The
GALE
ENCYCLOPEDIA
of
Genetic
The
GALE
ENCYCLOPEDIA
of
Genetic
Disorders
S T A C E Y L . B L A C H F O R D , E D I T O R V O L U M E
M-Z
APPENDIX GLOSSARYINDEX
The
GALE
ENCYCLOPEDIA
of
GENETIC DISORDERS
STAFF
Stacey L. Blachford,Associate Editor
Christine B. Jeryan,Managing Editor Melissa C. McDade,Associate Editor Ellen Thackery,Associate Editor
Mark Springer,Technical Training Specialist Andrea Lopeman,Programmer/Analyst
Barbara Yarrow,Manager, Imaging and Multimedia Content
Robyn Young,Project Manager, Imaging and Multimedia Content
Randy Bassett,Imaging Supervisor Robert Duncan,Senior Imaging Specialist
Pamela A. Reed,Coordinator, Imaging and Multimedia Content
Maria Franklin,Permissions Manager Ryan Thomason,Permissions Associate Lori Hines,Permissions Assistant
Kenn Zorn,Product Manager
Michelle DiMercurio,Senior Art Director
Mary Beth Trimper,Manager, Composition and Electronic Prepress
Evi Seoud,Assistant Manager, Composition Purchasing and Electronic Prepress
Dorothy Maki,Manufacturing Manager
Ronald D. Montgomery,Manager, Data Capture Gwendolyn S. Tucker,Project Administrator Beverly Jendrowski,Data Capture Specialist
Indexing provided by:Synapse. Illustrations created by:
Argosy, West Newton, Massachusetts
Electronic Illustrators Group, Morgan Hill, California
Since this page cannot legibly accommodate all copyright notices, the acknowledgments constitute an extension of the copyright notice.
While every effort has been made to ensure the reliability of the infor-mation presented in this publication, the Gale Group neither guarantees the accuracy of the data contained herein nor assumes any responsibil-ity for errors, omissions or discrepancies. The Gale Group accepts no payment for listing, and inclusion in the publication of any organiza-tion, agency, instituorganiza-tion, publicaorganiza-tion, service, or individual does not imply endorsement of the editors or publisher. Errors brought to the attention of the publisher and verified to the satisfaction of the publisher will be corrected in future editions.
This book is printed on recycled paper that meets Environmental Protection Agency standards.
The paper used in this publication meets the minimum requirements of American National Standard for Information Sciences-Permanence Paper for Printed Library Materials, ANSI Z39.48-1984.
This publication is a creative work fully protected by all applicable copyright laws, as well as by misappropriation, trade secret, unfair com-petition, and other applicable laws. The authors and editors of this work have added value to the underlying factual material herein through one or more of the following: unique and original selection, coordination, expression, arrangement, and classification of the information.
Gale Group and design is a trademark used herein under license.
All rights to this publication will be vigorously defended.
Copyright © 2002 Gale Group 27500 Drake Road
Farmington Hills, MI 48331-3535
All rights reserved including the right of reproduction in whole or in part in any form.
ISBN 0-7876-5612-7 (set) 0-7876-5613-5 (Vol. 1) 0-7876-5614-3 (Vol. 2)
Printed in the United States of America 10 9 8 7 6 5 4 3 2 1
Library of Congress Cataloging-in-Publication Data
The Gale encyclopedia of genetic disorders / Stacey L. Blachford, associate editor.
p. cm.
Includes bibliographical references and index.
Summary: Presents nearly four hundred articles describing genetic disorders, conditions, tests, and treatments, including high-profile diseases such as Alzheimer’s, breast cancer, and heart disease.
ISBN 0-7876-5612-7 (set : hardcover : alk.paper
1. Genetic disorders—Encyclopedias, Juvenile. [1. Genetic disorders—Encyclopedias. 2. Diseases—Encyclopedias.] I. Blachford, Stacey.
RB155.5 .G35 2001 616’.042’03—dc21
Machado-Joseph disease see
Azorean
disease
I
Macular degeneration—
age-related
Definition
Macular degeneration age-related (AMD) is one of the most common causes of vision loss among adults over age 55 living in developed countries. It is caused by the breakdown of the macula, a small spot located in the back of the eye. The macula allows people to see objects directly in front of them (called central vision), as well as fine visual details. People with AMD usually have blurred central vision, difficulty seeing details and colors, and they may notice distortion of straight lines.
Description
In order to understand how the macula normally functions and how it is affected by AMD, it is important to first understand how the eye works. The eye is made up of many different types of cells and tissues that all work together to send images from the environment to the brain, similar to the way a camera records images. When light enters the eye, it passes through the lens and lands on the retina, which is a very thin tissue that lines the inside of the eye. The retina is actually made up of 10 different layers of specialized cells, which allow the retina to function similarly to film in a camera, by record-ing images. The macula is a small, yellow-pigmented area located at the back of the eye, in the central part of the retina. The retina contains many specialized cells called photoreceptors that sense light coming into the eye and convert it into electrical messages that are then sent to the brain through the optic nerve. This allows the brain to “see” the environment.
The retina contains two types of photoreceptor cells: rod cells and cone cells. The rod cells are located prima-rily outside of the macula and they allow for peripheral (side) and night vision. Most of the photoreceptor cells inside of the macula, however, are the cone cells, which are responsible for perceiving color and for viewing objects directly in front of the eye (central vision). If the macula is diseased, as in AMD, color vision and central vision are altered. There are actually two different types of AMD: Dry AMD and Wet AMD.
Dry AMD
Approximately 90% of individuals with AMD have dry AMD. This condition is sometimes referred to as nonexudative, atrophic, or drusenoid macular degenera-tion. In this form of AMD, some of the layers of retinal cells (called retinal pigment epithelium, or RPE cells) near the macula begin to degenerate, or breakdown. These RPE cells normally help remove waste products from the cone and rod cells. When the RPE cells are no longer able to provide this “clean-up” function, fatty deposits called drusen begin to accumulate, enlarge and increase in number underneath the macula. The drusen formation can disrupt the cones and rods in the macula, causing them to degenerate or die (atrophy). This usually leads to central and color vision problems for people with dry AMD. However, some people with drusen deposits have minimal or no vision loss, and although they may never develop AMD, they should have regular eye exam-inations to check for this possibility. Dry AMD is some-times called “nonexudative”, because even though fatty drusen deposits form in the eye, people do not have leak-age of blood or other fluid (often called exudate) in the eye. In some cases, dry AMD symptoms remain stable or worsen slowly. In addition, approximately 10% of people with dry AMD eventually develop wet AMD.
Wet AMD
Around 10% of patients with AMD have wet AMD. This form of AMD is also called subretinal
ization, choroidal neovascularization, exudative form or disciform degeneration. Wet AMD is caused by leakage of fluid and the formation of abnormal blood vessels (called “neovascularization”) in a thin tissue layer of the eye called the choroid. The choroid is located underneath the retina and the macula, and it normally supplies them with nutrients and oxygen. When new, delicate blood vessels form, blood and fluid can leak underneath the macula, causing vision loss and distortion as the macula is pushed away from nearby retinal cells. Eventually a scar (called a disciform scar) can develop underneath the macula, resulting in severe and irreversible vision loss.
Genetic profile
AMD is considered to be a complex disorder, likely caused by a combination of genetic and environmental factors. This type of disorder is caused by multifactorial
inheritance, which means that many factors likely inter-act with one another and cause the condition to occur. As implied by the words “age-related”, the aging process is one of the strongest risk factors for developing AMD. A number of studies have suggested that genetic suscepti-bility also plays an important role in the development of AMD, and it has been estimated that the brothers and sis-ters of people with AMD are four times more likely to also develop AMD, compared to other individuals.
Genetic factors
Determining the role that genetic factors play in the development of AMD is a complicated task for scientists. Since AMD is not diagnosed until late in life, it is diffi-cult to locate and study large numbers of affected people in the same family. In addition, although AMD seems to run in families, there is no clear inheritance pattern (such as dominant or recessive) observed when examin-ing families. However, many studies have supported the observation that inheritance plays some role in the devel-opment of AMD.
One method scientists use to locate genes that may increase a person’s chance to develop multifactorial ditions like AMD is to study genes that cause similar con-ditions. In 1997, this approach helped researchers identify changes (mutations) in the ATP-binding cassette transporter, retina-specific (ABCR) genein people diag-nosed with AMD. The process began after genetic research identified changes in the ABCR gene among people with an autosomal recessive macular disease called Stargardt macular dystrophy. This condition is phenotypically similar to AMD, which means that people with Stargardt macular dystrophy and AMD have similar symptoms, such as yellow deposits in the retina and decreased central vision.
Macular degeneration—age-related
KEY TERMS
Central vision—The ability to see objects located directly in front of the eye. Central vision is neces-sary for reading and other activities that require people to focus on objects directly in front of them.
Choroid—A vascular membrane that covers the
back of the eye between the retina and the sclera and serves to nourish the retina and absorb scat-tered light.
Drusen—Fatty deposits that can accumulate
underneath the retina and macula, and sometimes lead to age-related macular degeneration (AMD). Drusen formation can disrupt the photoreceptor cells, which causes central and color vision prob-lems for people with dry AMD.
Genetic heterogeneity—The occurrence of the
same or similar disease, caused by different genes among different families.
Macula—A small spot located in the back of the eye that provides central vision and allows people to see colors and fine visual details.
Multifactorial inheritance—A type of inheritance pattern where many factors, both genetic and environmental, contribute to the cause.
Optic nerve—A bundle of nerve fibers that carries visual messages from the retina in the form of elec-trical signals to the brain.
Peripheral vision—The ability to see objects that are not located directly in front of the eye. Peripheral vision allows people to see objects located on the side or edge of their field of vision.
Photoreceptors—Specialized cells lining the
innermost layer of the eye that convert light into electrical messages so that the brain can perceive the environment. There are two types of photore-ceptor cells: rod cells and cone cells. The rod cells allow for peripheral and night vision. Cone cells are responsible for perceiving color and for central vision.
Retina—The light-sensitive layer of tissue in the back of the eye that receives and transmits visual signals to the brain through the optic nerve.
The ABCR gene maps to chromosome 1p22, and people who have Stargardt macular dystrophy have muta-tions in each of their two alleles (gene copies). However, the researchers who found mutations in the ABCR gene among people with AMD located only one allele with a mutation, which likely created an increased susceptibility to AMD. They concluded that people with an ABCR
gene mutation in one allele could have an increased chance to develop AMD during their lifetime if they also had inherited other susceptibility genes, and/or had con-tact with environmental risk factors. Other scientists tried to repeat this type of genetic research among people with AMD in 1999, and were not able to confirm that the ABCR gene is a strong genetic risk factor for this condi-tion. However, it is possible that the differing research results may have been caused by different research meth-ods, and further studies will be necessary to understand the importance of ABCR gene mutations in the develop-ment of susceptibility to AMD.
In 1998, another genetic researcher reported a fam-ily in which a unique form of AMD was passed from one generation to the next. Although most families with AMD who are studied do not show an obvious inheri-tance pattern in their family tree, this particular family’s pedigree showed an apparently autosomal dominant form of AMD. Autosomal dominant refers to a specific type of inheritance in which only one copy of a person’s gene pair (i.e. one allele) needs to have a mutation in order for it to cause the disease. An affected person with an auto-somal dominant condition thus has one allele with a mutation and one allele that functions properly. There is a 50% chance for this individual to pass on the allele with the mutation, and a 50% chance to pass on the working allele, to each of his or her children.
Genetic testingdone on the family reported in 1998 showed that the dominant gene causing AMD in affected family members was likely located on chromosome 1q25-q31. Although the gene linked to AMD in this fam-ily and the ABCR gene are both on chromosome 1, they are located in different regions of the chromosome. This indicates that there is genetic heterogeneity among dif-ferent families with AMD, meaning that difdif-ferent genes can lead to the same or similar disease among different families. It is also possible that although one particular gene may be the main cause of susceptibility for AMD, other genes and/or environmental factors may help alter the age of onset of symptoms or types of physical changes seen by examining the eye. Some studies have shown that other medical conditions or certain physical characteristics may be associated with an increased risk for AMD. Some of these include:
• Heart disease • High blood pressure
• Cataracts
• Farsightedness
• Light skin and eye color
However, not all studies have found a strong rela-tionship between these factors and AMD. Further research is needed to decipher the role that both genetic and environmental factors play in the development of this complex condition.
Environmental factors
Determining the role that environmental factors play in the development of AMD is an important goal for researchers. Unlike genetic factors that cannot be con-trolled, people can often find motivation to change their behaviors if they are informed about environmental risk factors that may be within their control. Unfortunately, identifying environmental factors that clearly increase (or decrease) the risk for AMD is a challenging task. Several potential risk factors have been studied. These include:
• Smoking
• High fat/high cholesterol diet
• Ultraviolet (UV) exposure (sunlight)
• Low levels of dietary antioxidant vitamins and minerals
Although research has identified these possible risk factors, many of the studies have not consistently shown strong associations between these factors and the devel-opment of AMD. This makes it difficult to know the true significance of any of these risk factors. One exception, however, is the relationship between smoking and AMD. As of 1999, at least seven studies consistently found that smoking is strongly associated with AMD. This is one more important reason for people to avoid and/or quit smoking, especially if they have a family history of AMD. Further research is needed to clarify the signifi-cance of the factors listed above so people may be informed about lifestyle changes that may help decrease their risk for AMD.
Demographics
Among adults aged 55 and older, AMD is the lead-ing cause of vision loss in developed countries. The chance to develop AMD increases with age, and although it usually affects adults during their sixth and seventh decades of life, it has been seen in some people in their forties. It is estimated that among people living in devel-oped countries, approximately one in 2,000 are affected by AMD. By age 75, approximately 30% of people have early or mild forms of AMD, and roughly 7% have an advanced form of AMD. Since the number of people in the United States aged 65 years or older will likely
ble between 1999 and 2024, the number of people affected also should increase. Although AMD occurs in both sexes, it is slightly more common in women.
The number of people affected with AMD is differ-ent in various parts of the world and it varies between dif-ferent ethnic groups. Some studies suggest that AMD is more common in Caucasians than in African Americans; however, other reports suggest the numbers of people affected in these two groups are similar. Some studies of AMD among Japanese and other Asian ethnic groups have shown an increasing number of affected individuals. Further studies are needed to examine how often AMD occurs in other ethnic groups as well.
Signs and symptoms
During eye examinations, eye care specialists may notice physical changes in the retina and macula that make them suspect the diagnosis of AMD. However, affected individuals may notice:
• Decreased visual acuity (ability to see details) of both up-close and distant objects
• Blurred central vision
• Decreased color vision
• Distorted view of lines and shapes
• A blind spot in the visual field
The majority of people with AMD maintain their peripheral vision. The severity of symptoms depends
upon whether a person has dry or wet AMD. In addition, the degree of vision loss and physical symptoms that can be seen by an eye exam change over time. For example, people with dry AMD usually develop vision loss very slowly over a period of many years. Their vision may change very little from one year to the next, and they usu-ally do not lose central vision completely. However, indi-viduals with wet AMD usually have symptoms that worsen more quickly and they have a greater risk to develop severe central vision loss, sometimes in as little as a two-month period. Since people diagnosed with dry AMD may go on to develop wet AMD, it is important for them to take note of any changes in their symptoms and to report them to their eye care specialist.
The physical symptoms of AMD eventually impact people emotionally. One study published in 1998 reported that people with advanced stages of AMD feel they have a significantly decreased quality of life. In addition, they may have a limited ability to perform basic daily activities due to poor vision, and as a result, they often suffer psychological distress. Hopefully, improved treatment and management will eventually change this trend for affected individuals in the future.
Diagnosis
Eye care specialists use a variety of tests and exam-ination techniques to determine if a person has AMD. Some of these include:
• Acuity testing—Involves testing vision by determining a person’s ability to read letters or symbols of various sizes on an “eye chart” from a precise distance away with specific lighting present.
• Color testing—Assesses the ability of the cone cells to recognize colors by using special pictures made up of dots of colors that are arranged in specific patterns.
• Amsler grid testing—Involves the use of a grid printed on a piece of paper that helps determine the health of the macula, by allowing people to notice whether they have decreased central vision, distorted vision, or blind spots.
• Fluorescein angiography—Involves the use of a fluo-rescent dye, injected into the bloodstream, in order to look closely at the blood supply and blood vessels near the macula. The dye allows the eye specialist to exam-ine and photograph the retina and macula to check for signs of wet AMD (i.e. abnormal blood vessel forma-tion or blood leakage).
As of 2001, there are no genetic tests readily avail-able to help diagnose AMD. Genetic research in the com-ing years will hopefully help scientists determine the genetic basis of AMD. This could help diagnose people
Macular degeneration—age-related
A retinal photograph showing macular degeneration.
with increased susceptibility before they have symptoms, so they may benefit from early diagnosis, management and/or treatment. This knowledge may also allow people who are at a genetically increased risk for AMD to avoid environmental risk factors and thus preserve or prolong healthy vision.
Treatment and management
Treatment
There is no universal treatment available to cure either wet or dry forms of AMD. However, some people with wet AMD can benefit from laser photocoagulation therapy. This treatment involves the use of light rays from a laser to destroy the abnormal blood vessels that form beneath the retina and macula and prevent further leak-age of blood and fluid. Previously lost vision cannot be restored with this treatment, and the laser can unfortu-nately damage healthy tissue as well, causing further loss of vision.
In April 2000, the FDA approved the use of a light-activated drug called Visudyne to help treat people with wet AMD. Visudyne is a medication that is injected into the bloodstream, and it specifically attaches to the abnor-mal blood vessels present under the macula in people with AMD. When light rays from a laser land on the blood vessels, the Visudyne is activated and can destroy the abnormal vessels, while causing very little damage to nearby healthy tissues. Although long term studies are needed to determine the safety and usefulness of this medication beyond two years, early reports find it an effective way to reduce further vision loss.
Researchers have been trying to identify useful treat-ments for dry AMD as well. Laser photocoagulation treatments are not effective for dry AMD since people with this form do not have abnormal blood or fluid leak-age. Although many drugs have been tested, most have not improved visual acuity. However, one study pub-lished in October 2000, reported that people with dry AMD who received a medication called Iloprost over a six-month period noted improvements in visual acuity, daily living activities and overall quality of life. Follow-up studies will be needed to determine how safe and use-ful this medication will be over time.
Management
Although no treatments can cure AMD, a number of special devices can help people make the most of their remaining vision. Some of these include:
• Walking canes
• Guide dogs • Audiotapes
• Magnifying lenses
• Telescopes
• Specialized prisms
• Large print books
• Reading machines
• Computer programs that talk or enlarge printed infor-mation
People with AMD may also find it useful to meet with low-vision specialists who can help them adapt to new lifestyle changes that may assist with daily living. Eye care specialists can help people locate low-vision specialists. There are also a number of nationwide and international support groups available that provide edu-cation and support for individuals and families affected by AMD.
Prognosis
People can live many years with AMD, although the physical symptoms and emotional side effects often change over time. The vision problems caused by dry AMD typically worsen slowly over a period of years, and people often retain the ability to read. However, for peo-ple who develop wet AMD, the chance to suddenly develop severe loss of central vision is much greater. Regular monitoring of vision by people with AMD (using an Amsler grid) and by their eye care specialists, may allow for early treatment of leaky blood vessels, therefore reducing the chance for severe vision loss. As physical symptoms worsen, people are more likely to suffer emo-tionally due to decreasing quality of life and independ-ence. However, many low-vision devices and various support groups can often provide much needed assistance to help maintain and/or improve quality of life.
Resources
BOOKS
D’Amato, Robert, and Joan Snyder. Macular Degeneration: The Latest Scientific Discoveries and Treatments for
Preserving Your Sight.New York: Walker & Co., 2000.
Solomon, Yale, and Jonathan D. Solomon. Overcoming Macular Degeneration: A Guide to Seeing Beyond the
Clouds.New York: Morrow/Avon, 2000.
PERIODICALS
Bressler, Neil M., and James P. Gills. “Age related macular degeneration.” British Medical Journal 321, no. 7274 (December 2000): 1425–1427.
Fong, Donald S. “Age-Related Macular Degeneration: Update for Primary Care.”American Family Physician61, no. 10 (May 2000): 3035–3042.
“Macular degeneration.”Harvard Women’s Health Watch6, no. 2 (October 1998): 2–3.
“Researchers set sights on vision disease.” Harvard Health
Letter23, no.10 (August 1998):4–5.
“Self-test for macular degeneration.” Consumer Reports on
Health12, no.12 (December 2000): 2.
ORGANIZATIONS
AMD Alliance International. PO Box 550385, Atlanta, GA 30355. (877) 263-7171. ⬍http://www.amdalliance.org⬎. American Macular Degeneration Foundation. PO Box 515,
Northampton, MA 01061-0515. (413) 268-7660. ⬍http://www.macular.org⬎.
Foundation Fighting Blindness Executive Plaza 1, Suite 800, 11350 McCormick Rd., Hunt Valley, MD 21031. (888) 394-3937. ⬍http://www.blindness.org⬎.
Macular Degeneration Foundation. PO Box 9752, San Jose, CA 95157. (888) 633-3937. ⬍http://www.eyesight.org⬎. Retina International. Ausstellungsstrasse 36, Zürich, CH-8005.
Switzerland (⫹41 1 444 10 77). ⬍ http://www.retina-international.org⬎.
Pamela J. Nutting, MS, CGC
Madelung deformity see
Leri-Weill
dyschondrosteosis
Maffuci disease see
Chondrosarcoma
I
Major histocompatibility
complex
Definition
In humans, the proteins coded by the genes of the major histocompatibility complex (MHC) include human leukocyte antigens (HLA), as well as other proteins. HLA proteins are present on the surface of most of the body’s cells and are important in helping the immune system distinguish ‘self’ from ‘non-self’.
Description
The function and importance of MHC is best under-stood in the context of a basic understanding of the func-tion of the immune system. The immune system is responsible for distinguishing ‘self’ from ‘non-self’, pri-marily with the goal of eliminating foreign organisms and other invaders that can result in disease. There are several levels of defense characterized by the various stages and types of immune response.
Natural immunity
When a foreign organism enters the body, it is encountered by the components of the body’s natural
immunity. Natural immunity is the non-specific first-line of defense carried out by phagocytes, natural killer cells, and components of the complement system. Phagocytes are specialized white blood cells capable of engulfing and killing an organism. Natural killer cells are also spe-cialized white blood cells that respond to cancer cells and certain viral infections. The complement system is a group of proteins called the class III MHC that attack antigens. Antigens consist of any molecule capable of triggering an immune response. Although this list is not exhaustive, antigens can be derived from toxins, protein, carbohydrates, DNA, or other molecules from viruses, bacteria, cellular parasites, or cancer cells.
Acquired immunity
The natural immune response will hold an infection at bay as the next line of defense mobilizes through acquired, or specific immunity. This specialized type of immunity is usually needed to eliminate an infection and is dependent on the role of the proteins of the major his-tocompatibility complex. There are two types of acquired immunity. Humoral immunity is important in fighting infections outside the body’s cells, such as those caused by bacteria and certain viruses. Other types of viruses and parasites that invade the cells are better fought by cellular immunity. The major players in acquired immu-nity are the antigen-presenting cells (APCs), B-cells, their secreted antibodies, and the T-cells. Their functions are described in detail below.
Humoral immunity
In humoral immunity, antigen-presenting cells, including some B-cells, engulf and break down foreign organisms. Antigens from these foreign organisms are then brought to the outside surface of the antigen-pre-senting cells and presented in conjunction with class II MHC proteins. The helper T-cells recognize the antigen presented in this way and release cytokines,proteins that signal B-cells to take further action. B-cells are special-ized white blood cells that mature in the bone marrow. Through the process of maturation, each B-cell develops the ability to recognize and respond to a specific antigen. Helper T-cells aid in stimulating the few B-cells that can recognize a particular foreign antigen. B-cells that are stimulated in this way develop into plasma cells,which secrete antibodies specific to the recognized antigen. Antibodies are proteins that are present in the circulation, as well as being bound to the surface of B-cells. They can destroy the foreign organism from which the antigen came. Destruction occurs either directly, or by ‘tagging’ the organism, which will then be more easily recognized and targeted by phagocytes and complement proteins. Some of the stimulated B-cells go on to become memory
cells,which are able to mount an even faster response if the antigen is encountered a second time.
Cellular immunity
Another type of acquired immunity involves killer T-cells and is termed celluar immunity. T-T-cells go through a process of maturation in the organ called the thymus, in which T-cells that recognize ‘self’ antigens are elimi-nated. Each remaining T-cell has the ability to recognize a single, specific, ‘non-self’ antigen that the body may encounter. Although the names are similar, killer T-cells are unlike the non-specific natural killer cells in that they are specific in their action. Some viruses and parasites quickly invade the body’s cells, where they are ‘hidden’ from antibodies. Small pieces of proteins from these invading viruses or parasites are presented on the surface of infected cells in conjunction with class I MHC pro-teins, which are present on the surface of most all of the body’s cells. Killer T-cells can recognize antigen bound to class I MHC in this way, and they are prompted to release chemicals that act directly to kill the infected cell. There is also a role for helper T-cells and antigen-pre-senting cells in cellular immunity. Helper T-cells release cytokines, as in the humoral response, and the cytokines stimulate killer T-cells to multiply. Antigen-presenting cells carry foreign antigen to places in the body where additional killer T-cells can be alerted and recruited.
The major histocompatibility complex clearly per-forms an important role in functioning of the immune system. Related to this role in disease immunity, MHC is important in organ and tissue transplantation, as well as playing a role in susceptibility to certain diseases. HLA typing can also provide important information in parent-age, forensic, and anthropologic studies. These various roles and the practical applications of HLA typing are discussed in greater detail below.
Genetic profile
Present on chromosome 6, the major histocompati-bility complex consists of more than 70 genes, classified into class I, II, and III MHC. There are multiple alleles, or forms, of each HLA gene. These alleles are expressed as proteins on the surface of various cells in a co-domi-nant manner. This diversity is important in maintaining an effective system of specific immunity. Altogether, the MHC genes span a region that is four million base pairs in length. Although this is a large region, 99% of the time these closely-linked genes are transmitted to the next generation as a unit of MHC alleles on each chromosome 6. This unit is called a haplotype.
Class I
Class I MHC genes include HLA-A, HLA-B, and HLA-C. Class I MHC are expressed on the surface of
almost all cells. They are important for displaying antigen from viruses or parasites to killer T-cells in cellular immu-nity. Class I MHC is also particularly important in organ and tissue rejection following transplantation. In addition to the portion of class I MHC coded by the genes on chro-mosome 6, each class I MHC protein also contains a small, non-variable protein component called beta-2 microglobu-lin coded by a gene on chromosome 15. Class I HLA genes are highly polymorphic, meaning there are multiple forms, or alleles, of each gene. There are at least 57 HLA-A alleles, 111 HLHLA-A-B alleles, and 34 HLHLA-A-C alleles.
Class II
Class II MHC genes include HLA-DP, HLA-DQ, and HLA-DR. Class II MHC are particularly important in humoral immunity. They present foreign antigen to helper T-cells, which stimulate B-cells to elicit an anti-body response. Class II MHC is only present on antigen presenting cells, including phagocytes and B-cells. Like class I MHC, there are hundreds of alleles that make up the class II HLA gene pool.
Class III
Class III MHC genes include the complement sys-tem (i.e. C2, C4a, C4b, Bf). Complement proteins help to activate and maintain the inflammatory process of an immune response.
Demographics
There is significant variability of the frequencies of HLA alleles among ethnic groups. This is reflected in anthropologic studies attempting to use HLA-types to determine patterns of migration and evolutionary rela-tionships of peoples of various ethnicity. Ethnic variation is also reflected in studies of HLA-associated diseases. Generally speaking, populations that have been subject to significant patterns of migration and assimilation with other populations tend to have a more diverse HLA gene pool. For example, it is unlikely that two unrelated indi-viduals of African ancestry would have matched HLA types. Conversely, populations that have been isolated due to geography, cultural practices, and other historical influences may display a less diverse pool of HLA types, making it more likely for two unrelated individuals to be HLA-matched.
Testing
Organ and tissue transplantation
There is a role for HLA typing of individuals in var-ious settings. Most commonly, HLA typing is used to establish if an organ or tissue donor is appropriately matched to the recipient for key HLA types, so as not to
elicit a rejection reaction in which the recipient’s immune system attacks the donor tissue. In the special case of bone marrow transplantation, the risk is for graft-versus-host disease (GVHD), as opposed to tissue rejection. Because the bone marrow contains the cells of the immune system, the recipient effectively receives the donor’s immune system. If the donor immune system recognizes the recipient’s tissues as foreign, it may begin to attack, causing the inflammation and other complica-tions of GVHD. As advances occur in transplantation medicine, HLA typing for transplantation occurs with increasing frequency and in various settings.
Disease susceptibility
There is an established relationship between the
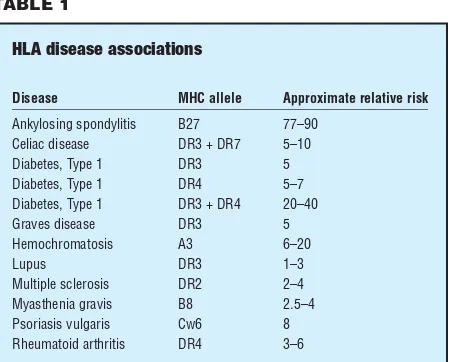
inheritance of certain HLA types and susceptibility to specific diseases. Most commonly, these are diseases that are thought to be autoimmune in nature. Autoimmune diseases are those characterized by inflammatory reac-tions that occur as a result of the immune system mistak-enly attacking ‘self’ tissues. The basis of the HLA association is not well understood, although there are some hypotheses. Most autoimmune diseases are charac-terized by the expression of class II MHC on cells of the body that do not normally express these proteins. This may confuse the killer T-cells, which respond inappropri-ately by attacking these cells. Molecular mimicry is another hypothesis. Certain HLA types may ‘look like’ antigen from foreign organisms. If an individual is infected by such a foreign virus or bacteria, the immune system mounts a response against the invader. However, there may be a ‘cross-reaction’ with cells displaying the HLA type that is mistaken for foreign antigen. Whatever the underlying mechanism, certain HLA-types are known factors that increase the relative risk for developing spe-cific autoimmune diseases. For example, individuals who carry the HLA B-27 allele have a relative risk of 77–90 for developing ankylosing spondylitis—meaning such an individual has a 77- to 90-fold chance of developing this form of spinal and pelvic arthritis, as compared to some-one in the general population. Selected associations are listed below, together with the approximate correspon-ding relative risk of disease.
In addition to autoimmune disease, HLA-type less commonly plays a role in susceptibility to other diseases, including cancer, certain infectious diseases, and meta-bolic diseases. Conversely, some HLA-types confer a protective advantage for certain types of infectious ease. In addition, there are rare immune deficiency dis-eases that result from inherited mutations of the genes of components of the major histocompatibility complex.
Parentage
Among other tests, HLA typing can sometimes be used to determine parentage, most commonly pater-nity, of a child. This type of testing is not generally done for medical reasons, but rather for social or legal reasons.
Forensics
HLA-typing can provide valuable DNA-based evi-dence contributing to the determination of identity in criminal cases. This technology has been used in domes-tic criminal trials. Additionally, it is a technology that has been applied internationally in the human-rights arena. For example, HLA-typing had an application in Argentina following a military dictatorship that ended in 1983. The period under the dictatorship was marked by the murder and disappearance of thousands who were known or suspected of opposing the regime’s practices. Children of the disappeared were often ‘adopted’ by mil-itary officials and others. HLA-typing was one tool used to determine non-parentage and return children to their biological families.
Anthropologic studies
HLA-typing has proved to be an invaluable tool in the study of the evolutionary origins of human popula-tions. This information, in turn, contributes to an
under-Major histocompatibility complex
HLA disease associations
Disease MHC allele Approximate relative risk
Ankylosing spondylitis B27 77–90 Celiac disease DR3 + DR7 5–10
Diabetes, Type 1 DR3 5
Diabetes, Type 1 DR4 5–7
Diabetes, Type 1 DR3 + DR4 20–40
Graves disease DR3 5
Hemochromatosis A3 6–20
Lupus DR3 1–3
Multiple sclerosis DR2 2–4 Myasthenia gravis B8 2.5–4 Psoriasis vulgaris Cw6 8 Rheumatoid arthritis DR4 3–6
The relative risks indicated in this table refer to the increased chance of a patient with an MHC allele to develop a disorder as compared to an individual without one. For example, a patient with DR4 is three to six times more likely to have rheumatoid arthritis and five to seven times more likely to develop type 1 diabetes than an individual without the DR4 allele.
standing of cultural and linguistic relationships and prac-tices among and within various ethnic groups.
Resources
BOOKS
Abbas, A.K., et al. Cellular and Molecular Immunology.
Philadelphia: W.B. Saunders, 1991.
Doherty, D.G., and G.T. Nepom. “The human major histocom-patibility complex and disease susceptibility.” In Emery and Rimoin’s Principles and Practice of Medical
Genetics.3rd ed. Ed. D.L. Rimoin, J.M. Connor, and R.E.
Pyeritz, 479–504. New York: Churchill Livingston, 1997. Jorde L.B., et al. “Immunogenetics.” In Medical Genetics.2nd
ed. St. Louis: Moseby, 1999. PERIODICALS
Diamond, J.M. “Abducted orphans identified by grandpaternity testing.”Nature327 (1987): 552–53.
Svejgaard, A., et al. “Associations between HLA and disease with notes on additional associations between a ‘new’ immunogenetic marker and rheumatoid arthritis.” HLA and Disease—The Molecular Basis. Alfred Benzon
Symposium.40 (1997): 301–13.
Trachtenberg, E.A., and H.A. Erlich. “DNA-based HLA typing for cord blood stem cell transplantation.” Journal of
Hematotherapy5 (1996): 295–300.
WEBSITES
“Biology of the immune system.” The Merck Manual
⬍http://www.merck.com/pubs/mmanual_home/sec16/176 .htm⬎.
Jennifer Denise Bojanowski, MS, CGC
Male turner syndrome see
Noonan
syndrome
Malignant fever see
Malignant
hyperthermia
Malignant hyperpyrexia see
Malignant
hyperthermia
I
Malignant hyperthermia
Definition
Malignant hyperthermia (MH) is a condition that causes a number of physical changes to occur among genetically susceptible individuals when they are exposed to a particular muscle relaxant or certain types of medications used for anesthesia. The changes may include increased rate of breathing, increased heart rate, muscle stiffness, and significantly increased body
tem-perature (i.e. hyperthermia). Although MH can usually be treated successfully, it sometimes leads to long-term physical illness or death. Research has identified a num-ber of genetic regions that may be linked to an increased MH susceptibility.
Description
Unusual response to anesthesia was first reported in a medical journal during the early 1960s, when physi-cians described a young man in need of urgent surgery for a serious injury. He was very nervous about exposure to anesthesia, since he had 10 close relatives who died dur-ing or just after surgeries that required anesthesia. The patient himself became very ill and developed a high tem-perature after he was given anesthesia. During the next decade, more cases of similar reactions to anesthesia were reported, and specialists began using the term
malignant hyperthermiato describe the newly recognized
condition. The word hyperthermia was used because peo-ple with this condition often rapidly develop a very high body temperature. The word malignant referred to the fact that the majority (70–80%) of affected individuals died. The high death rate in the 1960s occurred because the underlying cause of the condition was not understood, nor was there any known treatment (other than basically trying to cool the person’s body with ice).
Increased awareness of malignant hyperthermia and scientific research during the following decades improved medical professionals’ knowledge about what causes the condition, how it affects people, and how it should be treated. MH can be thought of as a chain reac-tion that is triggered when a person with MH susceptibil-ity is exposed to specific drugs commonly used for anesthesia and muscle relaxation.
Triggering drugs that may lead to malignant hyper-thermia include:
• halothane
• enflurane
• isoflurane
• sevoflurane
• desflurane
• methoxyflurane
• ether
• succinylcholine
Once an MH susceptible person is exposed to one or more of these anesthesia drugs, they can present with a variety of signs. One of the first clues that a person is sus-ceptible to MH is often seen when they are given a
cle relaxant called succinyl choline. This drug generally causes some stiffness in the masseter (jaw) muscles in most people. However, individuals with MH susceptibil-ity can develop a much more severe form of jaw stiffness called masseter spasmwhen they receive this drug. They may develop muscle stiffness in other parts of their bod-ies as well. When exposed to any of the trigger drugs listed above (inhalants for anesthesia), people with MH susceptibility can develop an increased rate of metabo-lism in the cells of their body, resulting in rapid breath-ing, rapid heartbeat, high body temperature (over 110°F), muscle stiffness, and muscle breakdown. If these signs are not recognized, treated, or able to be controlled, brain damage or death can occur due to internal bleeding, heart failure, or failure other organs.
The series of events that occur after exposure to trig-ger drugs is activated by an abnormally high amount of calcium inside muscle cells. This is due to changes in the chemical reactions that control muscle contraction and the production of energy. Calcium is normally stored in an area called the sarcoplasmic reticulum, which is a sys-tem of tiny tubes located inside muscle cells. This syssys-tem of tubes allows muscles to contract (by releasing cal-cium) and to relax (by storing calcal-cium) in muscle cells. Calcium also plays an important role in the production of energy inside cells (i.e. metabolism). There are at least three important proteins located in (or nearby) the sar-coplasmic reticulum that control how much calcium is released into muscle cells and thus help muscles contract. One of these proteins is a “calcium release channel” pro-tein that has been named the ryanodine receptor propro-tein, or RYR. This protein (as well as the genethat tells the body how to make it) has been an important area of research. For some reason, when people with MH sus-ceptibility are exposed to a trigger drug, they can develop very high levels of calcium in their muscle cells. The trig-ger drugs presumably stimulate the proteins that control the release of calcium, causing them to create very high levels of calcium in muscle cells. This abnormally high calcium level then leads to increased metabolism, muscle stiffness, and the other symptoms of MH.
The amount of time that passes between the expo-sure to trigger drugs and the appearance of the first symp-toms of MH varies between different people. Sympsymp-toms begin within 10 minutes for some individuals, although several hours may pass before symptoms appear in oth-ers. This means that some people do not show signs of MH until they have left the operating room and are recov-ering from surgery. In addition, some individuals who inherit MH susceptibility may be exposed to trigger drugs numerous times during multiple surgeries without any complications. However, they still have an increased risk to develop an MH episode during future exposures.
Malignant hyperthermia
KEY TERMS
Anesthesia—Lack of normal sensation (especially to pain) brought on by medications just prior to surgery or other medical procedures.
Genetic heterogeneity—The occurrence of the
same or similar disease, caused by different genes among different families.
Hyperthermia—Body temperature that is much
higher than normal (i.e. higher than 98.6°F).
Masseter spasm—Stiffening of the jaw muscles.
Often one of the first symptoms of malignant hyperthermia susceptibility that occurs after expo-sure to a trigger drug.
Metabolism—The total combination of all of the chemical processes that occur within cells and tis-sues of a living body.
Sarcoplasmic reticulum—A system of tiny tubes
located inside muscle cells that allow muscles to contract and relax by alternatively releasing and storing calcium.
Trigger drugs—Specific drugs used for muscle
relaxation and anesthesia that can trigger an episode of malignant hyperthermia in a suscepti-ble person. The trigger drugs include halothane, enflurane, isoflurane, sevoflurane, desflurane, methoxyflurane, ether, and succinylcholine.
This means that people who have an increased risk for MH susceptibility due to their family history cannot pre-sume they are not at risk simply because they previously had successful surgeries. Although MH was frequently a fatal condition in the past, a drug called dantrolene sodium became available in 1979, which greatly decreased the rate of both death and disability.
Genetic profile
Susceptibility to MH is generally considered to be inherited as an autosomal dominant trait. “Autosomal” means that males and females are equally likely to be affected. “Dominant” refers to a specific type of
have a child who is also susceptible is 50% for each preg-nancy. The same parent would also have a 50% chance to have a non-susceptible child with each pregnancy.
It is not unusual for people to not know they inher-ited a genetic change that causes MH susceptibility. This is because they typically do not show symptoms unless they are exposed to a specific muscle relaxant or certain anesthetics, which may not be needed by every person during his or her lifetime. In addition, people who inherit MH susceptibility do not always develop a reaction to trigger drugs, which means their susceptibility may not be recognized even if they do have one or more surgeries. Once MH susceptibility is diagnosed in an individual, however, it is important for his or her family members to know they also have a risk for MH susceptibility, since it is a dominant condition. This means that anyone with a family member who has MH susceptibility should tell their doctor about their family history. Since MH may go unrecognized, it is important that anyone who has had a close relative die from anesthesia notify the anesthesiol-ogist before any type of surgery is planned. People with a family history of MH susceptibility may choose to meet with a genetic counselor to discuss the significance of their family history as well. In addition, relatives of an affected person may consider having a test to see if they also inherited MH susceptibility.
Although there are many people who have the same symptoms of MH when exposed to trigger drugs, genetic research has shown that there are probably many genes, located on different chromosomes, that can all lead to MH susceptibility. This indicates that there is genetic het-erogeneity among different families with MH suscepti-bility, meaning that different genes can lead to the same or similar disease among different families. As of March 2001, researchers identified six different types of MH susceptibility. Although specific genes have been discov-ered for some of these types, others have been linked only to specific chromosomal regions.
Genetic classification of malignant hyperthermia:
• MHS1—Located on chromosome 19q13.1. Specific gene called RYR1. Gene creates the RYR protein.
• MHS2—Located on chromosome 17q11.2-24. Suspected gene called SCN4A.
• MHS3—Located on chromosome 7q21-22. Suspected gene called CACNA2DI. Gene creates part of the DHPR protein called the alpha 2/delta subunit.
• MHS4—Located on chromosome 3q13.1. Specific gene and protein unknown.
• MHS5—Located on chromosome 1q32. Specific gene called CACNA1S. Gene creates part of the DHPR pro-tein called the alpha 1 subunit.
• MHS6—Located on chromosome 5p. Specific gene and protein unknown.
Over half of all families with MH susceptibility are believed to have MHS1 (i.e. have changes in the RYR1 gene), while the rest have MHS2, MHS3, MHS4, MHS5, or MHS6. However, as of January 2000, only 20% of all families tested had specific genetic changes identified in the RYR1 gene. This is because there are many different types of genetic changes in the gene that can all lead to MH susceptibility, and many families have changes that are unique. As a result,genetic testingof the RYR1 gene is complicated, time consuming, and often cannot locate all possible genetic changes. In addition, genetic testing for families may become more complex as knowledge about MH grows. This issue was discussed in an article published by researchers in July 2000. The authors explained that although MH susceptibility has typically been described as an autosomal dominant trait caused by a single gene that is passed from one generation to the next, they believe MH susceptibility may actually depend upon various genetic changes that occur in more than one gene. Further research may clarify this issue in the future.
While specific genes have been identified for some of the MH susceptibility types (i.e. RYR1 and DHPR alpha 1 subunit), not all changes in these genes lead specifically to MH susceptibility. For example, although at least 20 different genetic changes have been identified in the RYR1 gene that can lead to MH susceptibility, some people who have certain types of these changes actually have a different genetic condition that affects the muscles called central core disease(CCD). Infants with this autosomal dominant condition typically have very poor muscle tone (i.e. muscle tension) as well as an increased susceptibility to MH. Among families who have CCD, there are some individuals who do not have the typical muscle changes, but have MH susceptibility instead. Hopefully, future research will help scientists understand why the same genetic change in the RYR1 gene can cause different symptoms among people belonging to the same family.
Demographics
The exact number of individuals who are born with a genetic change that causes MH susceptibility is not known. Until genetic research and genetic testing improves, this number will likely remain unclear. However, it is estimated that internationally one in 50,000 people who are exposed to anesthesia develop an MH reaction. Among children, it is estimated that one in 5,000 to one in 15,000 develop MH symptoms when exposed to anesthesia. MH has been seen in many coun-tries, although there are some geographic areas where it
occurs more often in the local populations, including parts of Wisconsin, North Carolina, Austria, and Quebec.
Signs and symptoms
Although the specific symptoms of malignant hyper-thermia can vary, the most common findings include:
• stiffness/spasms of jaw muscles and other muscles
• rapid breathing, causing decreased oxygen and increased carbon dioxide in the blood
• rapid or irregular heartbeat
• high body temperature (over 110°F)
• muscle breakdown (may cause dark or cola-colored urine)
• internal bleeding, kidney failure, brain damage, or death (if not treated successfully)
Diagnosis
The diagnosis of MH susceptibility can be made before or during a reaction to a triggering drug. Ideally, the diagnosis is made before a susceptible individual is exposed and/or develops a reaction. This is possible for people who learn they have an increased chance for MH because they have a relative with MH susceptibility. Testing these individuals requires a surgical procedure called a muscle biopsy, in which a piece of muscle tis-sue is removed from the body (usually from the thigh). Safe (i.e. non-triggering) anesthetics are used during the procedure. The muscle is taken to a laboratory and is exposed to halothane (a triggering anesthetic) and caf-feine, both of which cause any muscle tissue to contract, or tighten. Thus the test is called the caffeine halothane contracture test (CHCT). Muscle tissue taken from indi-viduals with MH susceptibility is more sensitive to caf-feine and halothane, causing it to contract more strongly than normal muscle tissue from non-susceptible people. This type of test is a very accurate way to predict whether a person has MH susceptibility or not. However, the test does require surgery, time to recover (typically three days), and it is expensive (approxi-mately $2,500). In the United States, many insurance companies will pay for the testing if it is needed. Although the test is not available in every state or coun-try, there are at least 40 medical centers worldwide that can perform the test.
Unfortunately, not all MH susceptible people will learn from their family histories that they have an increased risk for MH before they are exposed to a trig-ger drug. For these individuals, the diagnosis of MH sus-ceptibility is often made during surgery by the anesthesiologist (a physician specializing in anesthesia)
who is providing the anesthesia medications. Other health care specialists also may notice symptoms of MH during or after surgery. Symptoms such as rapid breath-ing, rapid heart rate, and high body temperature can usu-ally be detected with various machines or devices that examine basic body functions during surgery. Muscle stiffness of the jaw, arms, legs, stomach and chest may be noticed as well. These symptoms may happen during sur-gery or even several hours later. If the diagnosis is made during or after surgery, immediate treatment is needed to prevent damage to various parts of the body or death. If a person has a suspicious reaction to anesthesia, he or she may undergo a muscle biopsy to confirm MH suscepti-bility at a later date.
In spite of the fact that a number of important genes and genetic regions associated with MH susceptibility have been identified, testing a person’s DNA for all of the possible changes that may cause this condition is not easily done for affected individuals and their families. As of March 2001, existing genetic testing identifies some changes that have been seen among families with MHS1 and MHS6. Research studies may provide information for families with MHS2, MHS3, MHS4, and MHS5 as well. Sometimes the testing requires DNA from only one affected person, but in other cases, many samples are needed from a variety of family members. However, until genetic technology improves, the contracture test that is done on muscle tissue will likely remain the “gold stan-dard” for diagnosis of MH susceptibility.
Treatment and management
The early identification of an MH episode allows for immediate treatment with an “antidote” called dantrolene sodium. This medication prevents the release of calcium from the sarcoplasmic reticulum, which decreases mus-cle stiffness and energy production in the cells. If hyper-thermia develops, the person’s body can be cooled with ice. In addition, the anesthesiologist will change the anesthetic from a trigger drug to a non-trigger drug. Immediate treatment is necessary to prevent serious ill-ness and/or death.
Once a person with definite or suspected MH suscep-tibility is diagnosed (by an MH episode, muscle biopsy, or family history), prevention of an MH episode is possible. There are many types of non-triggering anesthetic drugs and muscle relaxants that can be used during surgical pro-cedures. The important first step in this process is for peo-ple with known or suspected MH susceptibility to talk with their doctors before any surgery, so that only non-triggering drugs are used. People with definite or sus-pected MH susceptibility should always carry some form of medical identification that describes their diagnosis in
case emergency surgery is needed. The Malignant Hyperthermia Association of the United States provides wallet-sized emergency medical ID cards for its members.
Prognosis
Early diagnosis and treatment of MH episodes with dantrolene sodium has dramatically improved the prog-nosis for people who develop MH during or just after surgery. When the condition was first recognized in the 1960s, no real treatment (other than cooling the per-son’s body) was available, and only 20–30% of people who developed MH survived. When the antidote (dantrolene sodium) became available in 1979, the sur-vival rate increased to 70–80%. However, 5–10% of people who develop MH after exposure to a trigger drug still may die even with proper medication and care. Among those who do survive, some are disabled due to kidney, muscle, or brain damage. The best prognosis exists for people with definite or suspected MH suscep-tibility who are able to prevent exposures to trigger drugs by discussing their history with their doctors. Improved genetic testing in the future may help identify most or all people with inherited MH susceptibility, so they too may prevent exposures that could trigger MH episodes.
Resources
BOOKS
Hopkins, Philip M., and F. Richard Ellis, eds. Hyperthermic and Hypermetabolic Disorders: Exertional Heat Stroke,
Malignant Hyperthermia and Related Syndromes. Port
Chester, NY: Cambridge University Press, 1996.
Morio, Michio, Haruhiko Kikuchi, and O. Yuge, eds. Malignant Hyperthermia: Proceedings of the 3rd International
Symposium on Malignant Hyperthermia, 1994.Secaucus,
NJ: Springer-Verlag, 1996.
Ohnishi, S. Tsuyoshi, and Tomoko Ohnishi, eds. Malignant
Hyperthermia: A Genetic Membrane Disease.Boca Raton,
FL: CRC Press, 1994. PERIODICALS
Denborough, Michael. “Malignant hyperthermia.”The Lancet
352, no. 9134 (October 1998): 1131–36.
Hopkins, P.M. “Malignant Hyperthermia: Advances in clinical management and diagnosis.”British Journal of Anesthesia
85, no. 1 (2000): 118–28.
Jurkat-Rott, Karin, Tommie McCarthy, and Frank Lehmann-Horn. “Genetics and Pathogenesis of Malignant Hyper-thermia.”Muscle & Nerve23 (January 2000): 4–17. ORGANIZATIONS
Malignant Hyperthermia Association of the United States. PO Box 1069, 39 East State St., Sherburne, NY 13460. (800) 98-MHAUS. ⬍http://www.mhaus.org⬎.
WEBSITES
Larach, Marilyn Green, MD, FAAP. “Making anesthesia safer: Unraveling the malignant hyperthermia puzzle.” Feder-ation of American Societies for Experimental Biology
(FASEB).⬍http://www.faseb.org/opar/mh/⬎.
“Malignant hyperthermia.” UCLA Department of
Anesthesi-ology.⬍http://www.anes.ucla.edu/dept/mh.html⬎.
Pamela J. Nutting, MS, CGC
Manic-depressive psychosis see
Bipolar
disorder
I
Mannosidosis
Definition
Mannosidosis is a rare inherited disorder, an inborn error of metabolism, that occurs when the body is unable to break down chains of a certain sugar (mannose) prop-erly. As a result, large amounts of sugar-rich compounds build up in the body cells, tissues, and urine, interfering with normal body functions and development of the skeleton.
Description
Mannosidosis develops in patients whose genes are unable to make an enzyme required by lysosomes (struc-tures within the cell where proteins, sugars, and fats are broken down and then released back into the cell to make other molecules). Lysosomes need the enzyme to break down, or degrade, long chains of sugars. When the enzyme is missing and the sugar chains are not broken down, the sugars build up in the lysosomes. The lyso-somes swell and increase in number, damaging the cell. The result is mannosidosis.
The enzyme has two forms: alpha and beta. Similarly, the disorder mannosidosis has two forms: alpha-mannosidosis (which occurs when the alpha form of the enzyme is missing) and beta-mannosidosis (which occurs when the beta form of the enzyme is missing). Production of each form of the enzyme is controlled by a different gene.
First described in 1967, alpha-mannosidosis is clas-sified further into two types. Infantile (or Type I) alpha-mannosidosis is a severe disorder that results in mental retardation, physical deformities, and death in childhood. Adult (or Type II) alpha-mannosidosis is a milder disor-der in which mental retardation and physical deformities develop much more slowly throughout the childhood and teenage years.
Beta-mannosidosis was identified nearly 20 years later in 1986. Patients with this form of the disorder are also mentally retarded but over a wide range of severity, from mild to extreme. Beta-mannosidosis is not well understood, in part because it is such a rare disease. It was discovered only because researchers searched for it: a deficiency of the beta form of the enzyme was known to cause disease in animals.
Genetic profile
The two forms of mannosidosis, alpha and beta, are caused by changes on two different genes. Mutations in the gene MANB, on chromosome 19, result in alpha-mannosidosis. This gene is also known as MAN2B1 or LAMAN. Defects in MANB cause alpha-mannosidosis in both infants and adults.
Beta-mannosidosis is caused by mutations in the gene MANB1 (also called MANBA). This gene is on chromosome 4.
Both genes, MANB and MANB1, are inherited as autosomal recessive traits. This means that if a man and woman each carry one defective gene, then 25% of their children are expected to be born with the disorder. Each gene is inherited separately from the other.
Demographics
Mannosidosis is a rare disorder, occurring in both men and women. The disorder does not affect any partic-ular ethnic group but rather appears in a broad range of people. Alpha-mannosidosis has been studied in Scandinavian, Western and Eastern European, North American, Arabian, African, and Japanese populations. Researchers have identified beta-mannosidosis in European, Hindu, Turkish, Czechoslovakian, Jamaican-Irish, and African families.
Signs and symptoms
The various forms and types of mannosidosis all have one symptom in common: mental retardation. Other signs and symptoms vary.
Infants with alpha-mannosidosis appear normal at birth, but by the end of their first year, they show signs of mental retardation, which rapidly gets worse. They develop a group of symptoms that includes dwarfism, shortened fingers, and facial changes. In these children, the bridge of the nose is flat, they have a prominent fore-head, their ears are large and low set, they have protrud-ing eyebrows, and the jaw juts out. Other symptoms include lack of muscle coordination, enlarged spleen and liver, recurring infections, and cloudiness in the back of the eyeball, which is normally clear. These patients often
Mannosidosis
KEY TERMS
Autosomal recessive—A pattern of genetic inheri-tance where two abnormal genes are needed to display the trait or disease.
Enzyme—A protein that catalyzes a biochemical
reaction or change without changing its own structure or function.
Lysosomal storage disease—A category of disor-ders that includes mannosidosis.
Lysosome—Membrane-enclosed compartment in
cells, containing many hydrolytic enzymes; where large molecules and cellular components are bro-ken down.
Mannose—A type of sugar that forms long chains in the body.
Mutation—A permanent change in the genetic
material that may alter a trait or characteristic of an individual, or manifest as disease, and can be transmitted to offspring.
have empty bubbles in their white blood cells, a sign that sugars are being stored improperly.
The adult form occurs in 10–15% of the cases of alpha-mannosidosis. The symptoms in adults are the same as in infants, but they are milder and develop more slowly. Patients with adult alpha-mannosidosis are often normal as babies and young children, when they develop mentally and physically as expected. In their childhood or teenage years, however, mental retardation and physi-cal symptoms become evident. These patients may also lose their hearing and have pain in their joints.
Beta-mannosidosis is characterized by symptoms that range from mild to severe. In all patients, however, the most frequent signs are mental retardation, lung infections, and hearing loss with speech difficulties. In mild cases, patients have red, wart-like spots on their skin. In severe cases, patients may have multiple seizures, and their arms and legs may be paralyzed. Because the symptoms of beta-mannosidosis vary so greatly, researchers suggest that the disorder may fre-quently be misdiagnosed.
Diagnosis
If doctors suspect that a pregnant woman may be carrying a child with mannosidosis, they can test cells in the fluid surrounding the baby for enzyme activity.
Treatment and management
There is no known treatment for mannosidosis. The symptoms—mental retardation and skeletal abnormali-ties—are managed by supportive care, depending on the severity. Patients with adult alpha-mannosidosis and beta-mannosidosis may show mild mental retardation or behavior problems (such as depression or aggression) and may be mainstreamed into society. Others may require institutionalization. Skeletal abnormalities may require surgery to correct them, and recurring infections are treated with antibiotics.
Research with animals suggests that mannosidosis can be treated by placing healthy cells without defective genes into the animals’ bones (bone marrow transplant). Other researchers have successfully treated mannosidosis in animals by inserting healthy genes into the unborn off-spring of a pregnant animal. These treatments have not been proven on humans, however.
Prognosis
The future for patients with mannosidosis varies with the form of their disorder. For infants with alpha-mannosidosis, death is expected between ages three and 12 years. For infants with beta-mannosidosis, death will come earlier, by the time they are 15 months old.
Patients with mild forms of alpha- and beta-man-nosidosis often survive into adulthood, but their lives are complicated by mental retardation and physical deterio-ration. They will generally die in their early or middle years, depending on the severity of their disorder.
Resources
BOOKS
Thomas, George. “Disorders of Glycoprotein Degradation: Alpha-Mannosidosis, Beta-Mannosidosis, Fucosidosis, and Sialidosis.” In The Metabolic and Molecular Bases of
Inherited Disease.Scriver, Charles R., et al., ed. Vol. II,
8th ed. New York: McGraw-Hill, 2001. PERIODICALS
Alkhayat, Aisha H., et al. “Human Beta-Mannosidase cDNA Characterization and First Identification of a Mutation Associated with Human Beta-Mannosidosis.” Human
Molecular Genetics7, no. 1 (1998): 75–83.
Berg, Thomas, et al. “Spectrum of Mutations in Alpha-Mannosidosis.”American Journal of Human Genetics64 (1999): 77–88.
Michalski, Jean-Claude, and Andre Klein. “Glycoprotein Lysosomal Storage Disorders: Alpha- and
Beta-Mannosidosis, Glucosidosis, and Alpha-N-Acetylgalacto-saminidase Deficiency.”Biochimica et Biophysica Acta:
Molecular Basis of Disease 1455, no. 2–3 (October 8,
1999): 69–84. ORGANIZATIONS
Arc (a National Organization on Mental Retardation). 1010 Wayne Ave., Suite 650, Silver Spring, MD 20910. (800) 433-5255. ⬍http://www.thearclink.org⬎.
Children Living with Inherited Metabolic Diseases. The Quadrangle, Crewe Hall, Weston Rd., Crewe, Cheshire, CW1-6UR. UK 127 025 0221. Fax: 0870-7700-327. ⬍http://www.climb.org.uk⬎.
International Society for Mannosidosis and Related Diseases. 3210 Batavia Ave., Baltimore, MD 21214. (410) 254-4903. ⬍http://www.mannosidosis.org⬎.
National MPS Society. 102 Aspen Dr., Downingtown, PA 19335. (610) 942-0100. Fax: (610) 942-7188. info @mpssociety.org. ⬍http://www.mpssociety.org⬎. WEBSITES
Web Site for Rare Genetic Diseases in Children: Lysosomal Storage Diseases. ⬍http://mcrcr2.med.nyu.edu/murphp01/ lysosome/lysosome.htm⬎.
Linnea E. Wahl, MS
I
Marfan syndrome
Definition
Marfan syndrome is an inherited disorder of the con-nective tissue that causes abnormalities of the patient’s eyes, cardiovascular system, and musculoskeletal system. It is named for the French pediatrician, Antoine Marfan (1858-1942), who first described it in 1896. Marfan syn-drome is sometimes called arachnodactyly, which means “spider-like fingers” in Greek, since one of the character-istic signs of the disease is disproportionately long fin-gers and toes. It is estimated that one person in every 3,000-5,000 has Marfan syndrome, or about 50,000 peo-ple in the United States. Marfan syndrome is one of the more common inheritable disorders.
Description
Marfan syndrome affects three major organ systems of the body: the heart and circulatory system, the bones and muscles, and the eyes. The genetic mutation respon-sible for Marfan was discovered in 1991. It affects the body’s production of fibrillin, which is a protein that is an important part of connective tissue. Fibrillin is the pri-mary component of the microfibrils that allow tissues to stretch repeatedly without weakening. Because the