VALIDASI METODE SPEKTROFOTOMETRI VISIBEL UNTUK
PENETAPAN KADAR AMOKSISILIN MENGGUNAKAN
PEREAKSI ASETILASETON DAN FORMALIN
SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi
oleh: Margareta Sunarto
NIM : 038114004
FAKULTAS FARMASI UNIVERSITAS SANATA DHARMA
God didn’t promise day without pain,
Laughter without sorrow, sun without rain…
But He promises strength for the day,
Comfort for the tears, and light for the way.
Disappointments are like road humps, they slow you.
Down a bit, but you’ll enjoy the smooth road afterwords.
Don’t stay on the humps too long, move on!
When you feel down, because you didn’t get what you want,
Just sit tight and be happy.
Because God must has something better to be given to you.
When something happens to you,
Good or bad, consider what it means.
There’s always a purpose in life’s events,
To teach you how to laugh more,
Or not to cry too hard.
Kupersembahkan karya ini untuk Papa dan Mama Sebagai rasa terima kasih dan baktiku
KATA PENGANTAR
Syukur dan terima kasih kepada Bapa di surga, Tuhan Yesus, dan Bunda Maria atas berkat dan penyertaan-Nya, penulis dapat menyelesaikan penelitian yang berjudul ” Validasi Metode Spektrofotometri Visibel Untuk Penetapan Kadar Amoksisilin Menggunakan Pereaksi Asetilaseton dan Formalin”. Skripsi ini disusun untuk memenuhi salah satu syarat dalam mencapai gelar Sarjana Farmasi (S.Farm.) pada Fakultas Farmasi Universitas Sanata Dharma.
Pada kesempatan ini, penulis ingin menyampaikan terima kasih yang tulus kepada:
1. Rita Suhadi, M.Si., Apt. selaku Dekan Fakultas Farmasi Universitas Sanata Dharma.
2. Prof. Dr. Sudibyo Martono, M.S., Apt. selaku dosen pembimbing. Terima kasih untuk masukan, bimbingan, dorongan, waktu, pengertian dan perhatian yang begitu besar, serta semangat yang selalu diberikan selama penelitian dan penyusunan skripsi ini
3. Dra. A. Nora Iska Harnita, M.Si., Apt. selaku dosen penguji, terima kasih atas dukungan, saran, dan waktu yang diberikan.
4. Drs. Sulasmono, Apt. selaku dosen penguji, terima kasih atas dukungan saran, dan waktu yang diberikan.
6. Papa, Mama, Andre, Ita, buat semua doa, cinta, dukungan, perhatian, pengertian, kesabaran, canda, dan tawa yang buat Cici selalu kuat. Makasih banyak ya.. I love you all...
7. Pak Bambang dan Bu Kis, laboratorium analisis obat dan makanan Fakultas Farmasi Universitas Gadjah Mada, yang selalu menemani dan membantu selama penelitian. Terima kasih banyak ya Pak, Bu…
8. Segenap laboran di Universitas Gadjah Mada dan Universitas Sanata Dharma. Terima kasih untuk waktu dan bantuannya.
9. Arnie, Eta, teman seperjuanganku. Inget slogan kita: gagal itu biasa, tetapi berhasil itu luar biasa… Makasih banyak teman buat dukungan, bantuan, dan kerjasamanya.
10.Mas Isun, kakak dan sahabatku, terima kasih untuk semua keceriaan, kesedihan, semangat, harapan, kekecewaan, dan semua hal yang pernah aku alami dengan adanya persahabatan kita. Hope our friendship will last forever. Aku belajar banyak hal dari persahabatan kita.. Overall, thanks for eveything.. I Love you brother..
11.Temen-temen Eternal Choir dan Koor Gregorius Caecilia buat semua kebersamaan, kegilaan, kekompakkan, keceriaan, dan pengertiannya. Thanks a lot..
12.Gurit buat editan dan ilmu-ilmu komputernya, Leli buat terjemahannya. Makasih ya..
14.Temen-temen kelompok A dan temen-temen Kelas A 2003, buat dukungan, kekompakan dan kebersamaan kita selama ini.
15.Mas Bowo, Mas Fahrul, buat diskusinya.
16.Semua pihak yang tidak dapat saya sebutkan satu per satu yang telah membantu dalam penelitian dan penyusunan skripsi ini.
Penulis menyadari bahwa skripsi ini sangat sederhana dan masih banyak kekurangannya. Oleh karena itu, saran serta kritik yang membangun sangat diharapkan untuk kesempurnaan skripsi ini di masa yang akan datang.
Yogyakarta, Januari 2007
INTISARI
Amoksisilin memiliki kemiripan struktur dengan sefaleksin yang juga memiliki gugus amin primer. Oleh karena itu, metode spektrofotometri visibel untuk penetapan kadar sefaleksin menggunakan pereaksi asetilaseton dan formalin diharapkan dapat juga digunakan untuk penetapan kadar amoksisilin.
Penelitian ini merupakan jenis penelitian non eksperimental dengan rancangan penelitian deskriptif. Pada penelitian ini dilakukan optimasi waktu reaksi, pH, dan volume yang menghasilkan serapan maksimum. Hasilnya kemudian digunakan dalam validasi metode. Selain itu, dilakukan pula aplikasi metode penetapan kadar tersebut pada sediaan tablet amoksisilin.
Hasil penelitian menunjukkan bahwa reaksi mulai stabil setelah menit ke-50 selama 30 menit, volume optimum pereaksi adalah 7 ml dan pH optimum adalah 4. Warna kuning yang terbentuk memberikan serapan maksimum pada panjang gelombang 401 nm. Untuk validasi metode, didapat data sebagai berikut: koefisien variansi sebesar 0,56%, perolehan kembali sebesar 104,09%, dan koefisien korelasi (r) persamaan garis linier kurva baku sebesar 0,9995. Aplikasi metode penetapan kadar pada sediaan amoksisilin menunjukkan hasil yang baik dengan kadar rata-rata amoksisilin dalam tablet adalah 589,56 mg. Dari seluruh data yang diperoleh, dapat disimpulkan bahwa metode penetapan kadar amoksisilin secara spektrofotometri visibel menggunakan pereaksi asetilaseton dan formalin memiliki akurasi dan presisi yang baik, namun akurasinya kurang baik.
ABSTRACT
The structure of amoxicillin is similar to that of cephalexin which is also has primary amine groups. For that reason, it is hoped that visible spectrophotometric method in determining the amount of cephalexin by using acetylacetone and formalin can also be used to determine the amount of amoxicillin.
This research is a non-experimental descriptive research. In this research, the reaction time, the pH and the volume of the reagent have been optimized to obtain the maximum absorption. Then, its result is used in the validation method. In addition, the method’s developed is applied to determine the amount of amoxicillin in the tablets.
The research result shows that the reaction begin to stable from the fiftieth minutes for 30 minutes, the optimal volume of the reagent is 7 ml, and the optimal pH is 4. The yellow chromophore is scanned and showed 401 nm as a maximum wavelength. From the validation method, the coefficient of variation is 0.56%, the recovery is 104.09%, and the correlation coefficient (r) is 0.9995. The application of the method’s developed in determining amoxicillin in tablet showed a good result with the average amount of amoxicillin is 589.56 mg/tablet. From the result it can be concluded that visible spectrophotometric method to determine the amount of amoxicillin by using acethylacetone and formalin gives a good precision and linearity, but not the accuracy.
DAFTAR ISI
HALAMAN JUDUL ... i
HALAMAN PERSETUJUAN PEMBIMBING ... ii
HALAMAN PENGESAHAN... iii
HALAMAN PERSEMBAHAN ... iv
KATA PENGANTAR ... v
PERNYATAAN KEASLIAN KARYA ... viii
INTISARI... ix
ABSTRACT ... x
DAFTAR ISI ... xi
DAFTAR TABEL ... xiv
DAFTAR GAMBAR ... xv
DAFTAR LAMPIRAN ... xvi
BAB I PENGANTAR ... 1
A. Latar Belakang ... 1
1. Perumusan masalah ... 3
2. Keaslian penelitian ... 4
3. Manfaat penelitian ... 5
B. Tujuan Penelitian ... 5
BAB II PENELAAHAN PUSTAKA ... 6
A. Amoksisilin ... 6
C. Formalin ... 9
D. Spektrofotometri UV-Vis ... 10
1. Definisi spektrofotometri UV-Vis ... 10
2. Konsep dasar radiasi elektromagnetik ... 10
3. Tipe transisi elektron ... 11
4. Interaksi molekul dengan radiasi elektromagnetik ... 13
5. Analisis kuantitatif secara spektrofotometri UV-Vis... 13
6. Serapan suatu larutan ... 14
7. Kesalahan fotometrik ... 15
8. Syarat-syarat penggunaan Hukum Beer... 16
9. Penggunaan spektrofotometri UV-Vis dalam metode analisis ... 18
E. Pengembangan Metode Spektrofotometri... 18
F. Validasi, Kesalahan, dan Parameter Metode Analisis ... 20
1. Validasi metode analisis ... 20
2. Kesalahan metode analisis ... 22
3. Parameter-parameter validasi metode analisis ... 23
G. Landasan Teori ... 24
H. Hipotesis ... 24
BAB III METODOLOGI PENELITIAN ... 25
A. Jenis dan Rancangan Penelitian ... 25
B. Definisi Operasional ... 25
C. Alat-alat Penelitian ... 25
E. Tata Cara Penelitian ... 26
1. Pembuatan larutan uji ... 26
2. Optimasi penetapan kadar amoksisilin ... 27
3. Pembuatan kurva baku ... 28
4. Aplikasi metode penetapan kadar amoksisilin pada tablet AM . 29 5. Validasi metode... 30
F. Analisis Hasil ... 30
BAB IV HASIL DAN PEMBAHASAN ... 31
A. Pembuatan Larutan Baku Amoksisilin ... 31
B. Penetapan Waktu Reaksi dan Operating Time ... 31
C. Penetapan pH Optimum Pereaksi ... 36
D. Penetapan Volume Optimum Pereaksi ... 38
E. Penetapan Panjang Gelombang Serapan Maksimum ... 39
F. Pembuatan Kurva Baku ... 41
G. Penetapan Kadar Amoksisilin dalam Tablet ... 44
H. Validasi Metode Analisis ... 45
BAB V KESIMPULAN DAN SARAN ... 48
A. Kesimpulan ... 48
B. Saran ... 48
DAFTAR PUSTAKA ... 49
LAMPIRAN ... 52
DAFTAR TABEL
Halaman
Tabel I. Parameter Analisis yang Diperlukan Untuk Kesahihan Pengukuran 23
Tabel II. Hasil Penetapan Waktu Reaksi ... 35
Tabel III. Hasil Penetapan pH Optimum Pereaksi ... 38
Tabel IV. Hasil Penetapan Volume Optimum Pereaksi ... 39
Tabel V. Hasil Penetapan Kurva Baku Amoksisilin... 42
Tabel VI. Hasil Modifikasi Kurva Baku Amoksisilin... 43
Tabel VII. Hasil Penetapan Kadar Amoksisilin dalam Tablet AM ... 44
DAFTAR GAMBAR
Halaman
Gambar 1. Struktur Sefaleksin ... 2
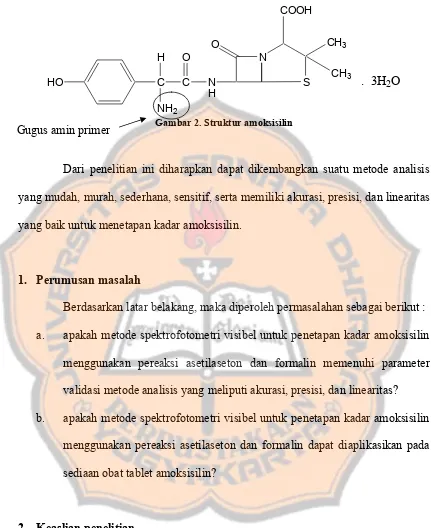
Gambar 2. Struktur Amoksisilin ... 3
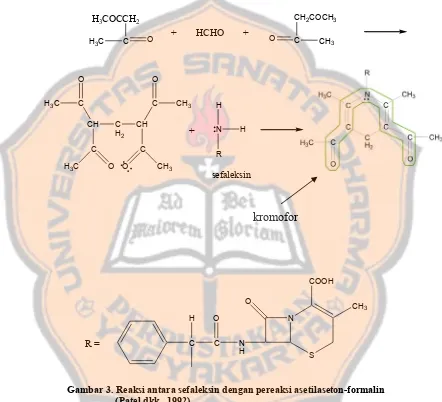
Gambar 3. Reaksi Antara Sefaleksin dengan Pereaksi Asetilaseton dan Formalin ... 8
Gambar 4. Struktur Asetilaseton ... 9
Gambar 5. Struktur Formalin ... 10
Gambar 6. Diagram Tingkat Energi Elektronik ... 12
Gambar 7. Usulan Mekanisme Reaksi Pembuatan Pereaksi Asetilaseton-Formalin ... 33
Gambar 8. Usulan Mekanisme Reaksi Antara Pereaksi Asetilaseton-Formalin dan Amoksisilin ... 35
Gambar 9. Hasil Penetapan Operating Time... 36
Gambar 10. Reaksi Eliminasi Pada Reaksi Antara Amoksisilin dengan Asetilaseton dan Formalin Pada Suasana Asam dan Basa ... 37
Gambar 11. Spektra Panjang Gelombang Serapan Maksimum Amoksisilin Konsentrasi 0,084 mg/ml (a), 0,117 mg/ml (b), dan 0,151 mg/ml (c) Hasil Reaksi dengan Asetilaseton dan Formalin ... 40
Gambar 12. Gugus pada senyawa hasil reaksi yang memberikan serapan pada panjang gelombang 335 nm ... 41
DAFTAR LAMPIRAN
Halaman
Lampiran 1. Data Penimbangan Baku Amoksisilin ... 52
Lampiran 2. Data Penetapan Kadar Sampel ... 53
Lampiran 3. Data Hasil Penetapan Perolehan kembali ... 54
Lampiran 4. Contoh Perhitungan... 55
BAB I
PENGANTAR
A. Latar Belakang
Saat ini, penggunaan antibiotik sebagai sarana pengobatan infeksi semakin berkembang dalam masyarakat terutama pada pengobatan penyakit yang disebabkan oleh mikroorganisme. Banyaknya penyakit yang ditimbulkan oleh mikroorganisme menyebabkan antibiotik menduduki peringkat yang tinggi dalam peresepan. Antibiotik merupakan suatu produk metabolik (zat kimia) yang dihasilkan oleh mikroorganisme tertentu, yang dalam jumlah amat kecil bersifat merusak atau menghambat mikroorganisme lain (Pelczar and Chan, 1988).
Agar dapat berefek maksimal, suatu obat harus memiliki dosis yang tepat. Oleh karena itu, harus dilakukan pengawasan untuk menjamin mutu, khasiat, dan keamanan penggunaan obat tersebut. Salah satu caranya adalah dengan melakukan penetapan kadar zat aktif untuk menjamin ketepatan dosis yang akan diterima oleh konsumen.
Amoksisilin dapat ditetapkan kadarnya dengan metode kromatografi cair kinerja tinggi (KCKT) dan metode titrasi iodometri (Anonim, 1995). Kedua metode tersebut memiliki kelebihan dan kekurangan. Metode KCKT memberikan hasil yang cepat karena tidak memerlukan pemisahan terlebih dahulu, tetapi operasionalnya mahal, sedangkan metode titrasi iodometri membutuhkan biaya yang lebih sedikit, tetapi kurang sensitif untuk analisis dalam jumlah kecil.
Menurut Patel dkk. (1992), sefaleksin (gambar 1) dapat ditetapkan kadarnya secara spektrofotometri visibel berdasarkan reaksi antara gugus amin primernya dengan hasil kondensasi antara 2 mol asetilaseton dan 1 mol formalin.
C
Gambar 1. Struktur sefaleksin
Gugus amin primer
HO C
Gambar 2. Struktur amoksisilin Gugus amin primer
Dari penelitian ini diharapkan dapat dikembangkan suatu metode analisis yang mudah, murah, sederhana, sensitif, serta memiliki akurasi, presisi, dan linearitas yang baik untuk menetapan kadar amoksisilin.
1. Perumusan masalah
Berdasarkan latar belakang, maka diperoleh permasalahan sebagai berikut : a. apakah metode spektrofotometri visibel untuk penetapan kadar amoksisilin
menggunakan pereaksi asetilaseton dan formalin memenuhi parameter validasi metode analisis yang meliputi akurasi, presisi, dan linearitas?
b. apakah metode spektrofotometri visibel untuk penetapan kadar amoksisilin menggunakan pereaksi asetilaseton dan formalin dapat diaplikasikan pada sediaan obat tablet amoksisilin?
2. Keaslian penelitian
1999) dan penetapan kadar penisilin sebagai pengotor pada produk obat yang beredar di pasaran secara KCKT-spektrometri massa (Takada dkk., 2005).
Selain itu, telah dilakukan beberapa penelitian tentang penetapan kadar yang mirip dengan metode penetapan kadar amoksisilin dengan pereaksi asetilaseton dan formalin secara spektrofotometri, antara lain penelitian Patel dkk. (1992) tentang penetapan kadar sefaleksin dalam berbagai sediaan secara spektrofotometri visibel menggunakan pereaksi asetilaseton dan formalin, penelitian Rianti (2005) tentang penetapan kadar sefadroksil secara spektrofotometri visibel menggunakan pereaksi etilasetoasetat dan formalin, penelitian Rofie (2005) tentang penetapan kadar sefadroksil secara spektrofotometri ultraviolet menggunakan pereaksi etilasetoasetat dan asetaldehid, penelitian Mirmayanti (2007) tentang penetapan kadar sefadroksil secara spektrofotometri visibel menggunakan pereaksi asetilaseton dan formalin, dan penelitian Roosita (2007) tentang penetapan kadar ampisilin secara spektrofotometri visibel menggunakan pereaksi asetilaseton dan formalin.
3. Manfaat penelitian
Penelitian ini diharapkan akan memberikan manfaat untuk pengembangan metode analisis yang mudah, murah, sederhana, sensitif, serta memiliki akurasi, presisi, dan linearitas yang baik untuk menetapkan kadar amoksisilin.
1. untuk mengetahui apakah metode spektrofotometri visibel untuk penetapan kadar amoksisilin menggunakan pereaksi asetilaseton dan formalin memenuhi parameter validasi metode analisis yang meliputi akurasi, presisi, dan linearitas. 2. untuk mengetahui apakah metode spektrofotometri visibel untuk penetapan kadar
amoksisilin menggunakan pereaksi asetilaseton dan formalin dapat diaplikasikan pada sediaan tablet amoksisilin.
PENELAAHAN PUSTAKA
A. Amoksisilin
Amoksisilin (gambar 2) adalah antibiotik golongan β-laktam turunan aminopenisilin yang bersifat bakterisid, bekerja dengan menghambat sintesis dinding sel bakteri (Petri, 2001). Tanpa adanya dinding sel, bakteri tidak dapat bertahan terhadap pengaruh luar. Selain itu, kerusakan membran dapat mengganggu pertukaran zat aktif yang penting untuk kehidupan bakteri (Wattimena dkk., 1997). Antibiotik ini mempunyai spektrum kerja yang luas, dapat mengalami absorpsi cepat dan sempurna dari saluran pencernaan, serta tahan dalam suasana asam sehingga dapat diberikan secara oral (Petri, 2001).
Amoksisilin berupa serbuk hablur, putih, praktis tidak berbau. Amoksisilin sukar larut dalam air dan metanol, tidak larut dalam benzen, dalam karbontetraklorida, dan dalam kloroform (Anonim, 1995). Larutan yang mengandung amoksisilin 2 mg/ml mempunyai pH antara 3,5 sampai 6,0 (Anonim, 2005). Baku pembanding yang digunakan adalah amoksisilin Baku Pembanding Farmakope Indonesia (BPFI), tidak boleh dikeringkan sebelum digunakan. Amoksisilin harus disimpan dalam wadah tertutup rapat pada suhu kamar terkendali (Anonim, 1995).
Tablet amoksisilin mengandung tidak kurang dari 90,0% dan tidak lebih dari 120,0% C16H19N3O5S jumlah yang tertera pada etiket. Tablet amoksisilin harus
1. Metode titrimetri yang meliputi iodometri dan potensiometri. Pada penetapan kadar menggunakan kedua metode tersebut, amoksisilin harus dihidrolisis terlebih dahulu menjadi asam penisiloat. Selanjutnya, pada metode iodometri hasil hidrolisis tersebut akan bereaksi dengan iodium atau kalium iodat, sedangkan pada metode potensiometri dengan litium-metoksid, asam perklorat, merkuri nitrat, atau kupri sulfat.
2. Metode spektrofotometri yang meliputi spektrofotometri ultraviolet dan visibel. Pada penetapan kadar menggunakan metode spektrofotometri ultraviolet, amoksisilin harus diderivatisasi agar memberikan serapan yang cukup. Sementara itu, pada metode spktrofotometri visibel amoksisilin akan bereaksi dengan suatu senyawa membentuk warna yang kemudian diukur serapannya pada daerah cahaya tampak. Salah satu contohnya adalah reaksi antara gugus karbonil pada cincin β-laktam amoksisilin dengan hidroksilamin dan ion ferri membentuk kompleks warna ungu yang kemudian diukur serapannya pada panjang gelombang 480 nm.
3. Metode kromatografi yang meliputi kromatografi lapis tipis, kromatografi cair kinerja tinggi menggunakan fase gerak campuran larutan kalium fosfat dalam air dan asetonitril (96:4) dan fase diam oktadesilsilan, serta kromatografi gas. Khusus untuk kromatografi gas, amoksisilin harus diderivatisasi terlebih dahulu agar mudah menguap dan stabil pada suhu tinggi.
Menurut Patel dkk. (1992), sefaleksin (gambar 1) dapat ditetapkan kadarnya secara spektrofotometri visibel berdasarkan reaksi antara gugus amin primernya dengan hasil kondensasi antara 2 mol asetilaseton dan 1 mol formalin. Reaksi secara singkat dapat dilihat pada gambar 3 berikut:
H3COCCH2
Gambar 3. Reaksi antara sefaleksin dengan pereaksi asetilaseton-formalin (Patel dkk., 1992)
Maka, amoksisilin (gambar 2) yang juga memiliki gugus amin primer diharapkan dapat ditetapkan kadarnya dengan cara tersebut.
Asetilaseton (gambar 4) atau CH3.CO.CH2.CO.CH3 (BM = 100,211)
merupakan cairan tidak berwarna atau kuning lemah, barbau harum, dan mudah terbakar. Satu bagian asetilaseton larut dalam delapan bagian air, dapat campur dengan alkohol, benzen, kloroform, eter, aseton, dan asam asetat glasial (Anonim, 1989). Asetilaseton mendidih pada suhu 138-139 oC (Anonim, 1995).
H3C C O
C H2
C O
CH3
Gambar 4. Struktur Asetilaseton
C. Formalin
Formalin merupakan larutan 37% uap formalin (gambar 5) atau HCHO (BM = 30,03) di dalam air. Formalin berupa cairan jernih, tidak berwarna atau hampir tidak berwarna, bau menusuk, serta memiliki uap yang merangsang selaput lendir hidung dan tenggorokan. Jika disimpan di tempat dingin formalin akan menjadi menjadi keruh. Formalin dapat bercampur dengan air, alkohol, dan aseton (Anonim, 1989). Sebaiknya disimpan dalam wadah tertutup baik, terlindung dari cahaya, pada suhu di atas 20o (Anonim, 1995).
H C
O
H Gambar 5. Struktur Formalin
1. Definisi spektrofotometri UV-Vis
Spektrofotometri UV-Vis adalah anggota teknik spektroskopik yang menggunakan sumber radiasi elektromagnetik ultraviolet dekat (190 – 380 nm) dan sinar tampak (380 – 780 nm) dengan instrumen spektrofotometer. Spektrofotometri UV-Vis melibatkan energi elektronik yang cukup besar pada molekul yang dianalisis sehingga spektrofotometri UV-Vis lebih banyak dipakai untuk analisis kuantitatif dibandingkan kualitatif (Mulja dan Suharman, 1995).
Secara umum, spektrofotometri UV-Vis terbagi menjadi dua metode, yaitu
direct spectrophotometry UV-Vis dan indirect spectrophotometry UV-Vis. Pada
direct spectrophotometry serapan energi cahaya didasarkan oleh ikatan rangkap
terkonjugasi pada senyawa tersebut. Sementara pada indirect spectrophotometry, pengukuran serapan energi cahaya dapat dilakukan setelah senyawa mengalami reaksi kimiawi atau modifikasi gugus kromofor (Schimer, 1982).
2. Konsep dasar radiasi elektromagnetik
terbalik dengan panjang gelombang radiasi. Rumusan energi sebuah foton dinyatakan sebagai (Mulja dan Suharman, 1995):
E = h . v = h .
λ
c
= h . c . v Keterangan:
E = energi yang diabsorpsi (J)
h = konsatante Planck sebagai faktor pembanding = 6,63 x 10-27 erg.detik atau 6,63 x 10-34 Joule detik v = frekuensi radiasi (Hz)
c = kecepatan cahaya = 3 x 1010 cm/detik
λ = panjang gelombang (cm)
v = bilangan gelombang (cm-1)
3. Tipe transisi elektron
Suatu senyawa dapat menyerap radiasi dalam daerah UV-Vis karena mempunyai elektron, baik berpasangan maupun sendiri, yang dapat dieksitasikan ke tingkat energi yang lebih tinggi (Skoog, 1985).
Ada tiga macam distribusi elektron di dalam suatu senyawa organik secara umum, yang selanjutnya dikenal sebagai orbital elektron pi (π), sigma (σ), dan elektron tidak berpasangan (n). Transisi yang dapat terjadi adalah (Skoog, 1985):
b. transisi n → σ*. Senyawa-senyawa jenuh yang mengandung atom-atom dengan elektron-elektron tak berpasangan (elektron non bonding) mempunyai kemampuan untuk mengadakan transisi n → σ*. Pasangan elektron bebas tersebut akan dieksitasikan ke tingkat energi yang lebih tinggi karena elektron non bonding tidak terikat terlalu kuat seperti elektron bonding σ, sehingga serapannya terjadi pada panjang gelombang yang lebih besar. Akibatnya, transisi ini memerlukan energi yang lebih kecil daripada transisi σ→σ* dan dapat disebabkan oleh radiasi di daerah antara 150-250 nm, dengan kebanyakan puncak absorpsi tampak pada panjang gelombang di bawah 200 nm.
c. transisi n →π* dan π→π*. Umumnya penggunaan spektroskopi serapan pada senyawa-senyawa organik didasarkan pada transisi elektron n dan π ke π*. Energi yang dibutuhkan cukup rendah yaitu pada daerah sekitar 200-700 nm.
Diagram tingkat energi elektronik dapat dilihat pada gambar 6 berikut: σ*
Anti bonding
π*
Anti bonding
E
n Non bonding
π Bonding
σ Bonding
Gambar 6. Diagram tingkat energi elektronik
4. Interaksi molekul dengan radiasi elektromagnetik
molekul maka dinamakan absorpsi (Pecsok dkk., 1976). Agar dapat mengabsorpsi radiasi UV-Vis, suatu molekul membutuhkan gugus yang dinamakan kromofor yang merupakan suatu gugus kovalen tak jenuh terkonjugasi yang bertanggungjawab untuk absorpsi radiasi UV-Vis (Fell, 1986). Selain itu, dikenal pula auksokrom yang merupakan gugus yang mengandung heteroatom yang memberikan transisi n → σ*. Terikatnya gugus auksokrom oleh gugus kromofor secara langsung akan mengakibatkan pergeseran pita absorpsi menuju ke λ yang lebih panjang, disertai peningkatan atau penurunan intensitas (Mulja dan Suharman, 1995).
5. Analisis kuantitatif secara spektrofotometri UV-Vis
Analisis kuantitatif zat tunggal dilakukan dengan mengukur nilai serapan (A) pada panjang gelombang yang memberikan serapan maksimum. Nilai serapan (A) digambarkan oleh suatu hukum yang disebut hukum Lambert-Beer.
a. Hukum Lambert menyatakan bahwa intensitas cahaya yang ditransmisikan menurun secara eksponensial sesuai dengan kenaikan tebal zat penyerap.
b. Hukum Beer menyatakan bahwa intensitas cahaya yang ditransmisikan menurun secara eksponensial sesuai dengan kenaikan konsentrasi zat penyerap.
Kombinasi kedua hukum tersebut menghasilkan hukum Lambert-Beer yang menyatakan hubungan antara logaritma intensitas sinar yang masuk dengan sinar yang keluar sebagai fungsi tebal zat penyerap dan konsentrasi zat penyerap, dirumuskan sebagai berikut:
Dengan: a = daya serap
c = konsentrasi larutan b = tebal kuvet
A = serapan
Io = intensitas energi yang mencapai cuplikan
I = intensitas pancaran yang dikeluarkan dari cuplikan.
Nilai a atau daya serap menggambarkan nilai serapan yang spesifik dan
sering disebut
A
cm
% 1
1 yang artinya serapan larutan dengan konsentrasi 1% b/v dengan pelarut tertentu pada kuvet setebal 1 cm adalah suatu angka yang spesifik (Fell, 1986).
6. Serapan suatu larutan
Serapan adalah karakteristik untuk suatu larutan senyawa pada suatu panjang gelombang. Hubungan serapan dengan daya serap molar digambarkan dengan rumus ε = a . M, dimana M adalah berat molekul senyawa (Silverstein dkk., 1991).
Penyinaran senyawa organik tidak selalu diikuti oleh eksitasi elektron baik dari orbital ikatan atau pasangan elektron bebas ke orbital non ikatan. Pernyataan ini dapat dituang dalam persamaan berikut ini:
ε = 0,87 x 1020 x P x a
keterangan:
P = probabilitas transisi elektron dengan nilai antara 0-1 a = panjang kromofor
serap molar (ε) yang kurang dari 1000 (Williams dan Fleming, 1980). Nilai serapan jenis adalah karakteristik penyerapan molekul pada pelarut dan panjang gelombang tertentu, dan tidak tergantung konsentrasi serta lamanya radiasi (Pecsok dkk., 1976).
7. Kesalahan fotometrik
Ketepatan dan ketelitian pembacaan intensitas sinar yang sampai pada detektor digambarkan sebagai nilai kesalahan fotometrik. Ketepatan fotometrik berkurang pada nilai serapan rendah maupun pada nilai serapan tinggi. Pada serapan yang rendah, intensitas sinar yang ditransmisikan baik ada maupun tidak ada sampel hampir sama sehingga kemungkinan terjadinya kesalahan sangat besar. Hal tersebut karena ada keterbatasan kepekaan detektor. Pada serapan yang tinggi, intensitas sinar yang sampai pada detektor sangat rendah sehingga tidak dapat diukur dengan tepat (Pecsok dkk., 1976).
Untuk pembacaan serapan (A) atau transmitan (T) pada daerah terbatas, kesalahan penentuan kadar hasil analisis dinyatakan sebagai:
C
ΔT adalah nilai rentang skala transmitan terkecil dari alat yang masih dapat terbaca
kesalahan analisis yang dapat diterima yaitu sebesar 0,5-1% untuk ΔT = 1 (Mulja dan Suharman, 1995).
Apabila pengukuran dilakukan di luar rentang A (0,2-0,8) atau %T (15-65%), maka sebaiknya dalam pengukuran digunakan panjang gelombang yang paling tepat dan menggunakan sel dengan pencahayaan paling tepat. Hal ini untuk menghindari besarnya kesalahan pembacaan serapan, yang berakibat pada kesalahan penetapan kadar (Pecsok, 1976).
8. Syarat-syarat penggunaan Hukum Beer (Skoog, 1985)
a. syarat konsentrasi.
Penyimpangan Hukum Beer dapat disebabkan dari nilai yang tergantung dari indeks bias larutan. Hubungan tersebut dapat dilihat dari persamaan berikut (Willard dkk., 1988):
Besar penyimpangan =
2 n = indeks bias larutan
Pada konsentrasi < 0,01 M, indeks bias larutan relatif konstan tetapi pada konsentrasi tinggi indeks bias ternyata berubah sehingga perlu dikoreksi agar diperoleh nilai serapan yang sesuai.
bergantung pada konsentrasi, maka peristiwa ini menyebabkan penyimpangan dari kelinieran hubungan antara absorpsi dengan konsentrasi. Pengaruh serupa kadang-kadang terjadi di dalam larutan yang mengandung konsentrasi zat pengabsorpsi yang rendah tetapi konsentrasi zat non-pengabsorpsinya tinggi, terutama elektrolit. Interaksi elektrostatis ion-ion yang berdekatan dengan zat pengabsorpsi akan mempengaruhi nilai absorptivitas molar. Pengaruh ini dapat dihindari dengan cara pengenceran. b. syarat kimia.
Zat pengabsorpsi tidak boleh terdisosiasi, berasosiasi, atau bereaksi dengan pelarut menghasilkan suatu produk pengabsorpsi spektrum yang berbeda dari zat yang dianalisis.
c. syarat cahaya.
Hukum Beer hanya berlaku untuk cahaya yang betul-betul monokromatik (cahaya yang mempunyai satu macam panjang gelombang). d. syarat kejernihan.
Kekeruhan larutan misalnya yang disebabkan oleh partikel-partikel koloid akan menyebabkan penyimpangan hukum Beer. Sebagian cahaya akan dihamburkan oleh partikel-partikel koloid akibatnya kekuatan cahaya yang diabsorpsi berkurang dari yang seharusnya.
9. Penggunaan spektrofotometri UV-Vis dalam metode analisis
a. analisis kuantitatif zat tunggal (analisis satu komponen)
b. analisis kuantitatif campuran dua macam zat (analisis dua komponen) c. analisis kuantitatif campuran tiga macam zat atau lebih (analisis multi
komponen).
Analisis kualitatif dengan metode spektrofotometri UV-Vis hanya dipakai untuk data sekunder atau data pendukung. Pada analisis kualitatif dengan metode spektrofotometri UV-Vis yang dapat ditentukan ada dua yaitu:
a. pemeriksaan kemurnian spektrum UV-Vis
b. penentuan panjang gelombang serapan maksimum.
(Mulja dan Suharman, 1995)
E. Pengembangan Metode Spektrofotometri
Salah satu bentuk pengembangan metode spektrofotometri adalah dengan reaksi pembentukan warna. Reaksi tersebut umumnya dilakukan dengan memodifikasi kromofor dari suatu molekul sehingga dapat dideteksi di daerah visibel (Fell, 1986). Kadarnya kemudian ditetapkan dengan membandingkan serapannya dengan kurva baku yang dibuat menggunakan baku pembanding (Rooth dan Blaschke, 1994).
Keuntungan utama reaksi pembentukan warna adalah bahwa metode ini dapat menambah sensitivitas dan selektivitas spektroskopi absorpsi (Fell, 1986).
Kriteria untuk reaksi pembentukan warna yang baik adalah sebagai berikut (Vogel, 1978):
Sangat sedikit reaksi yang spesifik untuk zat-zat tertentu. Oleh karena itu, harus diupayakan agar reaksi yang terjadi spesifik untuk zat tertentu. Caranya antara lain mereaksikan zat dengan reagen yang spesifik, mengubah kondisi percobaan, dan mengendalikan pH.
2. kesebandingan antara warna dan konsentrasi
Intensitas warna larutan hendaknya meningkat secara linier dengan naiknya konsentrasi zat yang akan ditetapkan.
3. kestabilan warna
Warna yang dihasilkan hendaknya cukup stabil dalam waktu tertentu untuk memungkinkan pembacaan yang tepat.
4. reprodusibilitas
Hasil yang didapat harus dapat diulang jika dilakukan pada kondisi yang sama.
5. kejernihan larutan
Larutan harus bebas dari endapan agar tidak menghamburkan ataupun menyerap cahaya.
6. kepekaan tinggi
Diharapkan reaksi warna sangat peka bahkan untuk zat dalam jumlah kecil.
F. Validasi, Kesalahan, dan Parameter Metode Analisis
1. Validasi metode analisis
Pedoman-pedoman validasi metode analisis didukung oleh parameter-parameter sebagai berikut :
a. ketepatan. Ketepatan (accuracy) berarti kedekatan hasil analisis yang diperoleh dengan menggunakan metode tersebut terhadap nilai sebenarnya. Accuracy dinyatakan dengan persen perolehan kembali (recovery) dari penambahan zat yang diketahui kadarnya (Anonim, 2005). Untuk kadar analit ≥ 10% biasanya disepakati perolehan kembali harus masuk dalam rentang 98-102% (Yuwono dan Indrayanto, 2005).
b. ketelitian. Ketelitian (precision) berarti ukuran kedekatan masing-masing hasil analisis dari beberapa pengukuran di bawah kondisi analisis yang sama. Ketelitian biasanya dinyatakan dengan standar deviasi atau relatif standar deviasi (koefisien variasi) (Anonim, 2005).
Presisi dapat dibedakan menjadi tiga, yaitu (Anonim, 2005):
1). repeatability adalah presisi yang dihasilkan dari pengujian suatu metode yang dilakukan oleh individu yang sama dengan menggunakan prosedur yang sama dan dikerjakan dalam waktu yang singkat.
2). intermediate precission adalah presisi yang dihasilkan dari pengujian suatu metode yang dilakukan oleh individu yang berbeda dengan menggunakan prosedur dan instrumen yang sama.
3). reproducibility adalah presisi yang dihasilkan dari pengujian suatu metode
analisis yang dikerjakan pada laboratorium yang berbeda.
c. limit of detection (LOD). Limit of Detection adalah kadar terkecil
analit yang dapat terdeteksi tetapi tidak perlu secara kuantitatif. Penentuan LOD dilakukan dengan cara membandingkan respon pengukuran analit dengan blangko. Rasio signal-to-noise yang diterima untuk LOD adalah 2:1 atau 3:1 (Anonim, 2005).
d. limit of quantitation (LOQ). Limit of Quantitation adalah konsentrasi
terkecil analit dalam sampel yang dapat diukur dengan ketelitian dan ketepatan yang diterima di bawah kondisi percobaan yang ditetapkan metode tersebut. Rasio signal-to-noise yang diterima untuk LOQ adalah 10:1 (Anonim, 2005).
e. spesifisitas. Spesifisitas merupakan kemampuan pengukuran analit secara akurat dan spesifik dengan kehadiran komponen lain (zat aktif, eksipien, pengotor, dan produk degradasi) dalam matriks sampel (Anonim, 2005).
f. linearity. Linearity adalah kemampuan suatu metode analisis untuk
secara langsung atau melalui perhitungan matematika mendapatkan hasil uji yang sebanding dengan kadar analit dalam sampel (Anonim, 2005).
g. range. Range suatu metode analisis diartikan sebagai interval antara kadar terendah sampai tertinggi analit yang dapat diukur secara kuantitatif menggunakan metode analisis tertentu dan menghasilkan ketelitian dan ketepatan, dan linearitas yang mencukupi (Anonim, 2005).
2. Kesalahan metode analisis
a. Kesalahan sistematik. Kesalahan ini merupakan hasil analisis yang menyimpang secara tetap dari nilai sebenarnya karena proses pelaksanaan prosedur analisis. Kesalahan sistematik ini dapat diketahui sebabnya sehingga dapat dikendalikan oleh peneliti. Beberapa cara untuk memperkecil kesalahan ini antara lain dengan melakukan kalibrasi instrumen secara berkala, pemilihan metode dan prosedur standar dari badan resmi, pemakaian bahan kimia dengan derajat untuk analisis, serta peningkatan pengetahuan dari peneliti yang bekerja di laboratorium analisis.
b. Kesalahan tidak sistematik. Kesalahan ini disebut juga penyimpangan tidak tetap dari hasil penentuan kadar menggunakan instrumen yang disebabkan fluktuasi dari instrumen yang dipakai (derau). Penyebab kesalahan ini tidak diketahui. Salah satu cara untuk mengatasi kesalahan ini adalah dengan menggunakan instrumen dengan kualitas yang baik.
3. Parameter-parameter validasi metode analisis
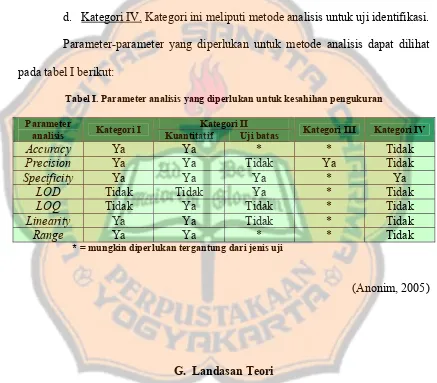
Uji yang paling umum dan prosedur pengukuran dapat dibagi menjadi empat kategori, yaitu (Anonim, 2005):
b. Kategori II. Kategori ini meliputi metode analisis untuk penentuan pengotor dalam obat atau senyawa degradasi dalam sediaan, termasuk pengukuran kuantitatif dan uji batas.
c. Kategori III. Kategori ini meliputi metode analisis untuk penentuan sifat-sifat fisik lain dari obat seperti uji disolusi dan uji pelepasan.
d. Kategori IV. Kategori ini meliputi metode analisis untuk uji identifikasi. Parameter-parameter yang diperlukan untuk metode analisis dapat dilihat pada tabel I berikut:
Tabel I. Parameter analisis yang diperlukan untuk kesahihan pengukuran
Kategori II Parameter
analisis Kategori I Kuantitatif Uji batas Kategori III Kategori IV
Accuracy Ya Ya * * Tidak
Precision Ya Ya Tidak Ya Tidak
Specificity Ya Ya Ya * Ya
LOD Tidak Tidak Ya * Tidak
LOQ Tidak Ya Tidak * Tidak
Linearity Ya Ya Tidak * Tidak
Range Ya Ya * * Tidak
* = mungkin diperlukan tergantung dari jenis uji
(Anonim, 2005)
G. Landasan Teori
amoksisilin dengan hasil kondensasi antara satu mol formalin dan dua mol asetilaseton membentuk warna kuning yang intensitasnya kemudian diukur menggunakan spektrofotometri visibel pada panjang gelombang serapan maksimum.
H. Hipotesis
Amoksisilin dapat ditetapkan kadarnya secara spektrofotometer visibel dengan menggunakan pereaksi asetilaseton dan formalin. Metode yang dikembangkan tersebut memenuhi parameter validasi yaitu akurasi, presisi, dan linearitas serta dapat diaplikasikan pada sediaan tablet amoksisilin.
BAB III
METODOLOGI PENELITIAN
Penelitian ini merupakan jenis penelitian non eksperimental dengan rancangan penelitian deskriptif, sebab pada penelitian ini tidak dilakukan manipulasi terhadap subjek uji. Penelitian hanya mendeskripsikan keadaan yang ada.
B. Definisi Operasional
1. Validasi metode analisis merupakan serangkaian prosedur yang digunakan untuk membuktikan apakah suatu metode analisis memenuhi persyaratan yang ditentukan, meliputi ketepatan, ketelitian, dan linearitas.
2. Spektrofotometri visibel adalah anggota teknik spektroskopik yang menggunakan sumber radiasi elektromagnetik sinar tampak (380 – 780 nm) dengan instrumen spektrofotometer.
3. Kadar amoksisilin ditetapkan dalam satuan mg/tablet.
C. Alat-alat Penelitian
Alat yang digunakan dalam penelitian ini adalah spektrofotometer ultraviolet – visibel (Spectronic Genesys 5, MILTON ROY), pH meter (Hanna Instrument pH 209), neraca analitik (Precisa 125 A.SCS Swiss Quality), penangas air,
termometer, kertas saring, dan alat-alat gelas yang lazim.
D. Bahan-bahan Penelitian
Asetilaseton, formalin, asam asetat glasial, natrium asetat (p.a., E. Merck), dan akuades (Fakultas Farmasi UGM).
E. Tata Cara Penelitian
1. Pembuatan larutan uji
a. pembuatan larutan natrium asetat 0,2 M.
Sebanyak 16,4 g natrium asetat ditimbang seksama, dimasukkan ke dalam labu ukur 1 liter kemudian dilarutkan dengan akuades sampai tanda.
b. pembuatan larutan asam asetat 0,2 M.
Sebanyak 12,5 ml asam asetat 96% dipipet, kemudian diencerkan dengan akuades sampai volume 1,0 liter.
c. pembuatan larutan NaOH 1M.
Ditimbang seksama 0,4 g NaOH kemudian dilarutkan dalam akuades bebas CO2 sampai volume 10,0 ml.
d. pembuatan larutan HCl 2M.
Sebanyak 17,0 ml asam klorida pekat dipipet, kemudian diencerkan dengan akuades sampai volume 100,0 ml.
e. pembuatan larutan pereaksi (Patel dkk., 1992).
Sebanyak 16,0 ml natrium asetat 0,2 M dan 34,0 ml asam asetat 0,2 M dicampur dengan 7,8 ml asetilaseton dan 15,0 ml formalin. Panaskan 5 menit di atas waterbath pada suhu 80 oC, dinginkan, pH diatur sampai (4,3), kemudian diencerkan dengan akuades sampai 100,0 ml.
Ditimbang seksama 209,7 mg baku amoksisilin kemudian dilarutkan dengan akuades sampai 100,0 ml hingga diperoleh konsentrasi 0,005 M.
2. Optimasi penetapan kadar amoksisilin (Patel dkk., 1992)
Pada penelitian ini dilakukan optimasi berbagai kondisi percobaan yaitu pH pereaksi, volume pereaksi, operating time, dan panjang gelombang serapan maksimum amoksisilin.
a. penentuan operating time.
Sebanyak 2,0 ml larutan baku amoksisilin 0,005 M dimasukkan ke dalam labu ukur 25 ml, ditambahkan larutan pereaksi pH 4 sebanyak 4 ml. Diencerkan dengan akuades sampai tanda. Diukur serapan larutan pada panjang gelombang 400 nm, sampai diperoleh serapan yang stabil pada rentang waktu tertentu. Dilakukan juga pengukuran blangko. b. penetapan nilai pH yang menghasilkan serapan maksimum.
Nilai pH larutan pereaksi dibuat bervariasi, yaitu pH 3, 4, 5, 6, dan 7. Untuk masing-masing nilai pH dipipet sebanyak 4 ml, dimasukkan ke dalam labu ukur 25 ml, ditambahkan 2,0 ml larutan baku amoksisilin 0,005 M, didiamkan selama operating time pada suhu 35 oC kemudian encerkan dengan akuades sampai tanda. Diukur serapan larutan pada panjang gelombang 400 nm. Dilakukan juga pengukuran blangko. Nilai pH optimum adalah pH larutan pereaksi yang menghasilkan serapan paling besar.
Dari larutan pereaksi dengan pH optimum dipipet masing-masing 1, 2, 3, 4, 5, 6, 7, 8, 9, dan 10 ml, dimasukkan ke dalam labu ukur 25 ml, ditambahkan 2,0 ml larutan baku amoksisilin 0,005 M, didiamkan selama operating time pada suhu 35 oC, dan diencerkan dengan akuades sampai tanda. Diukur serapan larutan pada panjang gelombang 400 nm. Dilakukan juga pengukuran blangko. Volume optimum adalah volume larutan pereaksi yang menghasilkan serapan paling besar.
d. Penentuan panjang gelombang serapan maksimum.
Sebanyak 1,0; 1,4; dan 1,8 ml larutan baku amoksisilin 0,005 M masing-masing dimasukkan ke dalam labu ukur 25 ml, ditambahkan larutan pereaksi dengan volume dan pH hasil optimasi. Diamkan selama operating time pada suhu 35 oC . Diencerkan dengan akuades sampai tanda. Larutan tersebut kemudian discan antara panjang gelombang 380 hingga 450 nm. Dilakukan juga pengukuran blangko. Panjang gelombang serapan maksimum adalah panjang gelombang yang memberikan serapan maksimum.
3. Pembuatan kurva baku (Patel dkk., 1992)
kurva hubungan kadar vs serapan dan ditentukan persamaan regresi linier serta koefisien korelasinya.
4. Aplikasi metode penetapan kadar amoksisilin pada tablet AM (Patel dkk.,
1992)
a. pengambilan sampel.
Sampel yang digunakan terdiri dari 1 merek tablet yang mengandung 500 mg amoksisilin yang beredar di pasaran (tablet AM). Tablet amoksisilin yang dipilih adalah tablet dengan nomor batch yang sama.
b. penentuan bobot rata-rata tablet.
Ditimbang 20 tablet satu persatu, kemudian dihitung bobot rata-rata tiap tablet.
c. penetapan kadar amoksisilin dalam tablet AM (Patel dkk., 1992).
Ditimbang seksama sejumlah serbuk dari 20 tablet yang setara dengan 209,7 mg amoksisilin. Dimasukkan ke dalam labu ukur 100 ml, diencerkan dengan akuades sampai tanda. Dipipet 1,0 ml, dimasukkan ke dalam labu ukur 25 ml, ditambahkan pereaksi dengan pH dan volume hasil optimasi. Didiamkan selama operating time pada suhu 35 oC, diencerkan dengan akuades sampai tanda. Kemudian diukur serapannya pada panjang gelombang serapan maksimum Dilakukan juga pengukuran blangko
5. Validasi metode
Ditimbang seksama sejumlah serbuk dari 20 tablet yang setara dengan 104,85 mg amoksisilin dan 104,85 mg baku amoksisilin. Dimasukkan ke dalam labu ukur 100 ml, diencerkan dengan akuades sampai tanda. Dipipet 1,0 ml, dimasukkan ke dalam labu ukur 25 ml, ditambahkan pereaksi dengan pH dan volume hasil optimasi. Didiamkan selama operating time pada suhu 35 oC, diencerkan dengan akuades sampai tanda. Kemudian diukur serapannya pada panjang gelombang serapan maksimum. Dilakukan juga pengukuran blangko. Setelah itu dihitung jumlah perolehan kembali sampel.
b. presisi (dinyatakan dengan koefisien variasi).
Penetapan koefisien variasi dilakukan dengan menggunakan data kadar amoksisilin dalam tablet AM.
c. linearitas (dinyatakan dengan koefisien korelasi).
Penetapan koefisen korelasi dilakukan dengan menggunakan koefisien korelasi korva baku amoksisilin.
F. Analisis Hasil
Analisis hasil penelitian berupa analisis validitas metode yang meliputi linearitas dengan taraf kepercayaan 99%, akurasi, dan presisi. Selain itu, dilakukan juga analisis kuantitatif berupa kadar amoksisilin yang dihitung dengan menggunakan persamaan kurva baku.
BAB IV
A. Pembuatan Larutan Baku Amoksisilin
Larutan baku yang digunakan dalam penelitian ini adalah larutan baku amoksisilin 0,005 M dalam akuades. Larutan ini dibuat dengan cara melarutkan 209,7 mg baku amoksisilin dalam 100,0 ml akuades. Pelarut yang digunakan adalah akuades karena amoksisilin larut dalam akuades (Anonim, 1995).
B. Penetapan Waktu Reaksi dan Operating Time (OT)
Waktu reaksi merupakan waktu yang dibutuhkan agar reaksi berlangsung sempurna, sehingga pada pengukuran yang terbaca adalah semua amoksisilin yang telah bereaksi.
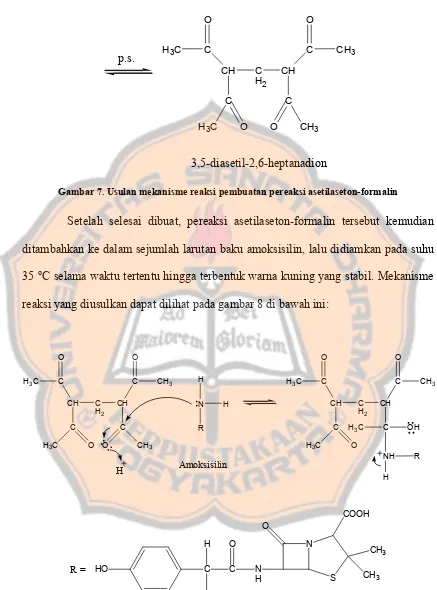
Pereaksi yang digunakan dalam penelitian ini adalah pereaksi campuran asetilaseton-formalin. Pereaksi tersebut dibuat dengan cara mencampurkan asetilaseton dan formalin di dalam bufer asetat yang terdiri dari asam asetat dan natrium asetat. Bufer asetat berfungsi menjaga pH pereaksi agar stabil di sekitar pH 4. Setelah dicampur, larutan dipanaskan selama 5 menit pada suhu 80 oC untuk mempercepat reaksi. Kemudian larutan didinginkan, dan dilakukan penyesuaian pH dengan menggunakan larutan HCl atau larutan NaOH.
H3C C
karbonil tak jenuh a,ß enol asetilaseton H
p.s.
Gambar 7. Usulan mekanisme reaksi pembuatan pereaksi asetilaseton-formalin
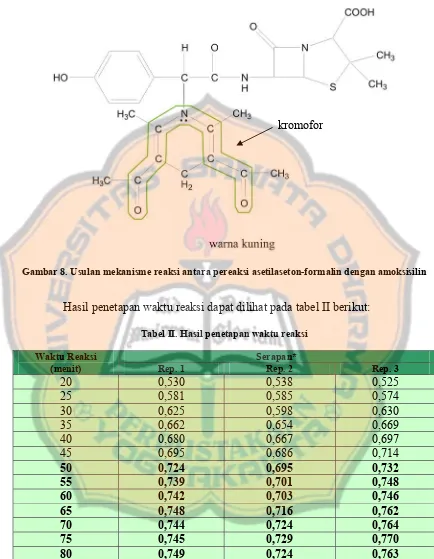
Setelah selesai dibuat, pereaksi asetilaseton-formalin tersebut kemudian ditambahkan ke dalam sejumlah larutan baku amoksisilin, lalu didiamkan pada suhu 35 oC selama waktu tertentu hingga terbentuk warna kuning yang stabil. Mekanisme reaksi yang diusulkan dapat dilihat pada gambar 8 di bawah ini:
kromofor
Gambar 8. Usulan mekanisme reaksi antara pereaksi asetilaseton-formalin dengan amoksisilin
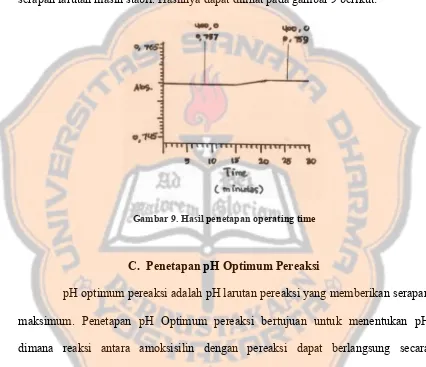
Hasil penetapan waktu reaksi dapat dilihat pada tabel II berikut: Tabel II. Hasil penetapan waktu reaksi
Serapan* Waktu Reaksi
(menit) Rep. 1 Rep. 2 Rep. 3
20 0,530 0,538 0,525
25 0,581 0,585 0,574
30 0,625 0,598 0,630
35 0,662 0,654 0,669
40 0,680 0,667 0,697
45 0,695 0,686 0,714
50 0,724 0,695 0,732
55 0,739 0,701 0,748
60 0,742 0,703 0,746
65 0,748 0,716 0,762
70 0,744 0,724 0,764
75 0,745 0,729 0,770
80 0,749 0,724 0,763
*) Serapan senyawa hasil reaksi antara amoksisilin dengan asetilaseton dan formalin
untuk mengetahui stabilitasnya , dilakukan penetapan OT yang merupakan rentang waktu saat suatu senyawa memberikan serapan yang stabil.
Setelah didiamkan selama 50 menit pada suhu 35 oC larutan dibaca serapannya menggunakan spektrofotometer selama 30 menit. Ternyata selama itu serapan larutan masih stabil. Hasilnya dapat dilihat pada gambar 9 berikut:
Gambar 9. Hasil penetapan operating time
C. Penetapan pH Optimum Pereaksi
pH optimum pereaksi adalah pH larutan pereaksi yang memberikan serapan maksimum. Penetapan pH Optimum pereaksi bertujuan untuk menentukan pH dimana reaksi antara amoksisilin dengan pereaksi dapat berlangsung secara optimum. Hal tersebut karena reaksi antara amoksisilin dengan pereaksi asetilaseton-formalin ini adalah reaksi yang sangat tergantung pada pH. Pada tahap pertama terjadi reaksi adisi amina pada gugus karbonil (gambar 8). Bila larutan terlalu asam, akan terjadi reaksi sebagai berikut:
Akibatnya, konsentrasi amina menjadi menjadi kecil sekali bahkan dapat diabaikan. Sehingga reaksi akan menjadi lambat. Tahap kedua dalam reaksi itu adalah eliminasi gugus H2O (gambar 8). Berbeda dengan reaksi tahap pertama, laju reaksi ini akan
bertambah dengan meningkatnya keasaman. Jika suasana larutan terlalu basa, gugus -OH2+ tidak akan terbentuk. Sebagai gantinya, akan terbentuk gugus –OH yang
merupakan gugus pergi yang kurang baik dibandingkan dengan gugus -OH2+
(gambar 10).
dak akan berlangsung sehingga Gambar 10. Reaksi eliminasi pada reaksi antara amoksisilin dengan asetilaseton da
maka reaksi tahap kedua ti Jika hal tersebut terjadi,
Hasil penetapan pH optimum pereaksi dapat dilihat pada tabel III berikut: Tabel III. Hasil penetapan pH optimum pereaksi
Serapan*
*) Serapa yawa hasil reaksi oksisilin denga seton dan forma
anjutnya, pH perea
D. Penetapan Volume Optimum Pereaksi
Volume n pereaksi yang
memberi
enambahkan pereaksi pH 4 dengan v
n sen antara am n asetila lin
Dari penelitian didapat bahwa pH optimum adalah pH 4. Untuk sel ksi yang digunakan adalah pH 4.
.
optimum pereaksi adalah volume laruta
kan serapan maksimum. Penetapan volume optimum pereaksi bertujuan untuk menentukan volume pereaksi agar semua amoksisilin dapat habis bereaksi. Jika pereaksi yang ditambahkan kurang, dikhawatirkan belum semua amoksisilin bereaksi sehingga pada saat pengukuran belum semua amoksisilin yang terbaca sehingga tidak menggambarkan kadar yang sebenarnya.
Penetapan volume pereaksi dilakukan dengan m
Tabel IV. Hasil penetapan volume optimum pereaksi
Serapan* Vol. Pereaksi (ml)
Rep. 1 Rep. 2 Rep. 3
*) Serapa wa hasil reaksi oksisilin denga seton dan forma
dengan 7
E. Penetapan Panjang Gelombang Serapan Maksimum (λmax)
g suatu senyawa
ingga 450 nm.
n senya antara am n asetila lin
Dari penelitian didapat bahwa serapan amoksisilin stabil saat direaksikan
ml hingga 10 ml pereaksi. Untuk selanjutnya volume pereaksi yang digunakan adalah 7 ml.
Panjang gelombang serapan maksimum adalah panjang gelomban
yang memberikan serapan yang paling besar. Pengukuran kadar dengan metode spektrofotometri umumnya dilakukan pada panjang gelombang serapannya maksimum. Hal tersebut karena pada panjang gelombang serapan maksimum perubahan serapan untuk setiap perubahan konsentrasi adalah paling besar sehingga menghasilkan sensitifitas dan akurasi yang lebih besar. Selain itu, pada panjang gelombang serapan maksimum absorptivitas molar senyawa relatif konstan sehingga didapat kurva kalibrasi konsentrasi vs serapan yang linear (Pecsok dkk., 1976).
asetilaseton akan menghasilkan senyawa berwarna kuning yang memberikan serapan paling besar pada panjang gelombang 400 nm. Gugus pada sefaleksin yang berperan dalam pembentukan senyawa berwarna tersebut adalah gugus amin primer (gambar 3). Penetapan kadar amoksisilin dalam penelitian ini juga didasarkan pada reaksi antara gugus amin primer amoksisilin dengan hasil kondensasi antara satu mol formalin dan dua mol asetilaseton (gambar 8). Dengan demikian, diperkirakan panjang gelombang serapan maksimum reaksi penetapan kadar ini juga berada di sekitar 400 nm.
Untuk
c. b.
a.
penentuan panjang gelombang serapan maksimum digunakan tiga konsentras
imum dapat dilihat pada gambar 1
Gambar 11. Spektra panjang gelombang serapan maksimum amoksisilin konsentrasi 0,084 mg/ml (a), 0,117 mg/ml (b), dan 0,151 mg/ml (c) hasil reaksi dengan
adalah 401,0 nm. Disamping itu, adanya perubahan konsentrasi tidak merubah panjang gelombang serapan maksimum gambar 11 (a, b, dan c).
i yang bertujuan untuk melihat apakah dengan perubahan konsentrasi akan terjadi perubahan panjang gelombang serapan maksimum.
Hasil penetapan panjang gelombang serapan maks 1 (a, b, dan c) berikut:
asetilaseton dan formalin
Selain itu, pada spektrum terlihat bahwa senyawa hasil reaksi juga memberikan serapan pada panjang gelombang nm. Diperkirakan, serapan tersebut adalah serapan gugus fenol pada amoksisilin (gambar 12).
C
Gambar 12. Gugus pada senyawa hasil reaksi yang diperkirakan memberikan serapan pada panjang gelombang 335 nm
F. Pembuatan Kurva Baku
si ang selanjutnya digunakan untuk menghitung kadar amoksisilin. Dalam pembuatan
kurva baku sebaiknya di amoksisilin baku dengan
konsen
Gugus yang memberikan serapan
Pembuatan kurva baku bertujuan untuk memperoleh persamaan garis regre y
gunakan suatu seri larutan
Pada penelitian ini, penetapan kurva baku dilakukan dengan menggunakan 5 seri konsentrasi amoksisilin yaitu 0,067; 0,084; 0,101; 0,117; dan 0,134 mg/ml dengan replikasi sebanyak tiga kali.
Adapun hasil kurva baku dari 3 kali replikasi dapat dilihat pada tabel V berikut:
Tabel V. Hasil penetapan kurva baku amoksisilin
Serapan*
*) Serapan senyawa hasil rea ksisilin de on dan for
Seluruh persamaan regresi linier pada tabel V di atas menghasilkan nilai l dengan taraf k
hampir membentuk garis tegak lurus dengan sumbu x. Oleh karena itu, ksi antara amo ngan asetilaset malin
koefisien korelasi (r) hitung yang lebih besar dari koefisien korelasi (r) tabe
epercayaan 99% dan derajat bebas 3 yaitu 0,959 (Cann, 2003). Dapat dikatakan ada korelasi bermakna antara serapan dan konsentrasi amoksisilin. Persamaan regresi yang digunakan untuk menghitung kadar amoksisilin dalam penelitian ini adalah persamaan y = 4,5516x – 0,0807 (replikasi 1) karena nilai r yang diperoleh paling mendekati satu dan nilai koefisien variasi fungsi (Vx0) yang
paling kecil.
dilakukan modifikasi satuan konsentrasi larutan baku sehingga didapat data yang dapat dilihat pada tabel VI berikut:
Tabel VI. Hasil modifikasi kurva baku amoksisilin
Konsentrasi Amoksisilin
*) Serapan hasil reaksi antara amok engan asetilaseton dan fo
Persamaan kurva baku hasil modifikas oleh dengan memplotkan konsentrasi r adalah
senyawa sisilin d rmalin
i diper
baku (mg/5 ml) vs serapan. Didapat: y = 0,9103x + 0,0807 dengan nilai
0,9995 dan α = 42,31o (gambar 13). Dengan demikian, persamaan garis tersebut dapat digunakan untuk menetapkan kadar amoksisilin yang direaksikan dengan asetilaseton dan formalin.
alin
Gambar 13. Hubungan konsentrasi amoksisilin dan serapan senyawa hasil reaksi antara amoksisilin dengan asetilaseton dan formalin
G. Penetapan Kadar Amoksisilin dalam Tablet
Penetapan kadar amoksisilin dalam tablet dilakukan dengan 3 kali penimbangan dan masing-masing penimbangan dilakukan replikasi pemipetan 3 kali. Replikasi pemipetan bertujuan untuk m
Tablet amoksisilin yang digunakan adalah tablet dari suatu pabrik dengan nomor batch yang sama. Dengan nomor batch yang sama, diharapkan variasi yang terjadi pada saat formulasi dapat diminimalkan sehingga jika keragaman hasil benar-benar menggambarkan ketelitian metode ini.
Dari hasil penelitian didapat data yang disajikan pada tabel VII berikut: Tabel VII Hasil penetapan kadar amoksisilin dalam tablet AM
engetahui reprodusibilitasnya.
Penimbangan
Sampel (mg) Serapan*
Kadar (mg) % kadar
KV (%) Per tablet Dlm tablet
150,0
*) Serapan senyawa hasil reaksi antara amoksisilin dengan asetilaseton dan formalin
Dari data didapat kadar rata-rata amoksisilin dalam tablet adalah 589,56 mg
atau s tablet
amoksisilin m dari 120,0%
jumlah yang terter (An ), karena menurut etiket, tablet amoksisilin yang dig blet andung zat aktif amoksisilin 500 mg/tablet.
ekitar 117,91%. Hasil tersebut masih memenuhi syarat karena engandung tidak kurang dari 90,0% dan tidak lebih
H. Validas e Analisi
Validasi met lisis diar agai sua dur yang digunakan untuk buktikan suatu m nalisis m i persyara ang ditentukan (Anonim, 2
netapan ka oksisilin kukan d elitian ini termasuk ke dalam kategori I yaitu ntuk peneta komponen terbesar dalam
enurut
i Metod s
ode ana tikan seb tu prose
mem apakah etode a emenuh tan y
005).
Pe dar am yang dila alam pen
u pan kadar sediaan.
M Anonim (2005), parameter yang perlu ditetapkan dalam analisis kategori I adalah akurasi, presisi, spesifisitas, linearitas, dan range.
Dalam penelitian ini, beberapa parameter yang ditetapkan adalah: 1. akurasi.
Akurasi adalah kedekatan hasil analisis yang diperoleh menggunakan suatu metode dengan nilai sebenarnya. Akurasi dinyatakan dengan perolehan kembali dari penambahan zat yang dike , 2005).
P
oksisilin. Kadar yang didapat kemudian dibandingkan dengan kadar yang sebenarnya.
tahui kadarnya (Anonim
Dari hasil penelitian didapat data yang disajikan pada tabel VIII berikut: Tabel VIII. Data hasil penetapan perolehan kembali
Kadar Sebanarnya
(mg/ml)
Serapan* Kadar didapat Perolehan Kembali (mg/ml) (%) *) Serapan senyawa hasil reaksi antara amoksisilin dengan asetilaseton dan formalin
ari data dapat dilihat bahwa rata-rata perolehan kembali yang didapat adalah 104,09%. Hal tersebut tidak memenuhi syarat karena untuk kadar analit 10% biasanya disepakati perolehan kembali harus masuk dalam rentang 98-102%
(Yu cara
spektrofotomet n dan formalin
mem urang baik.
2. presi D
≥
wono dan Indrayanto, 2005). Berarti metode penetapan amoksisilin se ri visibel menggunakan pereaksi asetilaseto
iliki akurasi yang k si.
Presisi adalah kedekatan masing-masing hasil analisis dari beberapa penguk di bawah k nalisis yang s Presisi biasany takan dengan persen simpangan baku atau simpanga u relatif (koef riasi) (Anon 05).
Pada penelitian ini, penetapan presisi dilakukan dengan menggunakan data
rata-uran ondisi a ama. a dinya
n bak isien va
im, 20
rata koefisien variasi yang didapat adalah 0,56%. Hal tersebut masih memenuhi syarat karena untuk kadar analit ≥ 10% biasanya disepakati koefisien variasi tidak boleh lebih dari 2,7% (Yuwono dan Indrayanto, 2005). Berarti metode penetapan amoksisilin secara spektrofotometri visibel menggunakan pereaksi asetilaseton dan formalin memiliki presisi yang baik.
linearitas. 3.
Linearitas ditentukan dengan melihat nilai koefisien korelasi (r) hitung pada per
dan derajat bebas 3 yaitu 0,959 (Cann, 2003). Dapat dikatakan ada
samaan regresi linier kurva baku. Dari hasil penentuan kurva baku, didapat persamaan y = 0,9103x + 0,0807 dengan r = 0,9995. Nilai koefisien korelasi (r) hitung tersebut lebih besar dari koefisien korelasi (r) tabel dengan taraf kepercayaan 99%
korelasi bermakna antara serapan dan konsentrasi amoksisilin. Selain itu, didapat nilai koefisien variasi fungsi (Vx0) sebesar 1,009%. Menurut Mulja dan
Hanwar (2003), nilai Vx0 tidak boleh lebih dari 2%. Berarti metode penetapan
BAB V
KESIMPULAN DAN SARAN
I. Kesimpulan
Berdasarkan hasil penelitian dapat disimpulkan sebagai berikut: metode spektrofotometri visibel untuk
1. penetapan kadar amoksisilin
menggunakan pereaksi asetilaseton dan formalin memiliki presisi dan linearitas yang baik, namun akurasinya kurang baik.
. aplikasi metode ini pada sediaan tablet amoksisilin memberikan hasil yang baik dengan kadar rata-rata amoksisilin dalam tablet sebesar 589,56 mg.
J. Saran
Perlu dilakukan penelitian lebih lanjut tentang suhu yang menghasilkan kecepatan reaksi dan serapan yang opt
2
DAFTAR PUSTAKA
Anonim, 1989, The Merck Index An Encyclopedia of Chemicals, Drugs, and
IV, 95-96, 1136, 1157, Departemen Kesehatan Republik Indonesia, Jakarta.
nonim, 2005, The United States Pharmacopeia, 28th ed., 138-139, 141, 143, 144, 146, United States Pharmacopeia Convention, Rockville.
ird, A. E., 1994, Amoxicillin in Analytical Profiles of Drug Substances and Excipients, 23, 4-44, Academic Press Inc., California.
ann, A. J., 2003, Maths from Scratch for Biologist, 213, John Wiley & Sons Ltd., England.
ell, A. F., 1986, Ultraviolet, Visible, and Flourescence in Clarke’s Isolation and Identification of Drugs in Pharmaceuticals Body Fluid and Post Mortem Material, 2nd ed., 221-232, The Pharmaceutical Press, London.
essenden, R. J. dan Fessenden, J. S., 1994, Kimia Organik, Diterjemahkan oleh Pudjaatmaka, A. H., Edisi ketiga, Jilid I, 23, 67-129, Penerbit Erlangga, Jakarta.
Harmita, 2004, Petunjuk Pela e dan Cara Perhitungannya, Majalah Ilmu Kefarmasian, I (3):117-135.
Biologicals, 11th ed., 81, 4261, Merck & Co. Inc., Rahway N. J., USA. Anonim, 1995, Farmakope Indonesia, Edisi
A
Hidayat, R., 1999, Titrasi Iodometri Hasil Hidrolisis Amoksisilin Secara Coulometri, http://fa.lib.itb.ac.id/go.php?id=jbptitbfa-gdl-sl-1999-rahmathida-223., diakses pada 8 April 2006.
Kusuma, resepkan dan Masuk di Beberapa
Apotek Wilayah Kotamadya Yogyakarta, Skripsi, Fakultas Farmasi
irmayanti, B., 2007, Validasi Metode Penetapan Kadar Sefadroksil dengan ogyakarta.
rmasi Airlangga, III (2): 71-76.
Mulja, M. M. dan Suharman, 1995, Analisis Instrumental, 6-10, 26-48, Airlangga University Press, Surabaya.
2000, Jenis-jenis Antibiotika yang Di Universitas Sanata Dharma, Yogyakarta. M
Pereaksi Asetilaseton dan Formaldehid secara Spektrofotometri Visibel, Skripsi, Fakultas Farmasi Universitas Sanata Dharma, Y
Patel, I. T., Devani, M. B., and Patel, T. M., 1992, Spectrophotometric Method for Determination of Cephalexin in Its Dosage Forms, J. of AOAC Int., 75 (6): 994-998.
Pelczar, M. J. and Chan, E. C. S., 1988, Elements of Microbiology, Ed III, 561, Diterjemahkan oleh Ratna Siri Hadi Oetomo, Penerbit UI, Jakarta.
ecsok, R. L., Shields L. D., Cairns, T. Mc. and William, T. G., 1976, Modern
is of raw-Hill Companies Inc., USA.
Mada, Yogyakarta.
Roosita, ar Ampisilin dengan Pereaksi
Asetilaseton dan Formaldehid secara Spektrofotometri Visibel, Skripsi,
yakarta. P
Methods Of Chemical Analysis, 2nd ed., 117, 139, 142-143, 226-235, John Wiley & Sons Inc., New York.
Petri, W. A., 2001, Antimicrobial Agents Penicillins, Cephalosporins, and Other β -Lactam Antibiotics in Goodman and Gilman’s The Pharmalogical Bas Therapeutics, 10th ed., Mc-G
Rianti, A., 2005, Penetapan Kadar Sefadroksil Secara Spektrofotometri Visibel dengan Pereaksi Etilasetoasetat dan Formaldehid, Skripsi, Fakultas Farmasi Universitas Gadjah Mada, Yogyakarta.
Rofie, F., 2005, Penetapan Kadar Sefadroksil dalam Kapsul Menggunakan Metode Spektrofotometri Ultraviolet dengan Pereaksi Etilasetoasetat dan Asetaldehid, Skripsi, Fakultas Farmasi Universitas Gadjah
A., 2007, Validasi Metode Penetapan Kad
Fakultas Farmasi Universitas Sanata Dharma, Yogyakarta.
Roth H. J. and Blaschke G., 1994, Pharmazeutische Analytik, diterjemahkan oleh Sarjono Kisman dan Slamet Ibrahim, 359-361, Gadjah Mada University Press, Yog
Schirmer, R. E., 1982, Modern Methods of Pharmaceuticals Analysis, I: 60-74, CRC Press Inc., Florida.
Silverstein, R. M., Bassler, G. C., and Morrill, T. C., 1991, Spectrometric Identification of Organic Compounds, 5th ed, 292, John Wiley & Sons Inc., Canada.
Takada, W., Adachi, T., Kihara, N., Kitamura, S., Kitagawa, T., Mifune, M., and
attimena, J. R., Sugiarso, W. C., Widianto, M. B., Sukandar E. Y., dan Setiadi, A.
illiams, D. H. dan Fleming, I., 1980, Spectroscopic Methods in Organic Chemistry,
illard, H. H., Merritt, L. L., Dean, J. A., and Settle, F. A., 1988, Instrumental
iratih, 2002, Gambaran Resep Antibiotik di Apotek-apotek yang Terletak di ta Dharma, Yogyakarta.
uwono, M. dan Indrayanto, G., 2005, Validation of Chromatographic Methods of Analysis, Profiles of Drug Substances, Excipients, and Related Methodology, 32: 243-259.
Saito, Y., 2005, Quantitative Determination Method for Trace Amount of Penicillin Contaminants in Comercially Available Drug Product by HPLC Coupled with Tandem Mass Spectrometry, Chem. Pharm. Bull., 53 (2): 172-176.
Vogel, A. I., 1978, A Textbook of Quantitative Inorganic Analysis, 4th ed., 809-810, 846-849, The English Language Book Society, Richard Clay Ltd., Bungay. W
R., 1997, Farmakodinamik dan Terapi Antibiotik, 56-61, 66, Gadjah Mada University Press, Yogyakarta.
W
3rd ed., 4, McGraw Hill Book Company, United Kingdom. W
Methods of Analysis, 7th ed., 162, Wadsworth Publishing Company,
Belmont, California. W
Perbatasan Bagian Utara Kotamadya Yogyakarta dan Kabupaten Sleman, Skripsi, Fakultas Farmasi Universitas Sana
LAMPIRAN
Lampiran 1. Data Penimbangan Baku Amoksisilin
Kertas + zat (mg) 506,6 Kertas + sisa (mg) 297,0
Lampiran 2. Data Penetapan Kadar Sampel
. Perhitungan Bobot Rata-Rata Tablet a
. Penimbangan Sampel
Lampiran 3. Data Hasil Penetapan Perolehan Kembali
dar amoksisilin dalam sampel a. Perhitungan ka
a penetapan kadar, rata-rata kad sisilin per 15 mpel dalah 123,64 mg
adar amoksisilin dalam sampel =
Dari dat didapat ar amok 0 mg sa
. Penimbangan baku + sampel b
Rep. 1 Rep. 2 Rep. 3
Baku Sampel Baku Sampel Baku Sampel Kertas+zat 0,4091 g 0,4596 g 0,4091 g 0,4593 g 0,4091 g 0,4602 g
adar sebenarnya = kadar amoksisilin dalam sampel + kadar baku
= 2,29 mg/ml K
= 123,73 mg + 104,8 mg = 228,53 mg
Lam n 4. r
a. Pembuatan Kurva Baku Amoksisilin
pira Contoh Pe hitungan
aquadest.
Konsentrasi baku amo
Perhitungan konsentrasi baku amoksisilin:
Baku amoksisilin yang ditimbang = 209,6 mg, dilarutkan dalam 100 ml
ksisilin =
Dibuat seri kurva baku dengan mempipet:
1,4 ml
Seri kadar tersebut kemudian diplotkan vs serapan yang diperoleh sehingga diperoleh persamaan kurva baku yang akan digunakan dalam penetapan kadar.
b. Penetapan Kadar Sampel 1,4 . 2,0
Serapan yang didapat = 0,533, dimasukkan ke dalam persamaan kurva baku 0,533 = 0,9103x + 0,0807
kadar anoksisilin d x 715,26 = 592,24 mg
% kadar amoksisilin dalam tablet = 500
24 , 592
c. Penetapan Recovery
Kadar sebenarnya = kadar amox dalam sampel + kadar amox baku Kadar sebenarnya = 123,73 mg + 104,8 mg
ilarutkan dalam aquadest 100 ml Kadar sebenarnya = 228,53 mg
Serbuk tersebut kemudian d
100 53 , 228
Kadar sebenarnya = = 2,29 mg/ml
bil 1 ml kemudian dimasukkan ke dalam labu 25 ml sehingga konsentrasinya:
1 . 2,29 = 25 . c2
2
emas am
persamaan kurva baku.
asukkan ke dalam persamaan kurva baku
% recovery =
dari larutan tersebut, diam
v1 . c = v2 . c2
c = 0,0916 mg/ml
Kadar yang didapat dihitung dengan m ukkan serapan yang diperoleh ke dal
c. Perhitungan Vx0
menggunakan rumus sebagai berikut (Harmita, 2004): Vx0 dapat dihitung
Persamaan Kurva baku replikasi 1: y = 4,5516x + 0,0807
BIOGRAFI
Penulis skripsi yang berjudul “Validasi Metode Spektrofotometri Visibel Untuk Penetapan Kadar Amoksisilin Menggunakan Pereaksi Asetilaseton dan Formalin” ini bernama Margareta Sunarto. Penulis lahir di Garut pada tanggal 14 Januari 1985. Anak pertama dari tiga bersaudara dari pasangan Sunarto Yosep Tjitrahadi dan Catharina Lena Tanzil ini mengawali pendidikannya di TK aya Susila Garut pada tahun 1988. Kemudian penulis melanjutkan pendidikannya i SD Daya Susila Garut pada tahun 1991. Selanjutnya, pada tahun 1997 penulis enempuh pendidikan di SLTP Yos Sudarso Garut. Setelah lulus, pada tahun 2000 enulis melanjutkan pendidikan di SMU Negeri 2 Yogyakarta. Lalu, pada tahun 003 penulis melanjutkan pendidikan di Fakultas Farmasi Universitas Sanata
harma Yogyakarta. Selama kuliah, penulis pernah menjadi asisten dosen untuk mata kuliah praktikum spektroskopi
D d m p 2 D