PENJAJARAN LOKAL SEKUEN DNA MENGGUNAKAN
ALGORITME SMITH-WATERMAN
FARIZ ASHAR HIMAWAN
DEPARTEMEN ILMU KOMPUTER
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM
INSTITUT PERTANIAN BOGOR
PERNYATAAN MENGENAI SKRIPSI DAN
SUMBER INFORMASI SERTA PELIMPAHAN HAK CIPTA
Dengan ini saya menyatakan bahwa skripsi berjudul Penjajaran Lokal Sekuen DNA menggunakan Algoritme Smith-Waterman adalah benar karya saya dengan arahan dari komisi pembimbing dan belum diajukan dalam bentuk apa pun kepada perguruan tinggi mana pun. Sumber informasi yang berasal atau dikutip dari karya yang diterbitkan maupun tidak diterbitkan dari penulis lain telah disebutkan dalam teks dan dicantumkan dalam Daftar Pustaka di bagian akhir skripsi ini.Dengan ini saya melimpahkan hak cipta dari karya tulis saya kepada Institut Pertanian Bogor.
Bogor, 3 September 2013
Fariz Ashar Himawan
ABSTRAK
FARIZ ASHAR HIMAWAN. Penjajaran Lokal Sekuen DNA Menggunakan Algoritme Smith-Waterman. Dibimbing oleh WISNU ANANTA KUSUMA.
Penjajaran lokal sekuen DNA bertujuan untuk mencari kemiripan dua buah sekuen DNA. Penelitian ini mengimplementasikan penjajaran lokal menggunakan algoritme Smith-Waterman. Pada data pertama sekuen DNA Ancylostoma duodenale (NC_003415.1) dan Necator americanus (NC_003416.2), hasil penjajaran menunjukkan similaritas 84%, dengan gap 3%. Data kedua sekuen DNA Human papillomavirus type 134 (NC_014956.1) dan Human papillomavirus type 132 (NC_014955.1), hasil penjajaran menunjukkan similaritas 62.1%, dengan gap 15.9%. Data ketiga sekuen DNA Chaetoceros lorenzianus DNA virus (NC_015211.1) dan Chaetoceros tenuissimus DNA virus
(NC_014748.1), hasil penjajaran menunjukkan similaritas 53.2% dengan gap 26.2%. Data terakhir sekuen DNA Thermoproteus tenax spherical virus 1
(NC_006556.1) dan Vesicular stomatitis indiana virus (NC_001560.1), hasil penjajaran menunjukkan similaritas 40.8%, dengan gap 47.6%. Hal ini menunjukkan bahwa penjajaran menggunakan algoritme Smith-Waterman menghasilkan beragam nilai kemiripan yang tergantung dari panjang sekuen, dan bentuk sekuennya. Smith-Waterman memiliki kompleksitas O(mn).
Kata kunci: penjajaran global, penjajaran sekuen, Smith – Waterman
ABSTRACT
FARIZ ASHAR HIMAWAN. Local Alignment of DNA Sequence Using Smith-Waterman Algorithm. Supervised by WISNU ANANTA KUSUMA.
The Local Alignment of DNA sequence objective is to determine similarity between two DNA sequences. This research implements the local alignment using Smith–Waterman algorithm. The first data of Sequences from Ancylostoma duodenale (NC_003415.1) and Necator americanus (NC_003416.2), the result shows that the similarity is 84%, with 3% gaps, Second data of DNA sequence from Human papillomavirus type 134 (NC_014956.1) and Human papillomavirus type 132 (NC_014955.1), the result shows that the similarity is 62.1%, with 15.9% gaps. Third data of DNA sequence from Chaetoceros lorenzianus DNA virus (NC_015211.1) and Chaetoceros tenuissimus DNA virus (NC_014748.1), the result shows that the similarity is 53.2%, with 26.2% gaps. Last data DNA sequence from Thermoproteus tenax spherical virus 1 (NC_006556.1) and Vesicular stomatitis indiana virus (NC_001560.1), the result shows that similarity is 40.8%, with 47.6% gaps. These result shows that alignment using Smith– Waterman can result in various similarity values, depending on the length and form of sequences. The complexity of Smith-Waterman is O(mn).
PENJAJARAN LOKAL SEKUEN DNA MENGGUNAKAN
ALGORITME SMITH-WATERMAN
Skripsi
sebagai salah satu syarat untuk memperoleh gelar Sarjana Komputer
pada
Departemen Ilmu Komputer
DEPARTEMEN ILMU KOMPUTER
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM
INSTITUT PERTANIAN BOGOR
BOGOR
2013
Judul Skripsi : Penjajaran Lokal Sekuen DNA Menggunakan Algoritme Smith-Waterman
Nama : Fariz Ashar Himawan NIM : G64104007
Disetujui oleh
Dr. Wisnu Ananta Kusuma, ST MT Pembimbing
Diketahui oleh
Dr. Ir Agus Buono, M Si M Kom Ketua Departemen
Tanggal Lulus:
Penguji :
PRAKATA
Puji dan syukur penulis panjatkan ke hadirat Allah subhanahu wata’ala, yang telah memberikan nikmat yang begitu banyak, sehingga penulis dapat menyelesaikan penelitian dan tulisan ini. Shalawat dan salam penulis sampaikan kepada Nabi Muhammad shallallahu ‘alaihi wasallam, keluarganya, sahabatnya, serta umatnya hingga akhir zaman. Tulisan ini merupakan hasil penelitian yang penulis lakukan sejak Agustus 2012 hingga Agustus 2013. Tulisan ini mengambil topik bioinformatika, dan bertujuan menerapkan algoritme Smith - Waterman pada penjajaran lokal sekuen DNA.
Tak lupa penulis mengucapkan terima kasih kepada seluruh pihak yang telah berperan dalam penelitian ini, yaitu:
1 Ayahanda Subakir Amin, Ibunda Siti Rodiyah, serta adik Resa Rizki Setiawan atas kasih sayang, doa, semangat, dan dorongan kepada penulis agar dapat segera menyelesaikan penelitian ini.
2 Bapak Dr. Wisnu Ananta Kusuma, ST, MT selaku dosen pembimbing, yang telah memberikan banyak ide, masukan, dan dukungan kepada penulis.
3 Bapak Irman Hermadi, S.Kom, MS dan Bapak Toto Haryanto, S.Kom, M.Si, yang telah bersedia menjadi penguji.
4 Bapak Beni dan Bapak Jamsy selaku atasan yang telah memberikan izin, mendukung dan memotivasi dalam menyelesaikan pendidikan di alih jenis Ilmu Komputer IPB.
5 Para sahabat : Yusrizal Ihya, Rizkina M. Syam, Dedi Kiswanto, Asep Haryono, Septiandi Wibowo, Alrasyid Trio, Wahyu Dias, Mas Syam, Rudi Rusdi, Silvia Rahmi, Putri Ayu Pramesti, Sri Rahayu Natasia, Yudi Haryanto serta seluruh rekan - rekan Ilkom Alih Jenis angkatan 5, May the odd return favor to all of us.
6 Rekan satu bimbingan : Agung Widyo Utomo, Bernita Sinurat, Alharis Tamsin, Fitria, dan Galih yang saling berbagi ide dan saling memotivasi selama pengerjaan skripsi.
7 Kakak sepupu Endah Nurhayati beserta suami, Sasmoyo dan keponakan Lintang Ayu Prameswari yang selalu memberikan doa dan dukungannya. 8 Kakak ipar sepupu Sujadmiko beserta istri Hesti dan keponakan yang telah
memberikan doa dan dukungannya.
9 Rekan - rekan Radar-Bogor Grup atas perhatian dan motivasinya. 10 Pihak-pihak lain yang tidak dapat penulis sebutkan satu persatu.
Penulis berharap penelitian dan tulisan ini dapat memberikan manfaat untuk kemajuan masyarakat Indonesia.
Bogor, 3 September 2013
DAFTAR ISI
DAFTAR TABEL vii
DAFTAR GAMBAR vii
DAFTAR LAMPIRAN viii
PENDAHULUAN 1
Latar Belakang 1
Tujuan Penelitian 2
Manfaat Penelitian 2
Ruang Lingkup Penelitian 3
METODE 3
Penyiapan Data 4
Algoritme Penjajaran Lokal Smith-Waterman 4
Analisis dan Evaluasi 7
Lingkungan Pengembangan 8
HASIL DAN PEMBAHASAN 8
Implementasi Algoritme Penjajaran Lokal Smith-Waterman 8
Analisis dan Evaluasi 16
SIMPULAN DAN SARAN 20
Simpulan 20
Saran 20
DAFTAR PUSTAKA 21
DAFTAR TABEL
Tabel 1 Hasil pengujian Ancylostoma duodenale dan Necator americanus 10 Tabel 2 Hasil pengujian Human papillomavirus type 134 dan Human
papillomavirus type 132 12
Tabel 3 Hasil pengujian Thermoproteus tenax spherical virus 1 dan Vesicular
stomatitis indiana virus 13
Tabel 4 Hasil pengujian Chaetoceros lorenzianus dan Chaetoceros tenuissimus 15 Tabel 5 Hasil perbandingan penjajaran Ancylostoma duodenale dan Necator
americanus 16
Tabel 6 Hasil perbandingan penjajaran Human papillomavirus type 134 dan
Human papillomavirus type 132 17
Tabel 7 Hasil Perbandingan Penjajaran Chaetoceros lorenzianus DNA virus dan
Chaetoceros tenuissimus DNA virus 17
Tabel 8 Hasil perbandingan penjajaran Thermoproteus tenax spherical virus 1
dan Vesicular stomatitis indiana virus 18
DAFTAR GAMBAR
Gambar 1 Contoh penjajaran global dan penjajaran lokal 1
Gambar 2 Diagram alur metode penelitian 3
Gambar 3 Contoh data berformat FASTA 4
Gambar 4 Contoh ilustrasi proses Inisialiasi 5 Gambar 5 Contoh ilustrasi proses pengisian matriks 6
Gambar 6 Contoh traceback 7
Gambar 7 Ilustrasi sekuens yang match dan mismatch 8 Gambar 8 Grafik similaritas Ancylostoma duodenale dan Necator americanus
dengan parameter match 5 mismatch -4 9 Gambar 9 Grafik similaritas Ancylostoma duodenale dan Necator americanus
dengan parameter match 9 mismatch -1 10 Gambar 10 Grafik similaritas Human papillomavirus type 134 dan Human
papillomavirus type 132 dengan parameter match 5 mismatch -4 11 Gambar 11 Grafik similaritas Human papillomavirus type 134 dan Human
papillomavirus type 132 dengan parameter match 9 mismatch -1. 11 Gambar 12 Grafik similaritas Thermoproteus tenax spherical virus 1 dan
Vesicular stomatitis indiana virus dengan parameter match 5
mismatch -4 12
Gambar 13 Grafik similaritas Thermoproteus tenax spherical virus 1 dan
Vesicular stomatitis indiana virus dengan parameter match 9
mismatch -1 13
Gambar 14 Grafik similaritas Chaetoceros lorenzianus DNA virus dan
Chaetoceros tenuissimus DNA virus dengan parameter match 5
mismatch -4 14
Gambar 15 Grafik similaritas Chaetoceros lorenzianusDNA virus dan
Chaetoceros tenuissimus DNA virus dengan parameter match 9
mismatch -1 14
DAFTAR LAMPIRAN
1 Pseudocode algoritme Smith-Waterman 22
1
PENDAHULUAN
Latar Belakang
Deoxyribo nucleic acid (DNA) mengandung informasi genetik dari makhluk hidup bersel satu hingga bersel banyak. Informasi ini yang menentukan karakteristik tertentu dari makhluk hidup, mulai dari karakteristik fisik hingga karakteristik perilaku. DNA terbentuk dari rantai ganda molekul sederhana (nukleotida) yang diikat bersama-sama dalam struktur double helix, tersusun atas empat basa nitrogen yaitu adenine, cytosine, guanine, dan thymine yang dinotasikan sebagai A, C, G, dan T (Annibal 2003).
Ada banyak cara untuk menganalisis struktur DNA, di antaranya penjajaran sekuen (sequence alignment), yaitu suatu cara untuk mencari sebanyak mungkin kecocokan dengan menjajarkan sepasang sekuen atau lebih. Dengan penjajaran ini didapatkan subsekuen yang identik untuk kemudian dilakukan analisis dan disimpulkan kemiripannya (Annibal 2003). Penjajaran sekuen DNA penting dilakukan sebagai dasar dari sequence analysis antara lain rekonstruksi sekuen DNA dari fragmen, pemetaan data fisik dan genetika (Agrawal 2009).
Selain itu basis data DNA yang terus berkembang membutuhkan suatu alat penganalisa yang dapat mengimbangi perkembangan data yang ada, dan salah satu alat yang digunakan untuk meneliti basis data genom yang ada di GenBank DNA adalah melalui penjajaran sekuen (Annibal 2003).
Penjajaran sekuen DNA dapat dilakukan dengan dua metode, yaitu penjajaran secara utuh (global) dan penjajaran sebagian (lokal). Penjajaran global dilakukan dengan melibatkan keseluruhan sekuen dan mencari nilai kemiripan minimal sepanjang sekuen terkecil. Sedangkan penjajaran lokal dilakukan dengan melibatkan sebagian sekuen yang ditentukan dengan nilai densitas similaritas maksimum (Chan 2004). Perbedaan penjajaran lokal dan global dapat dilihat pada Gambar 1.
Gambar 1 Contoh penjajaran global dan penjajaran lokal
Dalam penelitian ini difokuskan kepada penjajaran lokal dengan algoritme Smith-Waterman yang ditemukan oleh TF Smith dan MS Waterman pada tahun 1981. Algoritme ini mencoba menemukan sebanyak mungkin kesamaan dari sepasang sekuen, dengan cara memberikan nilai negatif pada base pair yang tidak sama (mismatch), dan nilai positif pada base pair yang sama (match). Sehingga
Penjajaran Global
A T T G C T C G T T G G A | . . . | | | | . | A A A A C - C G T A - - A
Penjajaran Lokal
- - - - C T C G T - - - - | | | |
2
nantinya akan didapatkan nilai positif maksimum sebagai akhir dari penjajaran, dan nilai minimum sebagai awal penjajaran (Annibal 2003).
Penelitian mengenai penjajaran sekuen lebih berkembang kepada penjajaran global di antaranya penelitian yang dilakukan oleh Pantua (2011) dan Utomo (2013). Data yang digunakan untuk percobaan pertama adalah sekuen genom lengkap dari mitokondria Ancylostoma duodenale (NC_003415.1), dan sekuen genom lengkap dari mitokondria Necator americanus (NC_003416.2). Adapun data yang digunakan untuk percobaan kedua adalah sekuen genom lengkap dari
Human papillomavirus type 134 (NC_014956.1) dan sekuen genom lengkap dari
Human papillomavirus type 132 (NC_014955.1). Pada percobaan ini ditambahkan dua pasang data baru yaitu, Chaetoceros lorenzianus DNA virus (NC_015211.1) dan Chaetoceros tenuissimus DNA virus (NC_014748.1), serta yang terakhir
Thermoproteus tenax spherical virus 1 (NC_006556.1) dan Vesicular stomatitis indiana virus (NC_001560.1). Keempat pasang sekuen DNA itu akan dilakukan penjajaran lokal menggunakan algoritme Smith-Waterman.
Pantua (2011) menggunakan metode super pairwise alignment yang menggabungkan metode probabilitas dan analisis kombinatorial. Simpulan yang diperoleh adalah penggunaan metode super pairwise alignment memiliki kelemahan dalam pemilihan parameter yang mengakibatkan hasil berbeda – beda dan tidak optimal. Sementara itu, Utomo (2013) menggunakan metode Needleman-Wunsch, salah satu algoritme penjajaran klasik yang melakukan penjajaran secara global. Simpulan yang diperoleh, penggunaan algoritme Needleman-Wunsch menghasilkan similaritas yang lebih tinggi ketimbang metode
super pairwise alignment yang dilakukan oleh Pantua (2011).
Pada penelitian ini akan diterapkan algoritme Smith-Waterman dan dibandingkannya dengan hasil penelitian Utomo (2013), Selain itu dilakukan validasi similaritas dan gaps yang dihasilkan menggunakan EMBOSS-water sebagai software penjajaran lokal yang berbasis algoritme Smith-Waterman, sehingga diharapkan akan mendapatkan hasil penjajaran lokal yang optimal.
Tujuan Penelitian
Tujuan dari penelitian ini adalah untuk menerapkan algoritma Smith-Waterman pada penjajaran lokal sekuen DNA.
Manfaat Penelitian
Manfaat dari penelitian ini adalah untuk mendukung penelitian-penelitian di bidang Bioinformatika yang memerlukan informasi dari hasil penjajaran lokal DNA, seperti
3
Ruang Lingkup Penelitian
Pada penelitian ini dilakukan pembatasan masalah antara lain:
1 Data yang digunakan berasal dari GenBank NCBI berformat FASTA yang diambil dari data tahun 2010.
2 Sekuen yang dijajarkan adalah sekuen DNA yang dapat direpresentasikan sebagai string yang terdiri atas karakter A, C, G, dan T yang mewakili basa nitrogen dari adenin, sitosin, guanin dan timin.
3 Menggunakan data 4 pasang sekuen DNA yaitu Ancylostoma duodenale
(NC_003415.1), dan Necator americanus (NC_003416.2), Human papillomavirus type 134 (NC_014956.1) dan Human papillomavirus type 132 (NC_014955.1), Chaetoceros lorenzianus DNA virus (NC_015211.1) dan Chaetoceros tenuissimus DNA virus (NC_014748.1), serta yang terakhir
Thermoproteus tenax spherical virus 1 (NC_006556.1) dan Vesicular stomatitis indiana virus (NC_001560.1). Keempat pasang sekuen DNA akan dibandingkan dengan software lain berbasis Smith-Waterman, dalam hal ini EMBOSS.
4 Ukuran masing - masing sekuen DNA yang digunakan tidak harus sama.
METODE
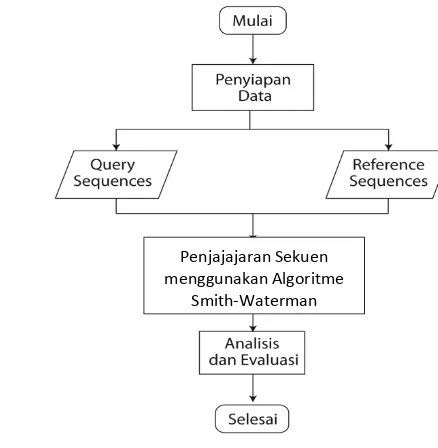
Tujuan penelitian ini adalah melakukan penjajaran dua buah sekuen yang berbeda. Sekuen pertama disebut reference sequence dan yang kedua disebut
query sequence. Tahapan proses penelitian disajikan dalam diagram alur pada Gambar 2.
Gambar 2 Diagram alur metode penelitian
Penjajajaran Sekuen menggunakan Algoritme
4
Penyiapan Data
Data yang digunakan pada penelitian ini berasal dari GenBank yang diunduh dari situs resmi National Centre for Biotechnology Information (NCBI). Data tersebut berformat FASTA seperti contoh pada Gambar 3. Data berformat FASTA terdiri atas baris pertama berupa simbol “ > ” yang diikuti identifier dari sekuen dan baris kedua berupa data sekuen yang akan digunakan.
Gambar 3 Contoh data berformat FASTA
Untuk membaca fail berformat FASTA, diperlukan proses parsing. Pada proses ini, program akan mengidentifikasi keberadaan simbol “ > ” sebagai tanda bahwa baris tersebut merupakan baris identifier. Kemudian program akan mengidentifikasi keberadaan baris baru sebagai tanda dimulainya baris data sekuen. Data sekuen inilah yang akan digunakan untuk penjajaran lokal sekuen DNA, yang berupa urutan string basa nitrogen (A, C, G, T).
Penelitian ini menggunakan data yang sama dengan penelitian Pantua (2011) dan Utomo (2013) yaitu sekuen genom lengkap dari mitokondria
Ancylostoma duodenale (NC_003415.1), dengan panjang sekuen 13721 bp,
mitokondria Necator americanus mitochondrion (NC_003416.2), dengan panjang sekuen 13605 bp, ditambah dengan data baru yakni Human papillomavirus type 134 (NC_014956.1) dengan panjang sekuen 7309 bp, dan Human papillomavirus type 132 (NC_014955.1) dengan panjang sekuen 7125 bp. Sebagai pembanding, dalam penelitian ini ditambahkan dua pasang sekuen DNA lain dengan panjang sekuen yang jauh berbeda, yaitu sekuen genom lengkap dari Thermoproteus tenax spherical virus 1 (NC_006556.1) dengan panjang sekuen 20933bp, dan sekuen genom lengkap dari virus Vesicular stomatitis indiana virus (NC_001560.1) dengan panjang sekuen 11161bp, pasangan sekuen ini dipilih, karena terdapat selisih yang besar dari sekuen, sehingga melalui percobaan akan diteliti apakah ada pengaruhnya dengan similaritas, gaps dan waktu eksekusi algoritme. Sekuen terakhir genom lengkap dari Chaetoceros lorenzianus DNA Virus (NC_015211.1) dengan panjang sekuen 5813bp, dan sekuen genom lengkap dari Chaetoceros tenuissimus DNA virus (NC_014748.1) dengan panjang sekuen 5639bp, pasangan ini dipilih karena merupakan sekuen DNA dari family yang sama, namun dengan
genus yang berbeda, sehingga dapat digunakan sebagai pembanding bagi pasangan sekuen Human papillomavirus type 134 (NC_014956.1) dan Human papillomavirus type 132 (NC_014955.1).
Algoritme Penjajaran Lokal Smith-Waterman
Algoritme Smith-Waterman ditemukan pada tahun 1981 oleh TF Smith dan MS Waterman. Algoritme ini digunakan untuk menemukan penjajaran lokal yang memiliki nilai optimal lokal dari dua buah sekuen. Algoritme Smith-Waterman
5
menghitung semua informasi yang terdapat pada dua sekuen sehingga jika sekuen
query berukuran j, dan sekuen reference berukuran i maka waktu kompleksitas saat proses inisialisasi adalah O(i+j). Hal ini karena waktu inisialisasi hanya membutuhkan pengisian vertikal dan horizontal saja. Sedangkan waktu kompleksitas pengisian matriks adalah O(ij) karena harus mengakses semua bagian sekuen, dan saat penentuan nilai maksimum adalah O(ij) karena harus menyimpan informasi sekuen baris sekuen sebelumnya, sehingga perhitungan waktu kompleksitas Algoritme adalah sebagai berikut :
O(i+j)+ O(ij) + O(ij) = O(ij) (Chan 2004)
Untuk mencari penjajaran lokal terbaik, pada algoritme ini digunakan matriks penskoran dengan menerapkan gaps sebagai bobot. Gaps adalah proses terjadinya delesi ataupun insersi, keduanya tidak bisa berjalan dalam base pair
yang sama, sehingga jika terjadi delesi di sekuen reference, maka bisa terjadi insersi di sekuen query, atau tidak terjadi apa-apa, namun tidak akan bisa terjadi delesi di sekuen tersebut, begitu juga sebaliknya. Gap diberikan simbol (-) sebagai tanda terjadi delesi atau insersi di salah satu sekuen. Perhitungan gap ini juga diperkenalkan oleh Smith-Waterman dalam penelitiannya dengan memberikan nilai gaps sebagai.
Wk = a + b*k (1) Dimana Wk, adalah gap penalty yang akan diberikan kepada sekuen indeks ke–k yang menyatakan nilai maksimal dari sekuen sebelumnya, a adalah gap open, dan b adalah gap extend (Smith dan Waterman 1981)
Dalam algoritma Smith-Waterman secara umum terdapat 3 tahap, antara lain, tahap inisialisasi, pengisian matriks, dan tahap penyusunan balik (traceback). Pada tahap kedua akan digunakan metode dynamic programming
untuk menentukan nilai maksimum yang akan menjadi titik dimulainya tahap ketiga. Adapun tahap tersebut adalah :
1 Inisialisasi
Pada tahap ini dilakukan pemberian nilai awal pada matriks penskoran M[i,j]. Jika panjang query sequence adalah j dan panjang reference sequence adalah i, maka matriks penskoran M[i,j] tersebut berukuran (i+1) * (j+1).
Gambar 4 Contoh ilustrasi proses Inisialiasi.
6
Selanjutnya baris dan kolom pertama diisi dengan nilai 0, sebagai pengganti titik awal sekuen. Adanya similaritas di akhir sekuen, sehingga relasi perulangan sedikit diubah karena string kosong adalah akhir dari tiap sekuen, yang mengakibatkan nilai 0 sebagai penanda akhir sekuen menggantikan string yang kosong (Annibal 2003). Contoh inisialisasi awal matriks penskoran dapat dilihat pada Gambar 4.
2 Pengisian Matriks
Pada tahap pengisian matriks, dihitung semua nilai matriks dengan ketentuan sebagai berikut:
0
M[i-k,j] + wk
M[i,j] = Max M[i,j-k] + wk (2) M[i-1,j-1] + S[i,j]
di mana S[i,j] adalah match / mismatch skor yang telah ditentukan. Pada penelitian ini digunakan dua tipe parameter skor untuk S[i,j], yaitu : 1 Nilai match = 5, nilai mismatch = -4
2 Nilai match = 9, nilai mismatch = -1
Tipe pertama digunakan oleh EMBOSS (European Molecular Biology Open Software Suite) sebagai software penjajaran sekuen yang berbasis Smith-Waterman (Palmenberg 2008). Sedangkan tipe kedua digunakan oleh percobaan sebelumnya yaitu Utomo (2013). Dengan wk adalah konstanta
gap penalty (1), k adalah panjang sekuen sebelum M[i,j], dan M[i,j] sendiri adalah matriks penskoran yang diisi nilai hasil dari persamaan (2). Contoh matriks penskoran yang telah terisi dapat dilihat pada Gambar 5.
Gambar 5 Contoh ilustrasi proses pengisian matriks.
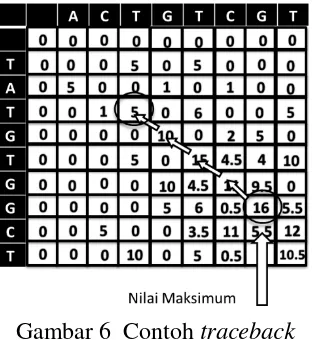
3 Penyusunan Balik (Traceback)
Penyusunan balik (Traceback) merupakan tahap menyusun jalur dari matriks penskoran (scoring matrix) yang telah berisi nilai-nilai pada langkah sebelumnya. Jalur tersebut disusun dari posisi base pair bernilai maksimum
A C T G T C G T
0 0 0 0 0 0 0 0 0
T 0 0 0 5 0 5 0 0 0
A 0 5 0 0 1 0 1 0 0
T 0 0 1 5 0 6 0 0 5
G 0 0 0 0 10 0 2 5 0
T 0 0 0 5 0 15 5 4 10
G 0 0 0 0 10 5 11 10 0
G 0 0 0 0 5 6 1 16 5.5
C 0 0 5 0 0 4 11 6 12
7
hingga tercapai base pair bernilai 0 atau nilai minimum. Arah panah yang dihasilkan akan menunjuk nilai terbesar diantara tiga region dengan mengabaikan apakah base-pair match atau mismatch. Contoh traceback
dapat dilihat pada Gambar 6.
Gambar 6 Contoh traceback
Keluaran dari implementasi algoritme Smith-Waterman berupa nilai dan visualisasi hasil penjajaran. Keluaran tersebut ditampilkan pada aplikasi yang selanjutnya digunakan untuk analisis dan evaluasi. Keluaran tersebut terdiri atas : 1 Length, yaitu panjang penjajaran yang terbentuk.
2 Similarity yang merepresentasikan jumlah sekuen yang match dan persentase kemiripan dua sekuen yang dijajarkan dari hasil panjang penjajaran yang terbentuk.
3 Gaps yang mewakili persentase kemunculan gaps dari panjang sekuen penjajaran yang terbentuk.
4 Score, yaitu total nilai hasil penjajaran.
5 Execution time, yaitu waktu yang dibutuhkan untuk mengeksekusi algoritme Smith-Waterman.
6 Visualisasi hasil penjajaran dua sekuen yang berupa bentuk pasangan penjajaran sekuen yang dipisahkan dengan simbol (“ | ”) untuk pasangan sekuen yang match (“ . “) untuk pasangan sekuen yang mismatch dan (“ “) untuk pasangan sekuen yang muncul gaps.
7 Grafik yang menampilkan hubungan match antara reference sequence dengan
query sequence.
Analisis dan Evaluasi
8
Lingkungan Pengembangan
Penelitian ini dibangun dengan menggunakan bahasa pemrograman ASP.NET dan C# yang didukung perangkat lunak dan perangkat keras dengan spesifikasi sebagai berikut :
• Perangkat lunak :
o Sistem operasi Microsoft Windows 7 o Microsoft Visual Studio 2010 X86 (32 Bit)
• Perangkat keras :
o AMD Turion X2 Dual-Core @ 2 Ghz o Memory 4 GB RAM
o Harddisk dengan kapasitas sisa 100 GB o Monitor resolusi 1366 * 768 pixel o Mouse dan keyboard
HASIL DAN PEMBAHASAN
Implementasi Algoritme Penjajaran Lokal Smith-Waterman
Pada tahap ini algoritme Smith-Waterman diimplementasikan ke dalam bentuk baris program untuk membangun aplikasi penjajaran lokal. Pseudo code
penjajaran lokal menggunakan Smith-Waterman disajikan pada Lampiran 1. User interface aplikasi yang telah dibangun dapat dilihat pada Lampiran 2. Pengujian pada aplikasi dilakukan dalam tiga jenis, yaitu pengujian untuk mencari hasil penjajaran tertinggi, pengujian kapasitas dengan mencoba panjang sekuen beragam, dan perbandingan hasil penjajaran tertinggi dengan software EMBOSS dan algoritme Needleman-Wunsch.
Pengujian akurasi juga terdiri dari empat jenis, yang melibatkan sepasang sekuen DNA. Dari tiap pengujian dibagi lagi menjadi dua percobaan dengan dua parameter inti yang berbeda, ada beberapa parameter yang digunakan dalam percobaan, diantaranya:
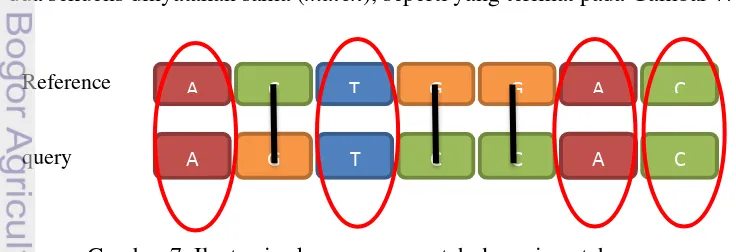
1.Nilai Match
Nilai match adalah nilai yang digunakan saat sepasang basa nitrogen dari dua sekuens dinyatakan sama (match), seperti yang terlihat pada Gambar 7.
Gambar 7 Ilustrasi sekuens yang match dan mismatch.
A C T G G A C
A G T C C A C
Reference
9
2.Nilai Mismatch
Nilai mismatch adalah nilai yang digunakan saat sepasang basa nitrogen dari dua sekuen dinyatakan tidak sama (mismatch), dalam Smith-Waterman, variabel mismatch haruslah negatif, karena berfungsi menjadi salah satu variabel yang akan mengurangi nilai penjajaran.
3.Gap Penalty
Gap Penalty adalah nilai yang diberikan saat terjadi insertion atau deletion
dalam basa nitrogen, gap penalty dihitung menggunakan persamaan (1).
Dalam pengujian, nilai match dan mismatch merupakan parameter inti, dan akan digunakan 2 pasang nilai match dan mismatch, pasangan pertama adalah nilai match = 5, nilai mismatch = -4, sedangkan pasangan kedua nilai match = 9 dan nilai mismatch = -1, pemilihan pasangan parameter pertama merujuk kepada
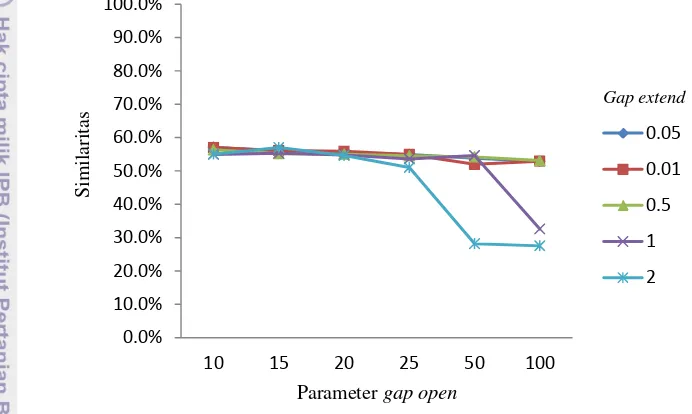
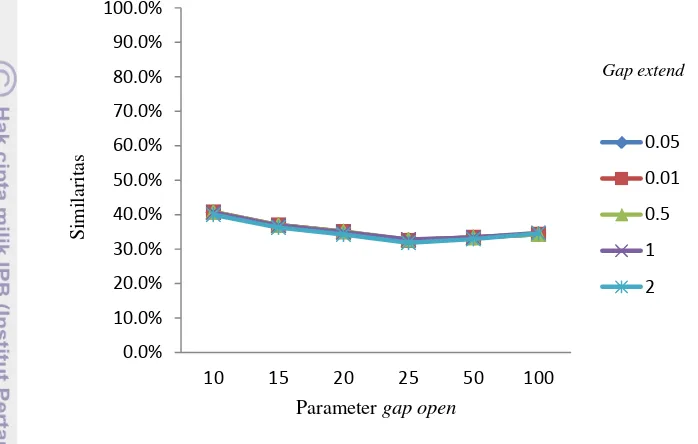
software EMBOSS yang menggunakan parameter pertama sebagai nilainya. Sedangkan pasangan parameter kedua, digunakan untuk membandingkan dua jenis penjajaran, yaitu penjajaran lokal dan global. Kedua pasang parameter dipilih untuk kemudian dibandingkan mana yang lebih efektif digunakan sebagai parameter dalam penjajaran lokal Smith-Waterman. Gap penalty yang akan digunakan adalah enam jenis gap open, yaitu 10, 15, 20, 25, 50, dan 100. Dan nilai gap extend 0.01,0.05,0.5,1, dan 2. Keduanya akan diuji coba ke empat pasang DNA sekuen dengan dua pasang parameter inti yang telah ditentukan.
Gambar 8 Grafik similaritas Ancylostoma duodenale dan Necator americanus
dengan parameter match 5 mismatch -4
Pengujian pertama dilakukan penjajaran sekuen genom lengkap dari se- pasang DNA sekuen, yang pertama adalah mitokondria Ancylostoma duodenale
(NC_003415.1)sebagai reference sequence dengan panjang sekuen 13721 bpdan sekuen genom lengkap dari mitokondria Necator americanus (NC_003416.2) sebagai query sequence dengan panjang sekuen 13605 bp. Percobaan dilakukan dengan cara memasangkan semua gap open dan gap extend, sehingga terdapat 60
10
data hasil percobaan, menghasilkan empat keluaran yaitu simmilarity, skor penjajaran, panjang hasil penjajaran, dan gaps, berikut adalah gambar-gambar grafik similaritas hasil penjajaran sekuen yang menggunakan parameter match = 5 dan mismatch = -4. Seperti yang terlihat pada Gambar 8.
Setelah menggunakan parameter inti pertama, maka dilakukan percobaan dengan pasangan sekuen DNA yang sama, namun menggunakan parameter inti kedua, yaitu nilai match = 9, dan mismatch = -1. Hasil similaritas dapat dilihat pada Gambar 9.
Gambar 9 Grafik similaritas Ancylostoma duodenale dan Necator americanus
dengan parameter match 9 mismatch -1
Dari kedua percobaan, dihasilkan kondisi maksimal dengan parameter gap penalty seperti pada Tabel 1.
Tabel 1 Hasil pengujian Ancylostoma duodenale dan Necator americanus
0.0%
Execution time 278 detik 263 detik
Gap extend
11
Pengujian kedua dilakukan penjajaran sekuen genom lengkap dari sepasang sekuen DNA, Sekuen pertama adalah Human papillomavirus type 134
(NC_014956.1) sebagai reference sequence dengan panjang sekuen 7309 bp dan sekuen DNA genom lengkap dari Human papillomavirus type 132 (NC_014955.1) sebagai query sequence dengan panjang sekuen 7125 bp. Pengujian menggunakan parameter match = 5 dan mismatch = -4. Hasil pengujian dapat dilihat pada Gambar 10.
Gambar 10 Grafik similaritas Human papillomavirus type 134 dan Human papillomavirus type 132 dengan parameter match 5 mismatch -4
Setelah menggunakan parameter inti pertama, maka dilakukan percobaan dengan pasangan sekuen DNA yang sama, namun menggunakan parameter inti kedua, yaitu nilai match = 9, dan mismatch = -1. Hasil similaritas pengujian dapat dilihat pada Gambar 11.
Gambar 11 Grafik similaritas Human papillomavirus type 134 dan Human papillomavirus type 132 dengan parameter match 9 mismatch -1.
12
Dari kedua percobaan, dihasilkan kondisi maksimal dengan parameter gap penalty seperti pada Tabel 2.
Tabel 2 Hasil pengujian Human papillomavirus type 134 dan Human papillomavirus type 132
Pengujian ketiga dilakukan penjajaran sekuen genom lengkap dari se- pasang DNA sekuen, sekuen ketiga adalah Thermoproteus tenax spherical virus 1
(NC_006556.1) dengan panjang sekuen 20933bp, dan sekuen genom lengkap dari
Vesicular stomatitis indiana virus (NC_001560.1) dengan panjang sekuen 11161bp. Percobaan dilakukan dengan cara memasangkan semua gap open dan
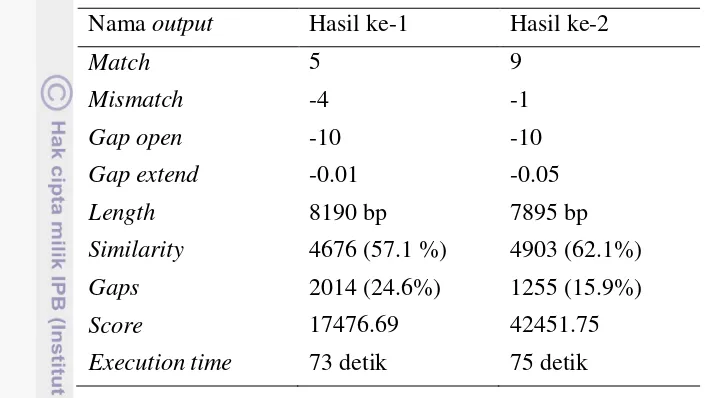
gap extend, sehingga terdapat 60 data hasil percobaan, menghasilkan empat keluaran yaitu simmilarity, skor penjajaran, panjang hasil penjajaran, dan gaps, hasil similaritas penjajaran menggunakan parameter match = 5 dan mismatch = -4 dapat dilihat pada Gambar 12.
Gambar 12 Grafik similaritas Thermoproteus tenax spherical virus 1 dan
Vesicular stomatitis indiana virus dengan parameter match 5
mismatch -4
Execution time 73 detik 75 detik
13
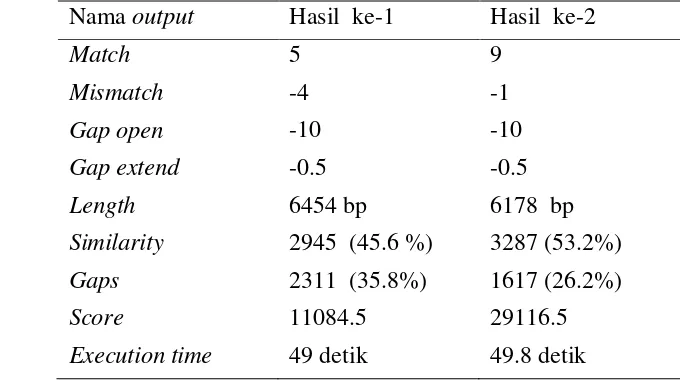
Setelah menggunakan parameter inti pertama, maka dilakukan percobaan dengan pasangan sekuen DNA yang sama, namun menggunakan parameter inti kedua, yaitu nilai match = 9, dan mismatch = -1. Hasil similaritas penjajaran dapat dilihat pada Gambar 13.
Gambar 13 Grafik similaritas Thermoproteus tenax spherical virus 1 dan
Vesicular stomatitis indiana virus dengan parameter match 9
mismatch -1
Dari kedua percobaan, dihasilkan kondisi maksimal dengan parameter gap penalty seperti pada Tabel 3.
Tabel 3 Hasil pengujian Thermoproteus tenax spherical virus 1 dan Vesicular stomatitis indiana virus
Execution time 287 detik 323 detik
14
Pengujian keempat dilakukan penjajaran sekuen genom lengkap dari sepasang sekuen DNA, yang pertama adalah Chaetoceros lorenzianus DNA virus
(NC_015211.1) dengan panjang sekuen 5813 bp sebagai reference, dan sekuen genom lengkap dari Chaetoceros tenuissimus DNA virus (NC_014748.1) dengan panjang sekuen 5639 bp sebagai query. berikut adalah gambar grafik hasil penjajaran sekuen yang menggunakan parameter match = 5 dan mismatch = -4. Hasil pengujian dapat dilihat pada gambar 14.
Gambar 14 Grafik similaritas Chaetoceros lorenzianus DNA virus dan
Chaetoceros tenuissimus DNA virus dengan parameter match 5
mismatch -4
Setelah menggunakan parameter inti pertama, maka dilakukan percobaan dengan pasangan sekuen DNA yang sama, namun menggunakan parameter inti kedua, yaitu nilai match = 9, dan mismatch = -1. Hasil similaritas penjajaran dapat dilihat pada Gambar 15.
Gambar 15 Grafik similaritas Chaetoceros lorenzianusDNA virus dan
Chaetoceros tenuissimus DNA virus dengan parameter match 9
15
Dari kedua percobaan, dihasilkan kondisi maksimal dengan parameter gap penalty seperti pada Tabel 4.
Tabel 4 Hasil pengujian Chaetoceros lorenzianus dan Chaetoceros tenuissimus
Setelah melakukan pengujian pada dua pasang sekuen DNA yang berbeda, dilakukan percobaan ketiga, yaitu pengujian ketahanan implementasi program dengan cara mencoba kombinasi berbagai jenis panjang base pair. Pengujian dilakukan dengan melibatkan sekuens diambil dari Azospirillum brasilense Sp245
(NC_016617.1) dan Azospirillum sp. B510 (NC_013854.1), kemudian diambil 5000 hingga 45000 bp dari tiap sekuen DNA, dan diujikan ke sistem yang telah dibangun. Hasil percobaan berada pada kondisi seperti pada Gambar 16.
Gambar 16 Grafik waktu eksekusi program Lokalsmith.
0.0
5000 10000 15000 20000 25000 30000 35000 40000 45000
W
16
Analisis dan Evaluasi
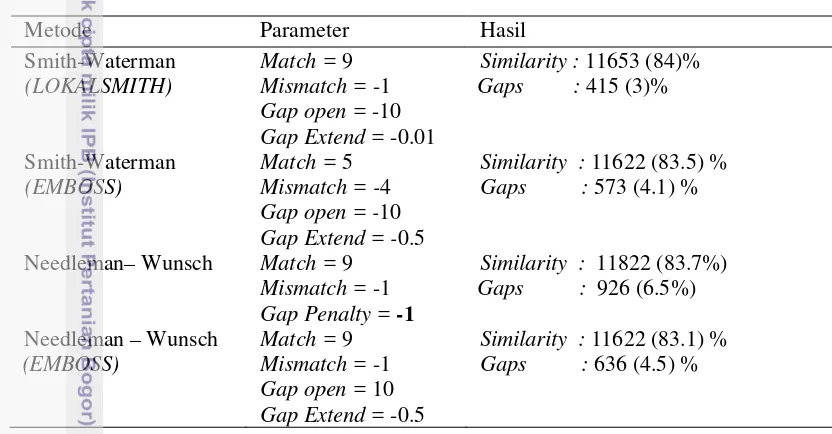
Pada tahap ini hasil pengujian yang telah dilakukan dianalisis satu persatu mulai dari pengujian parameter hingga pengujian waktu eksekusi, hasil yang didapat akan dibandingkan dengan software EMBOSS-Water dan hasil penelitian dari Utomo (2013), dari perbandingan Ancylostoma duodenale (NC_003415.1) dijajarkan dengan Necator americanus (NC_003416.2) menghasilkan nilai similaritas dan gaps seperti pada Tabel 5 di bawah ini.
Tabel 5 Hasil perbandingan penjajaran Ancylostoma duodenale dan Necator americanus
Metode Parameter Hasil Smith-Waterman
Similarity : 11653 (84)%
Gaps : 415 (3)%
Dari hasil perbandingan didapatkan hasil yang beragam, namun tidak berbeda jauh, dengan hanya selisih maksimum 1% dari tiap perbandingannya.
Ancylostoma duodenale (NC_003415.1) dijajarkan dengan Necator americanus
(NC_003416.2), menghasilkan similaritas 84% untuk penggunaan program Lokalsmith, pada software EMBOSS dihasilkan 83.5% untuk EMBOSS-Water
dan 83.1% untuk EMBOSS-Needle, sedangkan Needleman-Wunsch hasil percobaan dari Utomo (2013) menghasilkan similaritas 83.7%, selisih 0.3% dari percobaan menggunakan Lokalsmith, namun dari segi panjang sekuen yang dihasilkan Needleman-Wunsch lebih unggul, dikarenakan Needleman-Wunsch tidak memulai penjajaran dari nilai maksimum, akan tetapi dari ujung sekuen.
Similaritas yang tidak jauh berbeda juga disebabkan oleh bentuk sekuen yang serupa, terlihat dari nilai similaritas yang berada di atas 80%, atau mendekati 100% kemiripan. Namun dari gaps yang dihasilkan terdapat selisih yang cukup beragam, Lokalsmith menghasilkan gaps 2.99%, EMBOSS-water menghasilkan
gaps 4.1%, EMBOSS-Needle menghasilkan gaps 4.5% dan dari percobaan Utomo (2013) menghasilkan gaps tertinggi yaitu 6.5%. Gaps yang tinggi pada percobaan Needleman-Wunsch terjadi karena algoritme Needleman-Wunsch tidak menggunakan gap penalty seperti persamaan (1) .
Pada percobaan kedua, menjajarkan Human papillomavirus type 134
17
maksimal dari parameter kemudian dibandingkan dan menghasilkan perbandingan seperti yang tertera pada Tabel 6.
Tabel 6 Hasil perbandingan penjajaran Human papillomavirus type 134 dan
Human papillomavirus type 132
Metode Parameter Hasil Smith-Waterman
Dari hasil perbandingan didapatkan nilai similaritas 62.1% untuk percobaan menggunakan Lokalsmith, 57.1% untuk percobaan software EMBOSS-water, 56.8% menggunakan EMBOSS-Needle, dan percobaan Utomo (2013) menghasilkan 62.9%, berbeda tipis dengan hasil menggunakan program Lokalsmith, yaitu 0.8% selisihnya. Dari panjang penjajaran yang dihasilkan percobaan Utomo (2013) menghasilkan sekuen yang paling panjang diantara percobaan lainnya, yaitu 5146 bp. Hal ini dikarenakan algoritme Smith-Waterman memulai penjajarannya dari nilai maksimum yang telah dibentuk menggunakan persamaan (2), sedangkan pada algoritme Needleman-Wunsch akan melakukan penjajaran dari awal sekuen hingga akhir sekuen yang dijajarkan.
Similaritas yang jauh berbeda disebabkan oleh bentuk sekuen yang tidak serupa, terlihat dari nilai similaritas yang berada di atas 50%, namun berada di bawah 70%, dengan gaps yang dihasilkan juga beragam, Lokalsmith menghasilkan gaps 15.9%, EMBOSS-water menghasilkan gaps 24.9%, EMBOSS-Needle menghasilkan gaps 25.4% dan dari percobaan Utomo (2013) menghasilkan gaps tertinggi yaitu 23.5%. Gaps yang kecil dihasilkan oleh Lokalsmith, karena penggunaan parameter yang berbeda dengan EMBOSS-Water, sehingga mempengaruhi pembentukan gaps dari hasil penjajaran sekuen.
Pada percobaan ketiga, menjajarkan Chaetoceros lorenzianus DNA Virus
(NC_015211.1), dan sekuen genom lengkap dari virus Chaetoceros tenuissimus DNA virus (NC_014748.1). Hasil maksimal dari parameter kemudian dibandingkan dan menghasilkan perbandingan seperti yang tertera pada Tabel 7.
Tabel 7 Hasil Perbandingan Penjajaran Chaetoceros lorenzianus DNA virus dan
18
Metode Parameter Hasil Smith-Waterman
Dari hasil perbandingan didapatkan nilai similaritas 53.2% untuk percobaan menggunakan Lokalsmith, 45.5% untuk percobaan software EMBOSS-water, 39.9% menggunakan EMBOSS-Needle, data dicobakan pada program dari Utomo (2013) namun terjadi crash pada program sehingga tidak menghasilkan apa-apa. Karena itu hanya digunakan perbandingan software EMBOSS sebagai pembanding hasil penjajaran.
Tabel 8 Hasil perbandingan penjajaran Thermoproteus tenax spherical virus 1 dan Vesicular stomatitis indiana virus.
Metode Parameter Hasil Smith-Waterman
19
Lokalsmith sebesar 26.2%, EMBOSS-water menghasilkan gaps 40.9%, EMBOSS-Needle menghasilkan gaps 48.1%. Gaps yang kecil dihasilkan oleh Lokalsmith, karena penggunaan parameter yang berbeda dengan EMBOSS-water, sehingga mengakibatkan pengaruh pembentukan gaps dari hasil penjajaran sekuen, selain itu nilai gaps yang jauh berbeda juga membuktikan adanya pengaruh yang signifikan dari pemilihan parameter inti yang digunakan dalam penjajaran. Similaritas yang rendah juga membuktikan bahwa meskipun berasal dari jenis yang sama, namun sekuennya bisa jauh berbeda, sehingga similaritas yang rendah mengakibatkan perbedaan struktur dan fungsional antara sekuen.
Pada percobaan terakhir, dijajarkan Thermoproteus tenax spherical virus 1
(NC_006556.1) dan sekuen genom lengkap dari Vesicular stomatitis indiana virus
(NC_001560.1). Hasil maksimal dari parameter kemudian dibandingkan dengan software EMBOSS dan menghasilkan perbandingan seperti yang tertera pada Tabel 8.
Dari hasil perbandingan didapatkan nilai similaritas 40.8% untuk percobaan menggunakan Lokalsmith, 35.7% untuk percobaan software EMBOSS-water, 34.5% menggunakan EMBOSS-Needle, data dicobakan pada program dari Utomo (2013) namun terjadi crash pada program sehingga tidak menghasilkan apa-apa. Karena itu hanya digunakan perbandingan software EMBOSS sebagai pembanding hasil penjajaran.
Pada percobaan ini terlihat beragam similaritas yang jauh berbeda disebabkan oleh bentuk sekuen yang tidak serupa. Hal ini terlihat dari nilai similaritas yang berada di antara 40% hingga 30%, dengan gaps yang dihasilkan Lokalsmith sebesar 47.6%, EMBOSS-water menghasilkan gaps 55%, EMBOSS-Needle menghasilkan gaps 56.6%. Gaps yang kecil dihasilkan oleh Lokalsmith, karena penggunaan parameter yang berbeda dengan EMBOSS-Water, sehingga mengakibatkan pengaruh pembentukan gaps dari hasil penjajaran sekuen, pada percobaan ini terlihat besaran gaps yang besar yaitu antara 50% hingga 40%, hal ini disebabkan karena besarnya selisih antara panjang sekuen yang dijajarkan yaitu 9772 bp, sehingga mengakibatkan bertambahnya jumlah gaps, dan berkurangnya similaritas, karena terpisahkan oleh gaps yang panjang.
Dilihat dari waktu eksekusinya, algoritme Smith-Waterman memiliki waktu kompleksitas O(mn) atau mendekati kuadratik, hal ini karena adanya dua kali proses yang melibatkan semua bagian sekuen, yaitu proses penentuan nilai maksimal, dan penentuan arah jalur traceback. Pada gambar grafik waktu eksekusi terlihat bahwa pasangan sekuen dengan panjang yang sama akan memakan waktu yang lebih lama ketimbang panjang pasangan yang berbeda, hal ini dikarenakan waktu kompleksitas yang menjadi kuadratik saat sekuen query
dan reference memiliki panjang yang sama, sedangkan jika terjadi perbedaan panjang query dan reference, salah satu sekuen akan menjadi lebih panjang dari satu lainnya, sehingga m ≠ n, yang mengakibatkan berkurangnya waktu eksekusi program.
20
SIMPULAN DAN SARAN
Simpulan
Dari penelitian yang telah dilakukan dalam penjajaran lokal sekuen DNA menggunakan algoritme Smith-Waterman ini dapat disimpulkan sebagai berikut :
1 Pada penjajaran lokal sekuen, algoritme Smith-Waterman dapat diterapkan. 2 Penerapan algoritme Smith-Waterman pada penjajaran lokal sekuen DNA
memiliki hasil penjajaran paling optimal, karena memiliki smilaritas yang tinggi dibandingkan penggunaan algoritme Needleman-Wunsch.
3 Hasil penjajaran yang sangat beragam, karena perbedaan parameter yang digunakan, panjang sekuen dan bentuk sekuen yang dijajarkan.
4 Waktu kompleksitas dari algoritme Smith-Waterman adalah O(mn), karena terjadi dua kali proses yang melibatkan semua bagian sekuen, yaitu proses menemukan nilai maksimum dan proses penentuan arah traceback.
5 Parameter inti match = 9 dan mismatch = -1 adalah parameter terbaik, hal ini terbukti dari hasil similaritas yang tinggi, dan gaps yang kecil dihasilkan menggunakan parameter ini.
Saran
Untuk penelitian selanjutnya disarankan sebagai berikut:
1 Mengaplikasikan Parallel programming, sehingga menambah kemampuan maksimum panjang sekuen yang dijajarkan serta mempersingkat waktu eksekusi dari algoritme Smith-Waterman
21
DAFTAR PUSTAKA
Annibal S. 2003. Sequence alignment algorithms. London (UK): King's College. Agrawal A. 2009. Sequence-specific sequence comparison using pairwise
statistical significance[tesis] Iowa (US): Iowa State University Chan, A. 2004. An analysis of pairwise sequence alignment algorithm
complexities: Needleman-Wunsch, Smith-Waterman, FASTA, BLAST and gapped BLAST. [tesis] California(US) :Stanford University
Palmenberg A, Sgro JY. 2008. Biochemistry 711 : EMBOSS software for sequence analysis. Madison (US) : University of Wisconsin.
Pantua A. 2011. Implementasi super pairwise alignment pada global alignment
[skripsi]. Surabaya (ID): Intstitut Teknologi Sepuluh November.
Smith TF, and Waterman, MS. 1981. Identification of Common Molecular Subsequences. Journal of Molecular Biology. 147:195-197
22
Lampiran 1 Pseudocode algoritme Smith - Waterman
Pseudo Code
Initialization: F(0,j) = 0
F(i,0)= 0
Filling Matrix:
for each i,j = 0 to M,N { F(i,j) = max(0,
23
24
RIWAYAT HIDUP
Penulis dilahirkan di Malang pada tanggal 02 Mei 1986 sebagai anak pertama dari pasangan Subakir Amin dan Siti Rodiyah. Penulis merupakan lulusan SMA Negeri 8 Malang (2000 - 2003), Mts Negeri Malang (1997 - 2000) dan Madrasah Ibtidaiyah Negeri Malang (1991 - 1997).
Pada tahun 2004 penulis diterima sebagai mahasiswa Diploma III Program Keahlian Teknik Elektronika, Direktorat Program Diploma Politeknik Negeri Malang melalui jalur Undangan Seleksi Masuk Politeknik Negeri Malang. Setelah menyelesaikan pendidikan Diploma III pada tahun 2008, penulis kembali melanjutkan pendidikan Strata 1 (S1) melalui jalur Alih Jenis dan diterima sebagai mahasiswa Departemen Ilmu Komputer, Fakultas Matematika dan Ilmu Pengetahuan Alam, Institut Pertanian Bogor.