110001 menghasilkan 288 fitur.
Gambar 17 menunjukkan perbandingan fitness dari 3 kromosom. Kromosom 111111110001 menghasilkan fitness yang lebih tinggi dibandingkan dengan kromosom 111111111111. Penggunaan don’t care (0) dapat meningkatkan akurasi pengklasifikasian (ditunjukkan dengan nilai fitness).
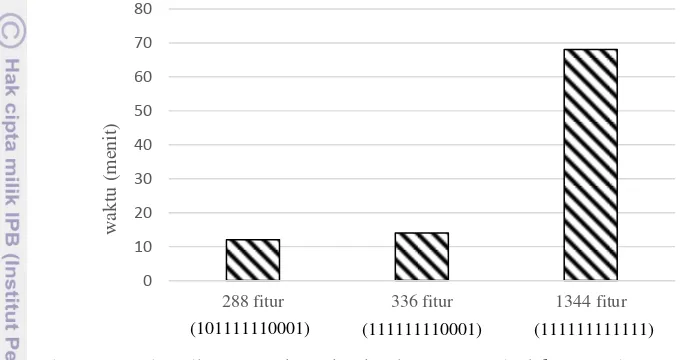
17 Selain itu, dilakukan perbandingan waktu eksekusi program dari 3 kromosom. Kromosom 101111110001 memerlukan waktu eksekusi yang lebih cepat yaitu 12 menit dibandingkan dengan kromosom 111111111111 yang memerlukan waktu eksekusi 68 menit. Oleh karena itu, don’t care (0) juga berperan untuk mereduksi jumlah fitur sehingga mempercepat waktu eksekusi program. Gambar 18 menunjukkan bahwa semakin sedikit jumlah fitur, maka waktu eksekusi program yang diperlukan semakin sedikit.
Gambar 18 Perbandingan waktu eksekusi program dari k-mers dan spaced k-mers frekuensi
Fitur Potensial
Telah dilakukan pengklasifikasian yang menggunakan fitur potensial. Fitur potensial adalah fitur yang memiliki nilai mean yang berbeda signifikan antara setiap genus. Fitur potensial dapat dilihat pada Gambar 14 dan Gambar 15. Klasifikasi menggunakan fitur 22 sampai 27 menghasilkan akurasi sebesar 78,83%. Sedangkan saat menggunakan fitur 38 sampai 43 menghasilkan akurasi sebesar 79,67%. Pada bagian ini dapat dilihat bahwa dengan menggunakan 6 fitur menghasilkan akurasi yang tidak jauh berbeda dengan yang melibatkan 336 fitur.
Hasil ini menunjukkan bahwa ada fitur yang berpengaruh dalam pengklasifikasian dan juga ada fitur yang tidak terlalu berpengaruh saat pengklasifikasian dilakukan. Sehingga dapat disimpulkan tidak semua fitur dapat mengklasifikasikan fragmen metagenome dengan akurat.
Keterbatasan Penelitian
Keterbatasan dalam penelitian ini adalah waktu eksekusi program yang memerlukan waktu lama. Dalam penelitian ini untuk 50 generasi memerlukan waktu 10 hari. Oleh karena itu, dibutuhkan eksekusi program secara paralel sehingga dapat mempercepat waktu eksekusi. Penggunaan waktu terbanyak dalam proses penelitian ini adalah proses ekstraksi fitur. Program yang dikembangkan telah menggunakan penyeleksi kromosom, dengan tujuan menghindari pengulangan pengekstraksian fitur terhadap kromosom yang telah dibangkitkan pada generasi sebelumnya. Namun, prosedur penyeleksi yang ada hanya bisa
0
288 fitur 336 fitur 1344 fitur
18
membandingkan kromosom pada generasi n dengan kromosom pada generasi n-1. Oleh karena itu, perlu dilakukan penyeleksian terhadap kromosom generasi n dengan cara membandingkan kromosom generasi ke n dengan seluruh kromosom yang telah dibangkitkan pada generasi-generasi sebelumnya. Apabila kromosom generasi ke n telah dibangkitkan pada generasi sebelumnya, maka ekstraksi fitur dan pengklasifikasian tidak perlu dilakukan lagi.
5
SIMPULAN DAN SARAN
Simpulan
Algoritme Genetika berhasil menemukan titik optimum model spaced k-mers terhadap posisi match (1) dan don’t care (0)pada spaced seed. Spaced seed optimum tersebut adalah kromosom 111111110001 dengan fitness 85,42 untuk fragmen dengan panjang 500 bp. Hasil penelitian ini menunjukkan penggunaan don’t care dapat meningkatkan akurasi pengklasifikasian. Selain itu, don’t care berperan mempercepat waktu eksekusi program.
Telah dilakukan pengklasifikasian terhadap fragmen-fragmen metagenome menggunakan 12 fitur potensial yang dibagi dalam 2 kelompok. Fitur-fitur potensial diambil dari kromosom 111111110001. Kelompok pertama melibatkan fitur ke- 22 sampai 27. Fitur ke- 38 sampai 43 merupakan kelompok kedua. Pengklasifikasian menggunakan fitur kelompok pertama menghasilkan akurasi sebesar 78,83%, sedangkan kelompok kedua menghasilkan akurasi sebesar 79,67%. Pada bagian ini dapat dilihat bahwa dengan menggunakan 6 fitur menghasilkan akurasi yang tidak jauh berbeda dengan yang melibatkan 336 fitur. Sehingga dapat disimpulkan tidak semua fitur dapat mengklasifikasikan fragmen metagenome dengan akurat.
Saran
Beberapa hal yang dapat dikembangkan lebih lanjut dari penelitian ini, yaitu pemrograman paralel untuk eksekusi program dan perlu dilakukan penyeleksian terhadap kromosom generasi n dengan cara membandingkan kromosom generasi ke n dengan seluruh kromosom yang telah dibangkitkan pada generasi-generasi sebelumnya. Apabila kromosom ke n telah dibangkitkan pada generasi sebelumnya, maka ekstraksi fitur dan pengklasifikasian tidak perlu dilakukan lagi.
Pembangkitan nilai random pada saat proses mutasi cukup dilakukan terhadap bit ke- 2, 5, 6, 9, 10, dan 11. Bit ke- 1, 3, 4, 7, 8, dan 12 tidak perlu dilakukan karena bit-bit tersebut harus tetap berisi biner 1. Hal ini tentunya akan mempercepat proses perbandingan probabilitas mutasi dengan nilai random untuk setiap bit.
Ucapan Terima Kasih
19
DAFTAR PUSTAKA
Altschul S, Gish W, Miller W, Myers E, Lipman D. 1990. Basic local alignment search tool. Journal of Molecular Biology. 215(3):403-410. doi:10.1016/S0022-2836(05)80360-2
Angelova M, Pencheva T. 2011. Tuning Genetic Algorithm parameter to improve converge time. International Journal of Chemical Engineering. doi:10.1155/2011/646917.
Bouchot JL, Trimble WL, Ditzler G, Lan Y, Essinger S, Rosen G. 2013. Advances in machine learning for processing and comparison of metagenomic data [internet]. [diunduh 15 September 2014]. Tersedia pada: http://www.sessinger.com/Publications/metagenomics_advances_ML_prepr int.pdf
Choi JH, Cho HG. 2002. Analysis of common k-mers for whole genome sequence using SSB-tree. Genome Information. 13: 30-41
Gen M, Cheng R. 1997. Genetic Algorithms and Engineering Design. New York (US): John Wiley & Sons, Inc.
Goldberg DE. 1989. Genetic Algorithms in search, optimization & machine learning. Indiana (US): Addison Wesley Longman.
Han J, Kamber M. 2001. Data Mining: Concepts and Techniques. San Francisco (US): Morgan Kaufmann Publishers.
Handelsman J. 2007. The new science of metagenomics: Revealing the secrets of our microbial planet. Washington (US): The National Academics Press. Huson DH, Auch AF, Schuster SC. 2007. MEGAN analysis of metagenomic data.
Genome Research. 17(3):337-386. doi: 10.1101/gr.5969107.
Kusuma WA. 2012. Combined approaches for improving the performance of denovo DNA sequence assembly and metagenomic classification of short fragments from next generation sequencer [disertasi]. Tokyo (JP): Tokyo Institute of Technology.
Ma B, Tromp J, Li M. 2002. PatternHunter: faster and more sensitive homology search. Bioinformatics. 18(3):440-445.
McHardy AC, Martin HG, Tsirigos A, Hugenholtz P, dan Rigoutsos I. 2007. Accurate Phylogenetic Classification of Variable-Length DNA Fragments. Nature Methods. 4(1):63-72. doi: 10.1016/j.mib.2007.08.004.
Meyerdierks A, Glockner FO. 2010. Metagenome Analysis. Advances in Marine Genomics 1. doi:10.1007/978-90-481-8639-6_2.
Overbeek MV, Kusuma WA, Buono A. 2013. Clustering metagenome fragments using growing self organizing map. Advanced Computer Science and Information Systems (ICACSIS): 285-289. doi: 10.1109/ICACSIS.2013. 6761590.
Richter DC, Ott F, Auch AF, Schmid R, Huson DH. 2008. MetaSim: a sequencing simulator for genomics and metagenomics. PloS one. 3(10):1-12. doi:10.1371/journal.pone.0003373.
20
Rosen G, Garbarine E, Caseiro D, Polikar R, Sokhansanj. 2008. Metagenome fragment classification using n-mer frequency profiles. Advances in Bioinformatics. doi:10.1155/ 2008/205969.
21 Lampiran 1 Pseudocode program optimasi pengekstraksi fitur spacedk-mers
frekuensi pada penglasifikasian fragmen metagenome
num_generation = 50 num_kromosom = 20 num_gen = 12
gnrt = 0 //generation /* A. Inisialisasi
for i=0 to num_kromosom {
while (gnrt < num_generation) {
for i=0 to num_kromosom {
str_kromosom str_kromosom + inttostr (kromosom [i][j]) }
K3 0
pola3 [i] str_kromosom str_kromosom ””
}
for i=0 to num_kromosom {
22
K4 0
pola4 [i] str_kromosom str_kromosom ””
}
for i=0 to num_kromosom {
str_kromosom str_kromosom + inttostr (kromosom [i][j]) }
/* 2.1 Pengecekan kromosom generation n yang sama dengan kromosom /* generation n-1
for i=0 to num_kromosom {
str_kromosom2 [generation] [i] = pola3 [i] + pola4 [i] + pola5 [i] for j=0 to num_kromosom
{
if (str_kromosom2 [gnrt][i] == str_kromosom [gnrt-1][j]) {
str_kromosom [gnrt][i] pola3 [i] + pola4 [i] + pola5 [i] fitness_kromosom [i] simp_akurasi [gnrt-1][j]
simp_akurasi [gnrt][i] fitness_kromosom [i] idx_kromosom [gnrt][i] true
} } }
for i=0 to num_kromosom {
if (idx_kromosom [gnrt][i] != true) {
ekstraksi (pola3[i], pola4[i], pola5[i], i, data, data_uji)
23 simp_akurasi [gnrt][i] fitness_kromosom[i]
} }
/* C. Seleksi menggunakan Roda Roullete for i=0 to num_kromosom
{
total_fitness total_fitness + fitness_kromosom [i] }
for i=0 to num_kromosom {
rasio_fitness [i] fitness_kromosom [i] / total_fitness }
area_pemilihan [0] rasio_fitness [0] for i = 1 to num_kromosom
{
area_pemilihan [i] area_pemilihan [i-1] + rasio_fitness [i] }
area_pemilihan [-1] = 0 for i=0 to num_kromosom {
for j=0 to num_kromosom {
if ((acak_seleksi[i] >= area_pemilihan [j-1]) and (acak_seleksi[i] <= area_pemilihan [j])) {
kromosom_tamp [idx_k_gen][j] = kromosom [idx_terpilih][j] }
24
for i=0 to num_kromosom {
for j=0 to num_kromosom {
kromosom [i][j] kromosom_tamp [i][j] }
}
/* D. Crossover one cut point for i=0 to num_kromosom {
acak_crossover [i] = rand() % 1001/1000.000 if (acak_crossover [i] < pc)
{
tukar (kromosom, index_parent [0], index_parent[1], cut_point) }
acak_mutasi [i][j] = rand() % 1001/1000.000 if (acak_mutasi [i][j] < pm)
25 Lampiran 2 Pseudocode prosedur Ekstraksi Fitur
Ekstraksi (string pola3, pola4, pola5, int a, int data, int data_uji ) {
for i=0 to length(pola3) {
if (pola3 [i] == „1‟)
{
++K3 }
}
for i=0 to length(pola4) {
if (pola4 [i] == „1‟)
{
++K4 }
}
for i=0 to length(pola5) {
if (pola5 [i] == „1‟)
{
++K5 }
}
for i=0 to (1 << (2*K3)) {
arrData3[i] fitur (i, K3) }
for i=0 to (1 << (2*K4)) {
arrData4[i] fitur (i, K4) }
for i=0 to (1 << (2*K5)) {
arrData5[i] fitur (i, K5) }
file_input (“data/data_latih_1000fragmen.fna”)
26
if (file_input.is_open()) {
while (getline (file_input, data_x)) {
while (n < array_size (arrData3)) {
for i=0 to length(data_baru) {
str2_3 data_baru.substr(i,length(pola3)) for j=0 to length(pola3)
{
if (str3_3 == isi_data_3) {
while (o < array_size (arrData4)) {
for i=0 to length(data_baru) {
str2_4 data_baru.substr(i,length(pola4)) for j=0 to length(pola4)
{
if (str3_4 == isi_data_4) {
27
while (p < array_size (arrData5)) {
for i=0 to length(data_baru) {
str2_5 data_baru.substr(i,length(pola5)) for j=0 to length(pola5)
{
if (str3_5 == isi_data_5) {
while (getline (file_uji, data_uji_x)) {
while (n_uji < array_size (arrData3)) {
for i=0 to length(data_baru_uji) {
str2_3_uji data_baru_uji.substr(i,length(pola3)) for j=0 to length(pola3)
28
if (pola3[j] == „1‟)
{
str3_3_uji str3_3_uji + str2_3_uji [j] }
}
if (str3_3_uji == isi_data_3_uji) {
++x_3_uji }
str3_3_uji “” }
data [d_uji][n_uji] x_3_uji n_uji++
}
while (o < array_size (arrData4)) {
for i=0 to length(data_baru_uji) {
str2_4_uji data_baru_uji.substr(i,length(pola4)) for j=0 to length(pola4)
{
if (pola4[j] == „1‟)
{
str3_4_uji str3_4_uji + str2_4_uji [j] }
}
if (str3_4_uji == isi_data_4_uji) {
++x_4_uji }
str3_4_uji “” }
data [d_uji][n_uji+o_uji] x_4_uji o_uji++
}
while (p_uji < array_size (arrData5)) {
for i=0 to length(data_baru_uji) {
str2_5_uji data_baru_uji.substr(i,length(pola5)) for j=0 to length(pola5)
{
if (pola5[j] == „1‟)
{
str3_5_uji str3_5_uji + str2_5_uji [j] }
29
if (str3_5_uji == isi_data_5_uji) {
++x_5_uji }
str3_5_uji “” }
data [d_uji][n_uji+o_uji+p_uji] x_5_uji p_uji++
30
Lampiran 3 Pseudocode prosedur Naïve Bayesian Classifier (NBC)
NBC (int a, b, tot_fitur, float fitness_kromosom[], int data[][], data_uji[][] ) {
x 10000 y tot_fitur x_uji 4500 y_uji tot_fitur
/* menjumlahkan setiap fitur untuk setiap kelasnya for i = 0 to tot_fitur
{ jumlahA [i] 0 for j = 0 to 3000
{ jumlahA [i] jumlahA [i] + data [j][i] }
}
for i = 0 to tot_fitur { jumlahB [i] 0
for j = 3000 to 7000
{ jumlahB [i] jumlahB [i] + data [j][i] }
}
for i = 0 to tot_fitur { jumlahC [i] 0
for j = 7000 to 10000
{ jumlahC [i] jumlahC [i] + data [j][i] }
31
/* menghitung mean setiap fitur untuk setiap kelas for i=0 to tot_fitur
{
meanA [i] jumlahA [i]/3000 }
for i=0 to tot_fitur {
meanB [i] jumlahB [i]/4000 }
for i=0 to tot_fitur {
meanC [i] jumlahC [i]/3000 }
/* menghitung standar deviasi setiap fitur untuk setiap kelas for i=0 to tot_fitur
{
tambah_pangkatA [i] tambah_pangkatA [i] + pangkatA }
}
for i=0 to tot_fitur {
tambah_pangkatB [i] tambah_pangkatB [i] + pangkatB }
}
for i=0 to tot_fitur {
tambah_pangkatC [i] tambah_pangkatC [i] + pangkatC }
32
for i=0 to tot_fitur {
stdA [i] tambah_pangkatA [i]/(3000-1) std_devA [i] pow (stdA[i]), 1.0/2) stdB [i] tambah_pangkatB [i]/(4000-1) std_devB [i] pow (stdB[i]), 1.0/2) stdC [i] tambah_pangkatC [i]/(3000-1) std_devC [i] pow (stdC[i]), 1.0/2) }
/* Pengujian menggunakan NBC sqrt_2kali pow(2*3.14,1.0/2) for i=0 to tot_fitur
{
for j=0 to x_uji {
x_krg_meanA [i] data_uji [j][i] – meanA [i];
like_pangkatA [j][i] x_krg_meanA [i] / std_devA [i] sqrtA [j][i] like_pangkatA [j][i]
pangkatnyaA [j][i] sqrtA [j][j] * (-0.5) penyebutA [i] sqrt_2kali * std_devA [i] exponenA [j][i] exp (pangkatnyaA[j][i]) satu_per_penyebutA [i] 1/penyebutA[i]
likelihoodA [j][i] satu_per_penyebutA [i] * exponenA [j][i] x_krg_meanB [i] data_uji [j][i] – meanB [i];
like_pangkatB [j][i] x_krg_meanB [i] / std_devB [i] sqrtB [j][i] like_pangkatB [j][i]
pangkatnyaB [j][i] sqrtB [j][j] * (-0.5) penyebutB [i] sqrt_2kali * std_devB [i] exponenB [j][i] exp (pangkatnyaB[j][i]) satu_per_penyebutB [i] 1/penyebutB[i]
likelihoodB [j][i] satu_per_penyebutB [i] * exponenB [j][i] x_krg_meanC [i] data_uji [j][i] – meanC [i];
like_pangkatC [j][i] x_krg_meanC [i] / std_devC [i] sqrtC [j][i] like_pangkatC [j][i]
pangkatnyaC [j][i] sqrtC [j][j] * (-0.5) penyebutC [i] sqrt_2kali * std_devC [i] exponenC [j][i] exp (pangkatnyaC[j][i]) satu_per_penyebutC [i] 1/penyebutC[i]
likelihoodC [j][i] satu_per_penyebutC [i] * exponenC [j][i] }
33
kelas_1_1 0 // jumlah individu kelas 1 yang terklasifikasi benar ke kelas 1 kelas_1_2 0 // jumlah individu kelas 1 yang terklasifikasi salah ke kelas 2 kelas_1_3 0 // jumlah individu kelas 1 yang terklasifikasi salah ke kelas3 kelas_2_1 0 // jumlah individu kelas 2 yang terklasifikasi salah ke kelas 1 kelas_2_2 0 // jumlah individu kelas 2 yang terklasifikasi benar ke kelas 2 kelas_2_3 0 // jumlah individu kelas 2 yang terklasifikasi salah ke kelas 3 kelas_3_1 0 // jumlah individu kelas 3 yang terklasifikasi salah ke kelas 1 kelas_3_2 0 // jumlah individu kelas 3 yang terklasifikasi salah ke kelas 2 kelas_3_3 0 // jumlah individu kelas 3 yang terklasifikasi benar ke kelas 3 for i=0 to x_uji
{
kali_likelihoodA [i] 1 kali_likelihoodB [i] 1 kali_likelihoodC [i] 1 for j=0 to tot_fitur {
kali_likelihoodA [i] kali_likelihoodA [i] * likelihoodA[i][j]; kali_likelihoodB [i] kali_likelihoodB [i] * likelihoodB[i][j]; kali_likelihoodC [i] kali_likelihoodC [i] * likelihoodC[i][j]; }
posteriorA [i] kali_likelihoodA[i] * 0.3 posteriorB [i] kali_likelihoodB[i] * 0.4 posteriorC [i] kali_likelihoodC[i] * 0.3 /* menghitung akurasi atau fitness kromosom
if ((posteriorA [i] > posteriorB [i] ) and (posteriorA [i] > posteriorC [i])) { kelas [i] „1‟
}
if ((posteriorB [i] > posteriorA [i] ) and (posteriorB [i] > posteriorC [i])) { kelas [i] „2‟
}
34
for (int i = 1500; i < 3000; i++) // 1500-2999 adalah data uji Bacillus { if (kelas[i]=="1")
{ kelas_2_1++; }
if (kelas[i]=="2") { kelas_2_2++; }
if (kelas[i]=="3") { kelas_2_3++; }
}
for (int i = 3000; i < 4500; i++) // 3000-4499 adalah data uji Staphylococcus
{ if (kelas[i]=="1") { kelas_3_1++; }
if (kelas[i]=="2") { kelas_3_2++; }
if (kelas[i]=="3") { kelas_3_3++; }
}
35 Lampiran 4 Pseudocode Fungsi pembentuk fitur
fitur (int i, K) {
alel “ACGT”
36
Lampiran 5 Pseudocode prosedur tukar pada operator crossover
tukar (int kromosom [][], int parent_1, parent_2, cut_point) {
for k=cut_point+1 to num_gen {
tampung [k] kromosom [parent_1][k]
kromosom [parent_1][k] kromosom [parent_2][k] kromosom [parent_2][k] tampung [k]
37 Lampiran 6 Percobaan optimasi menggunakan Algoritme Genetika
A. Percobaan 1 (Pc = 0,650 dan Pm = 0,050)
B. Percobaan 2 (Pc = 0,650 dan Pm = 0,075)
0 5 10 15 20 25 30 35 40 45 50 55 60 65 70 75 80 85 90
1 3 5 7 9 11 13 15 17 19 21 23 25 27 29 31 33 35 37 39 41 43 45 47 49
38
C. Percobaan 3 (Pc = 0,650 dan Pm = 0,100)
D. Percobaan 4 (Pc = 0,700 dan Pm = 0,050)
39 F. Percobaan 6 (Pc = 0,700 dan Pm = 0,100)
G. Percobaan 7 (Pc = 0,750 dan Pm = 0,050)
40
42
RIWAYAT HIDUP
Penulis dilahirkan di Kupang, Nusa Tenggara Timur pada tanggal 29 Mei 1990. Penulis merupakan anak pertama dari dua bersaudara pasangan Ir Marthen Yunus, MP dan Adrifina Ndjurumbaha, SSos. Penulis mengenyam pendidikan dasar di SD Negeri Angkasa Penfui Kupang (1996-2002). Kemudian, penulis melanjutkan pendidikan menengahnya di SMP Negeri 1 Kupang (2002-2005). Pada tahun 2008, penulis menamatkan pendidikan di SMA Negeri 1 Kupang. Penulis berkesempatan melanjutkan studi di Universitas Nusa Cendana (Undana) Kupang melalui jalur penelusuran minat dan kemampuan (PMDK) di Jurusan Ilmu Komputer, Fakultas Sains dan Teknik. Selama kuliah pada jenjang strata 1, penulis mendapatkan beasiswa peningkatan prestasi akademik (PPA) dari DIKTI. Pada tahun 2013 penulis melanjutkan pendidikan ke jenjang strata 2 di Program Studi Ilmu Komputer, Fakultas Matematika dan Ilmu Pengetahuan Alam Institut Pertanian Bogor (IPB). Perkuliahan S2 ini disponsori oleh DIKTI melalui beasiswa BPP-DN.