Distribusi Pembawa Sifat Thalassemia (
α
&
β
) dan Hemoglobin-E pada
Penduduk Medan
Ratna Akbari Ganie
Departemen Patologi Klinik, Fakultas Kedokteran USU, Medan, Sumatera Utara
α β
α β
α β
α β
variant were commonly found in Medan as 3,35%, 4,07% and 0, 26% respectively. From the public health of view, this finding seems to be important as basic recommendation for hereditary blood disorders management based on preventive effort both premarital genetic counseling or prenatal diagnosis. Premarital genetic counseling and prenatal diagnosis should be socialized in the near future to prevent the upcoming new high risk couples who could potentially produce new thalassemia babies.
Keywords: hereditary blood disorders, thalassaemia carrier, hemoglobin variants, premarital genetic conseling, prenatal diagnosis
PENDAHULUAN
Seperti Kota besar lainnya, Medan yang terletak di Sumatera Utara, mempunyai penduduk yang heterogen terdiri dari berbagai suku antara lain suku Batak, Melayu, Jawa,
variant lainnya (Cavalli Sforza, et al., 1994;
Bowie LJ, et al, 1997).1
Sama seperti daerah endemik malaria lainnya, diduga populasi di Medan juga mempunyai seleksi positif berbagai gen unggul terhadap invasi Plasmodium, seperti kelainan hemoglobin;
thalassemia-α, thalassemia-β dan
hemoglobin-E (Hb-hemoglobin-E) maupun kelainan eritrosit lainnya seperti Defisiensi enzim Glucose-6-Phosphat Dehydrogenase (G-6-PD) dan ovalositosis (Lie-Injoe, 1959; Flazt, 1967; Luzatto, 1979).2,3,4
Penyakit Thalassemia-α, Thalassemia-β
dan Hb-E adalah kelainan genetic paling umum dijumpai pada penduduk Asia Tenggara termasuk Indonesia (Weatherall and Clegg, 2001). Wong (1983) memperkirakan
frekuensi pengemban sifat (carrier)
thalassemia-α pada populasi Indonesia secara
keseluruhan sebesar 0,5%, thalasemia-β
sebesar 3,5%, dan Hb-E sebesar 4%.6
Penelitian yang lebih komprehensif telah dilakukan pada 17 populasi di Indonesia oleh Lanni (2002), mendapatkan nilai yang lebih tinggi pada beberapa populasi seperti Palembang, Melayu Sumatera pengemban
sifat thalassemia-β yaitu > 7% demikian pula
dengan pengemban gen Hb-E pada beberapa Populasi di Sunda Kecil mencapai 20% bahkan pada penduduk Sumba Timur
mencapai 30%.7
Walaupun penelitian sebelumnya telah pernah melaporkan keberadaan kelainan darah herediter terkait malaria pada populasi Medan
(Lie-Injoe, 1959; Flazt, 1967),2,3
namun seberapa besar frekuensinya di antara kelompok etnik penghuni kota Medan belum pernah dilaporkan. Apalagi secara terpisah telah dilaporkan bahwa kelompok etnik seperti yang Batak, Melayu, Cina, India, Jawa
mempunyai risiko tinggi untuk carrier gen
thalassemia-α, thalassemia-β dan Hb-E. Lanni
et al., (2004) telah melaporkan prevalensi
carrier thalassemia-β dan Hb-E untuk masyarakat Batak sebesar 1,5% dan 0%,
Melayu 5,2% dan 4,3%, Jawa 3,2% dan 4,8%.7
Selain thalassemia-α, jumlah pembawa sifat
thalassemia-β pada masyarakat China daratan
juga cukup tinggi berkisar antara 2,6 %, sampai 5%, sedangkan pembawa sifat
thalassemia-α dijumpai berkisar antara 3,8 %
sampai 14,95% (Lie et al., 1982; Yang et al,
1985).2,8
Prevalensi penyakit genetik memang
erat hubungannya dengan etnik atau ethnic
related genetic seperti yang ditunjukkan pada
penelitian di atas (Flint et al., 1993; Weiss,
1993).9,10
Dengan demikian maka dapat dipastikan bahwa penduduk kota Medan yang menurut Sensus Penduduk tahun 2000, sebagian besar terdiri dari kelompok etnik di atas, sangat berpotensial menjadi pengemban kelainan darah heriditer. Bertolak pada latar belakang permasalahan di atas, maka dilakukan penelitian terhadap 1.521 sampel darah penduduk kota Medan yang berasal dari berbagai kelompok suku untuk mengetahui
jumlah pengemban sifat thalassemia-α,
thalassemia-β dan hemoglobin-E. Hasil
penelitian ini sangat penting sebagai acuan untuk menetapkan perioritas pelayanan kesehatan di era MDGs dengan pendekatan
race-related medicine (Ruel, 2006) dengan melakukan konseling genetik pranikah maupun prenatal diagnosis untuk menurunkan insidensi penyakit darah herediter di Indonesia
khususnya di kota Medan.11
BAHAN DAN CARA PENELITIAN Populasi dan Sampel
Sampel darah dikoleksi dari darah vena 1.512 individu dewasa sehat, pendonor darah dengan kisaran umur 18 – 59 tahun, terdiri dari 1.306 laki-laki dan 215 perempuan. Kadar hemoglobin probandus di atas 12g%. Komposisi jumlah sampel wakil tiap suku diambil sedemikian rupa sehingga mendekati keadaan sebenarnya dari komposisi penduduk kota Medan berdasarkan data Sensus Penduduk tahun 2000.
Cara Penelitian
Terhadap semua sampel darah di atas dilakukan penapisan awal berdasarkan indeks hematologis yang meliputi kadar Hb, RBC, WBC, HCT, MCV, MCH. MCHC dengan
electronic cell counter Cell-Dyn 3500. Nilai MCV < 80% dan MCH < 27% sebagai kreteria untuk penegakkan diagnosis Hemoglobinopati dan Thalassemia. Selanjutnya semua sampel yang tersaring sebagai Hemoglobinopati dan Thalassemia dilakukan pemeriksaan sediaan apus darah tepi dengan pengecatan Giemsa untuk mendapatkan gambaran morfologi eritrosit mikrositer hipokrom.
transferin untuk menapis kemungkinan anemia defisiensi besi. Pemeriksaan kadar
HbA2 juga dilakukan dengan elektroforesis
hemoglobin pada pH alkali dalam media membran selulosa asetat (CAM) dengan memakai kit Helena dan kemudian diberi pewarnaan Ponceau. Fraksi Hb-A lebih ke
arah anoda dibandingkan dengan fraksi HbA2.
Fraksi hemoglobin secara relatif dapat diukur dengan alat densitometer dengan panjang
gelombang λ 525nm. Nilai kuantitasi HbA2
normal adalah 1,5% - 3,5%. Pada
thalassemia-α nilai kuantitasi adalah HbA2<3,5% dan pada
thalassemia-β HbA2>3,5%. Nilai kuantitasi
HbA2 dibedakan dari Hb-E heterizigot jika
nilai kuantitasi HbA2 terhitung > 10%. Untuk
memperkuat diagnosis thalassemia-α, selain
pemeriksaan kadar HbA2, dilakukan juga
pemeriksaan keberadaan badan inklusi secara mikroskopik pada preparat sediaan apus darah tepi.
Pemeriksaan kadar HbA2 dapat juga
dilakukan dengan cara kromatografi HPLC untuk menetapkan sampel pengemban
thalassemia-α, thalassemia-β dan Hb-E.
HASIL PENELITIAN
Hasil penelitian terhadap 1.521 sampel darah penduduk kota Medan menunjukkan hasil pada Tabel 2.
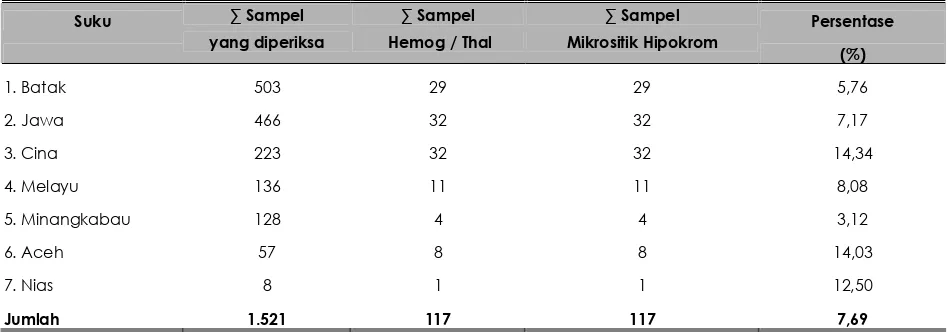
Penapisan indeks hematologis terhadap seluruh sampel darah, telah dijumpai 117 sampel di antaranya terdiagnosis sebagai Mikrositer Hipokrom dengan nilai MCV < 80 fl, dan MCH < 27 pg (Tabel.2). Selanjutnya setelah dilakukan pemeriksaan pemeriksaan mikroskopik morfologi eritrosit sediaan apus darah ternyata semua (117) sampel darah yang dinyatakan Mikrositer Hipokrom tersebut adalah carrier Hemoglobinopati / Thalassemia (Tabel 3).
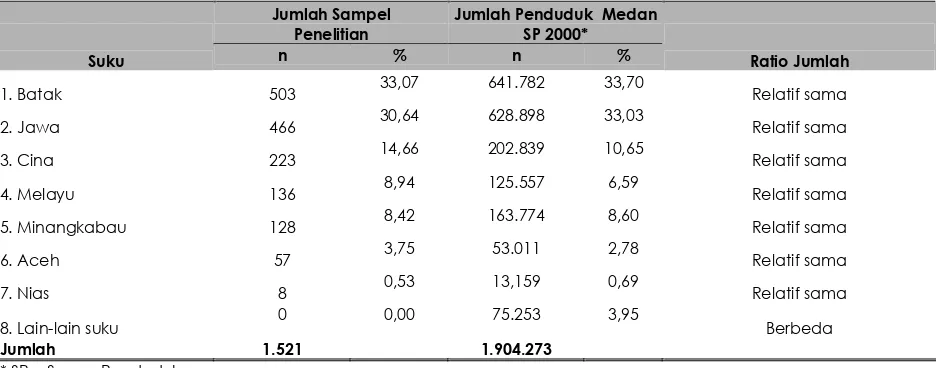
Tabel 1. Distribusi ratio pengambilan sampel tiap suku terhadap jumlah penduduk Kota Medan berdasarkan data sensus penduduk tahun 2000
Jumlah Sampel Penelitian
Jumlah Penduduk Medan SP 2000*
Suku n % n % Ratio Jumlah
1. Batak 503 33,07 641.782 33,70 Relatif sama
2. Jawa 466 30,64 628.898 33,03 Relatif sama
3. Cina 223 14,66 202.839 10,65 Relatif sama
4. Melayu 136 8,94 125.557 6,59 Relatif sama
5. Minangkabau 128 8,42 163.774 8,60 Relatif sama
6. Aceh 57 3,75 53.011 2,78 Relatif sama
7. Nias 8 0,53 13,159 0,69 Relatif sama
8. Lain-lain suku 0 0,00 75.253 3,95 Berbeda
Jumlah 1.521 1.904.273
* SP = Sensus Penduduk
Tabel 2. Distribusi sampel darah normal dan mikrositer hipokrom berdasarkan skrining indeks hematologis
dengan Electronic Cell Counter Cell Dyn 3500
Subjek Frekuensi HGB WBC RBC HCT MCV MCH MCHC RDW PLT
(g/dl) (k/ul) (M/ul) (%) (fl) (pg) (g/dl) (%) (k/ul)
MCV < 80 fl,
MCH < 27 pg 117 13,69 ± 6,26 ± 6,05 ± 41,27 ± 68,42 ± 22,75 ± 33,27 ± 15,45 ± 265,69 ± 1,24 2,38 0,75 4,45 4,71 1,79 1,79 1,73 90,34
MCV > 80 fl,
MCH > 27 pg 1.404 13,69 ± 6,22 ± 4,48 ± 41,87 ± 93,53 ± 30,69 ± 32,88 ± 15,59 ± 228,98 ± 1,05 1,91 0,45 4,42 5,91 2,27 2,44 1,73 56,93
Tabel 3. Distribusi sampel darah hemoglobinopati/thalassemia dan mikrositer hipokrom berdasarkan
Suku ∑ Sampel ∑ Sampel ∑ Sampel Persentase
Hasil pemeriksaan lanjutan terhadap kadar Serum Feritin dan Saturasi Transferin (Tabel 4 dan Tabel 5) menunjukkan kadar Feritin maupun Saturasi Transferin pada 117 sampel darah mikrositik hipokrom tersebut dalam kisaran normal. Artinya semua sampel (117) tersebut
adalah suspect Hemoglobinopati/Thalassemia
akibat kelainan hemoglobin herediter.
Hasil pemeriksaan lanjutan terhadap kadar
HbA2 menunjukkan dari 117 sampel suspect
kelainan hemoglobin herediter tersebut, 51
sampel di antaranya mempunyai kadar HbA2nya
kurang dari 3,5% (carrier thalassemia-α) 62
sampel dengan kadar HbA2nya > 3,5 < 15%
(carrier thalassemia-β) dan 4 sampel kadar
HbA2nya > 15% (carrier HbE) seperti tertera
pada Tabel 6.
PEMBAHASAN
Dari hasil penelitian ini menunjukkan bahwa pemeriksaan indeks hematologis
menggunakan Electronic Cell Counter dengan
patokan nilai MCV < 80fl dan MCH <27
cukup efektif untuk penapisan (screening)
awal kelainan hemoglobin herediter dalam populasi besar seperti yang telah direkomendasikan sebelumnya oleh WHO (1994).12
Hasil penelitian ini, diperkuat lagi dengan pemeriksaan morfologi eritrosit secara mikroskopik, kadar feritin serum dan saturasi transferin sebagai langkah penapisan kedua, ternyata hasilnya cukup signifikan karena seluruh sampel (117) yang terjaring pada penapisan indeks hematologis benar-benar
merupakan suspect kelainan hemoglobin
herediter.
Tabel 4. Hasil pemeriksaan nilai serum feritin pada 117 sampel darah mikrositer hipokrom
dengan kit abbot diagnostic
Jenis Kelamin Nilai
Serum Feritin Laki-Laki Perempuan
20 – < 110
Tabel 5. Hasil pemeriksaan nilai saturasi transferin
terhadap 117 sampel darah mikrositer hipokrom
Jenis Kelamin Nilai
Saturasi Transferin
Laki-Laki Perempuan
20 – < 30
Hasil penelitian ini juga menunjukkan
bahwa pemeriksaan kadar HbA2 cukup efektif
untuk membedakan antara carrier
thalassemia-α dari carrier thalassemia-β
maupun carrier Hb-E. Jumlah carrier
thalassemia-α, pada populasi Medan cukup
tinggi, mencapai 3,35% bahkan pada etnik Cina mencapai 6,72%. Hasil tersebut tidaklah mengejutkan karena sebelumnya Weatherall & Clegg (2001) telah memprediksi angka
pembawa sifat thalassemia-α pada berbagai
Tabel 6. Distribusi Carrier thalassemia-α , thalassemia-β dan Hb-E berdasarkan pemeriksaan kadar HbA2 dengan
Jumlah
Sampel ∑ Suspect ∑ Carrier ∑ Carrier ∑ Carrier
Suku yang diperiksa
Kelainan
populasi di Indonesia berkisar 1 – 10%, bahkan pada penduduk Cina sendiri jumlahnya mencapai 3,8 % sampai 14,95%
(Lie et al., 1982; Yang et al, 1985).8,13
Jumlah carrier thalassemia-β yang
teridentifikasi adalah 4,07% dan carrier Hb-E
sebesar 0,26%. Hemoglobin-E merupakan salah satu varian hemoglobin yang paling umum dijumpai pada populasi di kawasan Asia Tenggara (Fucharoen & Winichagoon, 1987).6,14
Secara umum prevalensi pengemban sifat (carrier) thalassemia-α, thalassemia-β dan Hb-E yang dijumpai dalam penelitian ini cukup representatif dan tidak jauh berbeda jika dibandingkan dengan laporan penelitian sebelumnya seperti Weatherall and Clegg (2001) yang memperkirakan keseluruhan
jumlah carrier thalassemia-β pada populasi
Indonesia adalah 3,7%, Hb-E sebesar 2,7%
dan thalassemia-α kira-kira 1% -10%. Data
lebih rinci tentang prevalensi carrier
thalassemia-β dan Hb-E juga dilaporkan oleh
Lanni et al., (2004) secara komprehensif pada
berbagai suku di Sumatera dan Jawa antara lain pada suku Batak di Medan sebesar 1,5% dan 0%; Minangkabau di Padang sebesar 3,7% dan 2,9%; Melayu di Pekanbaru sebesar 5,2% dan 4,3%; dan Jawa di Yogyakarta adalah 3,2% dan 4,8%.
Berdasarkan hasil penelitian di atas maka
prevalensi carrier thalassemia-α dan
thalassemia-β cukup tinggi pada populasi di
kota Medan. Keadaan ini juga mempunyai arti penting dalam manajemen kesehatan
masyarakat secara keseluruhan dalam konteks
race related medicine yang berbasis pada
ethnic related genetic (Wadman, 2005; Ruel, 2006).15,11
Seperti daerah Asia Tenggara dan
Indonesia lainnya, prevalensi carrier
thalassemia-α, thalassemia-β dan Hb-E cukup
tinggi memungkinkan terjadinya kasus thalassemia mayor cukup besar akibat
kombinasi antara sesama carrier thalassemia-α
atau dengan carrier thalassemia-β maupun
carrier Hb-E (Weatherall and Clegg, 2001).5
Kombinasi pada kasus di atas dapat menghasilkan bayi thalassemia mayor, dengan manifestasi klinis dapat dari ringan sampai
berat (Bunn and Forget, 1986; Bowie et al.,
1997; )16,17
Seperti negara berkembang lainnya, managemen klinis penyakit thalassemia di Indonesia belum memadai, sehingga penderita biasanya meninggal pada usia anak-anak dan jarang yang mencapai usia dewasa. Oleh karena itu tindakan preventif mutlak dilakukan sesuai dengan anjuran WHO (1994) untuk mengurangi insidensi thalassemia dan hemoglobinopati. Artinya dari hasil penelitian
ini yang menunjukkan prevalensi carrier
penyakit tersebut > 3%, merupakan alasan yang kuat untuk melakukan tindakan preventif di kota Medan baik melalui konseling genetik pranikah maupun prenatal diagnosis.
KESIMPULAN
1. Prevalensi carrier thalassemia α dan
thalassemia-β pada populasi Medan
cukup tinggi masing-masing 3,35% dan 4,07%.
2. Pembawa sifat thalassemia α pada etnik
Cina di Medan mencapai 6,72%
3. Hasil penelitian dapat dilakukan sebagai
acuan untuk melakukan usaha preventif untuk mengurangi insidensi penyakit thalassemia baik melalui Konseling Genetik Pranikah maupun Prenatal Diagnosis.
DAFTAR PUSTAKA
1. Cavalli-Sforza LL, Menozzi P and Piazza
A (1994). The History and Geography of
Human Genes. Princeton University Press. Princeton. New Jersey. 60-121.
2. Lie Injoe L E (1959). Phatological
Haemoglobin in Indonesia. In Abnormal Haemoglobins (eds. JHP Jonxis & JF. Delafresnaye) Blackwell Scientific Publication, Oxford. UK. p 210-216.
3. Flatz G (1967) Hemoglobin-E:
Distribution and Population Dynamics.
Hum. Genet. 3: 189-234.
4. Luzatto L (1079). Genetics of red cells
and susceptibility to malaria. Blood
54:961-976.
5. Weatherall DJ and Clegg JB (2001) The
Thalassemia Syndromes, 4th
eds. Blackwell Scientific Publ. Oxford. 422-439.
6. Wong, HB. Thalassemia as community
health in Southeast Asia. Naskah Lengkap
Kongres National PHDTI. Yogyakarta 24-26 September 1983.
7. Lanni F., Sofro ASM, Ismadi M, Marzuki
S (2004). ISVI-5 (GÆC): The most
Commom β-thalassemia mutation found
in the Island of Sumatera. Indonesian
Journal of Biotechnology 6: 571-577.
8. Yang TY, Yang XY and Chen WC (1985)
Thalassemia in China. Ann N.Y. Acad.
Sci 445: 92-97.
9. Flint J, Harding R, Clegg JB and Boyce A
(1993). Why are some genetic diseases so common? Distinguishing selection from other process by molecular analysis of
globin gene variants. Hum Genet.
91:91-117.
10. Weiss, KM (1993). Genetic Variation and
Human Disease. Cambridge University Press. UK.
11. Ruel MD (2006) Using race in clinical
research to develop tailored medications. Is the FDA encouraging discrimination or eliminating traditional disparities in health
care for African-Americans? J. Leg Med
27: 225-241.
12. WHO (1994) Guidelines for the control
of haemoglobin disorders report of the VIth Annual Meeting of the WHO Working Group on Haemoglobinopathies, Cagliari, Sardinia, 8-9 April 1989, World Health Organization, GenevaBowie LJ, Reddy PL and Beck KR (1997). Alpha thalassemia and its impact on other
clinical conditions. Clinics in Laboratory
Medicine. 17 (1) :97-108.
13. Li, AMC, Lee, FT and Tood D (1982)
The screening of Chinese blood cord
blood for hemoglobinopathies. Hum
Hered 32: 62-65.
14. Fucharoon S and Winichagoon P (1987)
Hemoglobinopathies in Southeast Asia: molecular biology and clinical medicine.
Hemoglobin 11:65-69.
15. Wadman M (2005) Drug targeting: is race
enough? Nature 435:1008-1009.
16. Bunn HF and Forget BG (1986)