TUGAS GENETIKA
OSTEOGENESIS IMPERFECTA
Disusun Oleh:
Retno Ayu Adhisty
NIM 0840709006
PROGRAM PASCA SARJANA PROGRAM STUDI ILMU BIOMEDIK
PROGRAM DOUBLE DEGREE ILMU KESEHATAN ANAK
2012
BAB I
PENDAHULUAN
Osteogenesis Imperfecta (OI) adalah kelainan pembentukan jaringan ikat bawaan dengan berbagai bentuk manifestasi klinis. Kelainan ini juga sering disebut dengan "brittle bone disease". Kelainan ini pada umumnya ditandai dengan tulang mudah patah, kelainan pada ligamen, kulit, sklera, gigi, ataupun tuli.Kelainan bentuk yang paling ringan biasanya ditandai dengan osteoporosis prematur, penderita bisa tidak mengalami patah tulang sampai masa dewasa, sedang untuk kelainan OI yang berat adalah ditandai dengan fraktur multipel dengan trauma ringan atau tanpa riwayat trauma sejak dalam kandungan. Disebutkan kurangnya asupan gizi saat hamil, paparan lingkungan/ ekosistem, dan ibu peminum alcohol meningkatkan risiko kejadian OI.1,2
EPIDEMIOLOGI
Osteogenesis Imperfecta diturunkan secara autosomal dominan. Kejadian OI diperkirakan 1 per 20.000 kelahiran hidup. Tidak ada perbedaan menurut ras dan jenis kelamin. Usia penderita saat gejala muncul bervariasi, terutama gejala mudah patahnya tulang. Pada kasus minoritas dapat ditemukan penurunan secara resesif yang disebabkan oleh mosaicism pada orangtua.1-3
KLASIFIKASI
Sejak tahun 1979, OI telah diklasifikasikan menjadi beberapa tipe oleh David Sillence. Sistim klasifikasi ini dikelompokkan berdasarkan keturunan, manifestasi klinis, dan gambaran radiologis. Klasifikasi OI bisa mencerminkan prognosis dari seorang penderita tetapi tidak bisa memprediksi batas akhir kemampuan seorang penderita. Klasifikasi awal ada 4 tipe yaitu tipe I sd. Tipe IV. Dan ada tiga tipe tambahan dari OI yaitu tipe V, VI, dan VII. Namun klasifikasi klinis lebih berguna berdasarkan manifestasi yang muncul pada bayi, anak-anak, dan orang dewasa dengan gejala yang ringan, sedang sampai berat, dan jenis yang mematikan.1-5
BAB II
TINJAUAN KEPUSTAKAAN ETIOLOGI
Hampir 90% bentuk klinis (tipe) OI disebabkan oleh kelainan struktural atau mutasi 2 gen yaitu COL1A1 dan COL1A2 yang mengkode rantai kolagen tipe 1 (prokolagen tipe 1), dimana COL1A1 dan COL1A2 merupakan komponen protein utama matriks ekstraselular tulang. Bentuknya yang beragam ini karena bisa terjadi pada lokus dan alel yang sangat heterogen. Manifestasi yang timbul tergantung dari rantai prokolagen tipe 1 yang terkena, jenisnya, dan lokasi mutasi dari lokusnya. Sekitar 10% kasus klinis yang tak jelas, terbukti tidak didapat kelainan biokimia dan molekul prokolagen. Hingga kini tidak diketahui dengan jelas apakah kasus ini dikarenakankemampuan deteksi kurang atau karena kelainan genetik yang heterogen.1,2,4,5
PATOGENESIS
Prokolagen tipe I adalah struktur protein utama yang menyusun matriks tulang dan jaringan fibrous lainnya, seperti kapsul organ, fasia, kornea, sklera, tendon, selaput otak dan dermis.2,5 Sekitar 30% berat badan manusia terdiri dari prokolagen tipe I.2 Secara struktural, molekul prokolagen tipe I berbentuk triple
helix, terdiri dari 2 rantai proα1(I) (disebut COL1A1, dikode pada kromosom 17)
dan 1 rantai proα2(I) (disebut COL1A2, dikode pada kromosom 7). Masing-masing rantai triple helix itu dibentuk oleh rangkaian 388 asam amino Gly-X-Y yang berulang. Prolin sering berada di posisi X, sedangkan hidroksiprolin atau hidroksilisin sering berada di posisi Y. Glisin (Gly) merupakan asam amino terkecil yang mempunyai struktur cukup padat dan berperan penting sebagai poros dari helix sehingga bila terjadi mutasi akan sangat mengganggu struktur dan produksi helix.Prokolagen yang abnormal akan membentuk cetakan yang tidak normal sehingga matriks pelekat tulang pun tak normal dan tersusun tak beraturan. Beberapa protein bukan kolagen dari matriks tulang juga berkurang. Hal ini menyebabkan adanya penurunan pembentukan tulang, osteopenia, dan terjadi kerapuhan sehingga meningkatkan angka kepatahan (fraktur). 1,2,4
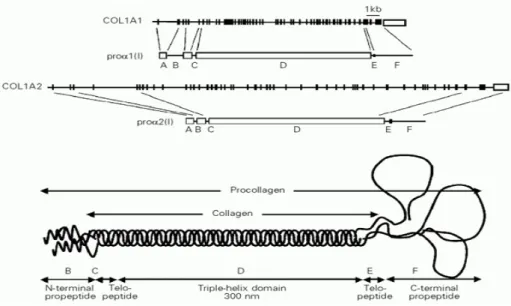
Gambar 1. Gen yang menyandi tipe I preprocollagen struktur rantai (atas) dan domain molekul procollagen (bawah). A, Sinyal urutan yang dibelah saat transit dari retikulum endoplasma kasar, B, propeptide N-terminal; C, non-triple-helix domain pendek, yang berisi situs pembelahan proteolitik peptida N-terminal; D, triple-helix domain yang penting, ditandai dengan mengulangi Gly-XY, E, Telopeptide, F, N-terminal propeptides yang mengandung ikatan disulfida. (Dikutip dari Beary JF, Chines A, dkk. Clinical features and diagnosis ofosteogenesis imperfecta
.
www.Uptodate.com)Lebih dari 200 mutasi yang berbeda mempengaruhi sintesis atau struktur prokolagen tipe I ditemukan pada penderita OI. Jika mutasi tersebut menurunkan produksi/ sintesis prokolagen tipe I, maka terjadi OI fenotip ringan (osteogenesis imperfecta tipe I), namun jika mutasi menyebabkan gangguan struktur prokolagen tipe I maka akan terjadi OI fenotip yang lebih berat (tipe II, III, dan IV). Kelainan struktur itu pada dasarnya terbagi menjadi dua macam, yaitu 85% karena point mutation akibat glisin digantikan oleh asam amino lain dan sisanya karena kelainan single exon splicing.Struktur normal prokolagen tipe I. Masing-masing rantai kolagen sebagai triple helix prokolagen, disekresikan ke ruang ekstraseluler. Domain amino- dan carboxyl-terminal dipecah di ruang ekstraseluler, mengalami maturitas, kemudian dirangkai, di tulang akan mengalami mineralisasi. 1,2,4
Mutasi terbanyak OI diturunkan secara autosomal dominan oleh gen kolagen tipe I, hanya sedikit yang resesif. Secara umum penyakit ini menggambarkan kompleksitas genetik, namun jika terjadi mutasi gen akan mempengaruhi struktur protein yang dirupakan dalam berbagai bentuk sub unit.
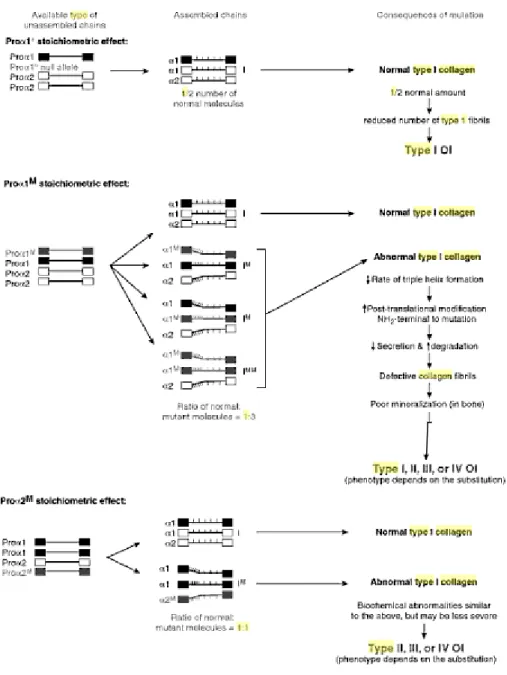
Bentuk phenotif yang ringan dari tipe I autosomal dominan, walaupun molekul normal yang terbentuk hanya separoh dari keseluruhan, secara kualitas akan tampak normal. Semakin berat mutasi yang terjadi akan tampak pada stoichiometri pada pembentukan defek rantai proα1(I) kolagen tipe I yang terdiri dari 2 rantai proα1 dan 1 rantai proα2. Jika separoh rantai proα1(I) adalah abnormal, 3 dari 4 kolagen tipe I akan mempunya minimal 1 rantai abnormal. Sebaliknya jika separoh rantai proα1(I) mengalami defek, maka yang terkena mutasi hanya 1 dari 2 molekul. Mutasi pada rantai proα1(I) yang tampak pada gambar 2 di bawah ini adalah dominant negative alelle (proα1M), karena mereka mempengaruhi kerusakan kedua rantai yaitu proα1 dan proα2. Dengan kata lain pengaruh mutan alel diperkuat dengan adanya betuk polimer alamiah dari molekul kolagen, khususnya untuk penyakit autosomal dominan seperti OI ini, akan lebih baik jika terjadi mutasi yang nantinya tidak memproduksi gene dari pada harus menghasilkan produk gen abnormal. Meskipun mutasi yang menghasilkan struktur abnormal rantai proα2 bisa menurunkan jumlah normal molekul kolagen tipe 1 hingga separoh, penurunan jumlah ini tidak selamanya jelek. Seperti pada kasus beberapa mutasi yang menyebabkan bentuk fenotip perinatal yang letal, kebanyakan bayi dengan OI tipe II, bentuk letal mempunyai dominasi mutasi yang baru, yang mana kaitannya dalam keluarga untuk terjadi kejadian berulang sangat rendah.2,4
Cartilage-associated protein (CRTAP) merupakan protein yang dibutuhkan untuk hidroksilasi prolil 3. Hilangnya CRTAP pada tikus menyebabkan osteochondroplasia yang dikarakterisasi dengan osteoporosis beratdan penurunan produksi osteoid. Pada manusia, mutasi CRTAP dapat menyebabkan modifikasi post-translasional dari kolagen yang berlebihan sehingga dapat menyebabkan gejala klinis yang mirip dengan OI.
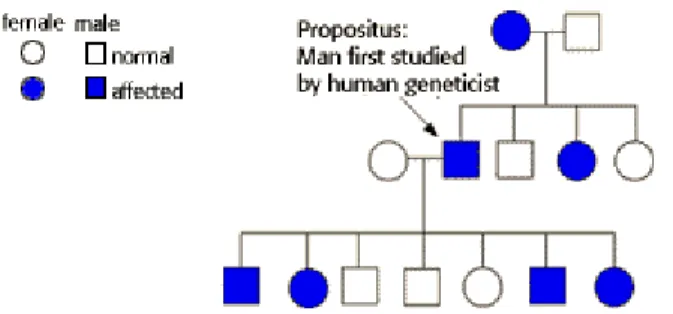
Gambar 2. Pedegree dari keluarga dengan Osteogenesis Imperfecta yang terbanyak diturunkan secara autosomal dominan. (Diambil dari www.carolguz.com)
Gambar 3. Patogenesis kelas mayor mutasi dari prokolagen tipe 1. ( Dikutip dari Nussbaum RL, McInnes RR, Willard HF. The molecular and biochemical basis of genetic disease. Dalam: Thompson and thompson genetic in medicine, edisi ke-6. Philadelphia: Saunders, 2004;342 )
MANIFESTASI KLINIS DAN DIAGNOSIS
Sistem klasifikasi yang paling sering dipakai untuk membedakan tipe OI adalah yang dibuat oleh Sillence dkk. Klasifikasi tersebut didasarkan pada gejala klinis, genetik, dan kriteria radiografi (tipe I sd. Tipe IV). Gejala klinisnya sangat bervariasi antar penderita walaupun dalam tipe yang sama. Dan ada tiga tipe tambahan dari OI yaitu tipe V, VI, dan VII. Namun klasifikasi klinis lebih berguna berdasarkan manifestasi yang muncul pada bayi, anak-anak, dan orang dewasa dengan gejala yang ringan, sedang sampai berat, dan jenis yang mematikan. 1-4
Tabel 1. Tipe-OI dari tinjauan Genetik & Biomolekuler sebagai berikut :
Tipe Fenotif Diturunkan secara
Defek Biomolekuler Defek Genetika Defek Produksi Kolagen Tipe I
I Ringan : sklera kebiruan, brittle bones , tetapi tdk ada deformitas tulang
Autosomal dominan
Biasanya : Susunan kolagen normal (alel normal), tetapi jumlah berkurang separoh
Biasanya: tidak punya alel, sehingga menggangu produksi rantai proα1(1), dan
mengganggu sintesis mRNA
Defek struktural Kolagen Tipe I II Perinatal letal:
abnormalitas tulang yg berat (fraktur, deformitas), sklera gelap, meninggal usia 1 bulan
Autosomal dominan
Biasanya: produksi molekul kolagen abnormal ok. Substitusi dari Gly-X-Y dari tripel helix dominan, dgn beberapa bias ke separoh protein C-terminal
Biasanya :kesalahan mutasi tulang dalam kodon glisin dari gene untuk rantai α1 dan α2
III Deformitas progresif: fraktur, sering saat kelahiran, deformitas tulang progresif, tumbuh terbatas, sklera biru
Autosomal dominan
Molekul kolagen abnormal : substitusi glisin dari berbagai tipe triple helix, berada di sepanjang protein
Kesalahan mutasi pada kodon glisin dari gene untuk rantai α1 dan α2
IV Sklera normal, deformitas tulang : derajat ringan – sedang, perawakan pendek, fraktur
Autosomal
dominan Abnormal molekul kolagen: substitusi glisin dari berbagai tipe triple helix, berada di sepanjang protein
Kesalahan mutasi pada kodon glisin dari gene untuk rantai α1 dan α2
Modifikasi dari Byers PH: Disorder of collagen biosynthesis and structur. In Scriver CR, Beaudert AL (eds): The Metabolic Basis of Inheritance Disease, 6th ed. New York, Mc Graw Hill, 1989 pp 2805-2842 and Bayers PH: Brittle bones-fragile molecules: disorder of collagen structure and expression. Trends Genet 6:293-300, 1990
1. Tipe I (Ringan)
Merupakan bentuk OI yang paling ringan dan paling banyak ditemukan, bahkan sering ditemukan dalam suatu pedigree keluarga yang besar. Diturunkan secara autosomal dominan dan disebabkan karena menurunnya produksi atau sintesis prokolagen tipe I (functional null alleles). Kebanyakan penderita tipe I mempunyai sklera berwarna biru, fraktur berulang pada masa anak-anak tapi tidak sering, dan tuli (30-60% pada usia 20-30 tahun). Fraktur terjadi karena trauma ringan–sedang dan menurun setelah masa pubertas. Ada 2 subtipe, yaitu subtipe A bila tidak disertai dentinogenesis imperfecta, dan subtipe B bila disertai dentinogenesis imperfecta. Kelainan lainnya yang bisa ditemukan antara lain mudah memar, kelemahan sendi dan otot, kifoskoliosis, serta perawakan pendek ringan dibanding anggota keluarga lainnya. 1-5
2. Tipe II (Sangat berat/ perinatal lethal)
Sering didapatkan bayi lahir mati atau meninggal pada tahun pertama kehidupan dengan berat lahir dan panjang badan kecil untuk masa kehamilan. Kematian disebabkan karena distres pernafasan, juga karena malformasi atau perdarahan sistem saraf pusat. Terjadi karena mutasi baru yang diturunkan secara autosomal dominan (jarang resesif) akibat penggantian posisi glisin pada
triple helix prokolagen tipe I dengan asam amino lain. Tulang rangka dan
jaringan ikat lain sangat rapuh. Terdapat fraktur multipel tulang panjang intrauterin yang terlihat sebagai crumpled appearance pada radiografi. Selain itu bisa terjadi pada tulang tengkorak/ vertebra. Tulang tengkorak tampak lebih besar dibanding ukuran tubuh dengan pembesaran fontanela anterior dan posterior. Fraktur multipel tulang iga membentuk gambaran manik-manik
(beaded appearance), thoraks yang sempit ikut berperan dalam terjadinya distres
pernafasan.Penderita mungkin mempunyai hidung yang kecil dan/ mikrognatia. Sklera warna biru gelap-keabuan. 1-5
3. Tipe III (Berat/Progresif)
Merupakan tipe dengan manifestasi klinis paling berat namun tidak mematikan dan menghasilkan gangguan fisik yang berat, berupa kelenturan sendi, kelemahan otot, nyeri tulang kronis berulang, dan deformitas tengkorak. Hal ini bisa terjadi karena point mutation atau frame shift mutation pada prokolagen tipe I yang diturunkan secara autosomal dominan atau resesif. Berat badan dan panjang lahir sering rendah. Fraktur sering terjadi dalam kandungan. Setelah lahir, fraktur sering terjadi tanpa sebab dan sembuh sendiri dengan sisa deformitas. Kebanyakan dari penderita berperawakan pendek, bentuk wajah relatif triangular dan makrosefali. Sklera bervariasi dari putih hingga biru. Sering dijumpai dentinogenesis imperfecta (80% pada anak usia < 10 tahun). Dari gambaran radiologis didapatkan disorganisasi matriks tulang menyebabkan gambaran popcorn pada metafisis.1-5
4. Tipe IV (Tak terdefinisi/ Moderately severe)
Terjadi karena point mutation atau delesi kecil pada prokolagen tipe I yaitu pada rantai COL1A2, kadang pada COL1A1. Merupakan tipe OI yang paling heterogen karena memasukkan temuan-temuan pada penderita yang tidak tergolong dalam 3 tipe sebelumnya. Fraktur bisa terjadi dalam rahim dengan tulang panjang bawah bengkok yang kelihatan sejak lahir. Sering terjadi fraktur berulang, kebanyakan penderita mempunyai tulang yang bengkok walau tidak sering mengalami fraktur. Frekuensi fraktur berkurang setelah mencapai pubertas. Penderita tipe ini memerlukan intervensi ortopedik dan rehabilitasi tetapi biasanya mereka dapat melakukan aktivitas sehari-hari. Penderita mengalami perawakan agak pendek. Warna sklera biasanya putih. Kadang dijumpai dentinogenesis imperfecta, sehingga beberapa penulis membedakan tipe ini menjadi 2 subtipe yaitu subtipe A bila tidak disertai dentinogenesis imperfecta dan subtipe B bila disertai dentinogenesis imperfecta. Gambaran osteoporotik dan kompresi vertebra didapatkan dari radiologiknya. 1-5
Penelitian beberapa tahun terakhir dengan menggunakan penelitian mikroskopik terhadap tulang penderita OI ditemukan tipe-tipe baru OI (tipe V, VI, VII). Yang tidak berasal dari mutasi tipe 1 gene kolagen. Penyebab mutasi pada tipe di atas belum dapat diidentifikasi, meskipun lokus dari OI tipe VII diketahui
berada di lengan pendek kromosom 3 dan diturunkan secara resesif. Bentuk lain diturunkan secara dominan autosomal dan dapat dibedakan secara klinis atau kelainan tulangnya tetapi gambarannya hampir sama dengan OI tipe IV. 2,4
Gambar 4. Wajah bulat dengan orbita dangkal, sclerae putih atau keabu-abuan, philtrum panjang, rhizomelic dari ekstremitas atas dan bawah dan posisi abduksi kaki.Di kutip dari Glorieux FH,. Experience With Bisphosphonates in Osteogenesis Imperfecta. Pediatrics 2007;119;S163-S165
(I) (II) (III)
Gambar 5. Gambaran radiologis (I) Fraktur mid diafise tampak obliq ri dari humerus kiri, (II) penipisan ringan dari tulang kepala, (III) fraktur proksimal femur bilateral dengan konsolidasi dan angulasi. Di kutip dari Glorieux FH,. Experience With Bisphosphonates in Osteogenesis Imperfecta.
Pediatrics 2007;119;S163-S165
KLASIFIKASI LANJUTAN OSTEOGENESIS IMPERFECTA
Tipe V: Deformitas derajat sedang (disebut juga congenital brittle bones
dengan formasi kalus redundant ). Manifestasi klinis : kerapuhan tulang derajat sedang, perawakan pendek ringan-sedang, sklera putih, tidak didapatkan dentinogenesis imperfecta, diturunkan secara dominan. Gambaran radiologis didapatkan dislokasi ujung tulang radius; mineralisasi membran interosseous; hiperplasi kallus.
Type VI: Deformitas derajat sedang-berat ( Disebut juga congenital britlle
bones dengan defek mineralisasi. Manifestasi klinis : kerapuhan tulang derajat
sedang, perawakan pendek derajat sedang, sklera putih, tidak didapatkan dentinogenesis imperfecta; tidak tampak skoliosis; tidak diketahui sifat keturunannya. Gambaran histologisnya tampak akumulasi osteoid di jaringan tulang, bentuk fish scale dari lamella tulang
Type VII: Deformitas derajat sedang (disebut juga congenital brittle bones dengan rhizomelia. Gambaran klinis : kerapuhan tulang derajat sedang, perawakan pendek derajat ringan, sklera putih, tidak didapatkan dentinogenesis imperfecta; autosomal resesif; ditemukan hanya pada kelompok suku asli Amerika Utara di northern Quebec. Gambaran radiologis tampaktulang humeri and femora yang pendek; coxa vara
Type VIII: Defisiensi Prolyl 3-hydroxylase 1. Gambaran klinis tampak sklera putih, wajah bulat, dada bentuk barrel chest pendek, tangan relatif lebih panjang dibandingkan kaki, tulang phalang panjang, tulang metakarpal pendek; diturunkan secara resesif. Gambaran radiologis tampak gracile, kadar mineral tulang iga dan tulang panjang yang rendah, fraktur multipe pada saat lahir, disorganisasi bulbus metafise dan matrixnya. 1-5
DIAGNOSIS
Diagnosis OI ditegakkan dari riwayat penyakit yang sama pada keluarga dan atau manifestasi klinis yang berbeda-beda tiap penderita, dari tipe ringan sampai berat, ditambah dengan beberapa pemeriksaan penunjang.Manifestasi klinis yang bisa ditemukan antara lain sering fraktur berulang, perawakan pendek, sklera berwarna biru, masalah gigi (dentinogenesis imperfecta), dan gangguan pendengaran yang makin progresif setelah masa pubertas. Pemeriksaan penunjang yang dapat dilakukan 1-4 :
1. Laboratorium biokimia dan molekular
Analisa sintesa kolagen didapat melalui kultur fibroblas dari biopsi kulit, terutama untuk mendeteksi osteogenesis imperfecta tipe I,III dan IV. Analisa mutasi DNA prenatal dilakukan pada kehamilan dengan resiko OI, melalui kultur villus korion. Pemeriksaan kombinasi antara analisa DNA dan biopsi kolagen akan mendeteksi hampir 90% dari semua tipe mutasi gen pengkode prokolagen tipe I. 1-4
2. Pencitraan Radiografi pada tulang skeletal setelah lahir (bone survey) Bentuk ringan (tipe I) tampak korteks tulang panjang yang menipis, tidak tampak deformitas tulang panjang. Bisa menunjukkan gambaran Wormian (Wormian bones) pada cranium. Bentuk sangat berat (tipe II) tampak gambaran manik-manik (beaded appearance) pada tulang iga, tulang melebar, fraktur multipel dengan deformitas tulang panjang. Bentuk sedang dan berat (tipe III dan IV) tampak metafisis kistik atau gambaran popcorn pada kartilago, tulang dapat normal atau melebar pada awalnya kemudian menipis, dapat ditemukan fraktur yang
Gbr.6. Warna sclera penderita OI. Di ambil dari www.uptodate.com
menyebabkan deformitas tulang panjang, sering disertai fraktur vertebra. Densitas mineral tulang (bone densitometry) diukur dengan Dual-Energy X-Ray Absorptiometry (DEXA) yang menghasilkan nilai rendah pada penderita. Ultrasonografi prenatal pada minggu 15-18 kehamilan untuk mendeteksi kelainan panjang tulang anggota badan. Yang tampak dapat berupa gambaran normal (tipe ringan) sampai dengan gambaran isi intrakranial yang sangat jelas karena berkurangnya mineralisasi tulang kalvaria atau kompresi kalvaria. Selain itu dapat juga ditemukan tulang panjang yang bengkok, panjang tulang berkurang (terutama tulang femur), dan fraktur iga multipel. USG prenatal ini terutama untuk mendeteksi OI tipe II. 1-4
DIAGNOSIS BANDING
1. Child abuse dan penelantaran anak
Pada OI tipe ringan paling sulit dibedakan dengan kasus penelantaran anak. Usia fraktur tulang yang berbeda-beda pada neonatus dan anak harus dicurigai karena kasus penelantaran anak. Selain itu pada penelantaran anak juga terdapat manifestasi klinis non skeletal, misalnya perdarahan retina, hematoma organ visera, perdarahan intrakranial, pankreatitis dan trauma limpa. Tipe fraktur pada penelantaran anak biasanya adalah fraktur sudut metafiseal yang jarang ditemukan pada OI. Densitas mineral tulang pada penelantaran anak juga normal, sedangkan pada OI rendah. 1-4
2. Osteoporosis juvenil idiopati (OJI)
Keadaan ini ditemukan pada anak yang lebih tua, terutama antara 8 – 11 tahun, yang mengalami fraktur dan tanda osteoporosis tanpa didasari penyakit lainnya. Gejala biasanya nyeri tulang belakang, paha, kaki, dan kesulitan berjalan. Fraktur khasnya berupa fraktur metafiseal, meski dapat juga terjadi pada tulang panjang. Sering terjadi fraktur vertebra yang menyebabkan deformitas dan perawakan pendek ringan. Tulang tengkorak dan wajah normal. OJI akan membaik spontan dalam 3-5 tahun, namun deformitas vertebra dan gangguan fungsi dapat menetap. Jika didapat riwayat keluarga dengan keluhan yang sama maka harus dipikirkan suatu OI tipe ringan 1-4
3. Achondroplasia
Merupakan penyakit yang diturunkan secara autosomal dominan akibat mutasi pada gen FGFR3. Gen ini bertanggung jawab pada pembentukan protein yang berperan dalam pertumbuhan, perkembangan dan pemeliharaan tulang (osifikasi) dan jaringan otak. Klinis didapat sejak lahir berupa perawakan pendek, termasuk tulang belakang, lengan dan tungkai terutama lengan dan tungkai atas, pergerakan siku terbatas, makrosefali dengan dahi yang menonjol. Kejadian fraktur berulang tak pernah terjadi. 1-4
4. Riketsia
Merupakan gangguan kalsifikasi dari osteoid akibat defisiensi metabolit vitamin D. Walau jarang terjadi, riketsia juga bisa karena kekurangan kalsium dan fosfor dalam diet. Klinis yang ditemukan antara lain hipotoni otot, penebalan tulang tengkorak yang menyebabkan dahi menonjol, knobby deformity pada metafisis dan dada (rachitic rosary), bisa terjadi fraktur terutama tipe greenstick
fracture. Hasil pemeriksaan laboratorium ditemukan kadar 25-hidroksi-vitamin D
serum, kalsium dan fosfor yang rendah, serta alkalin fosfatase meningkat. Beberapa penyakit malabsorpsi intestinal berat, penyakit hati atau ginjal menimbulkan gambaran klinis dan biokimia sekunder riketsia nutrisional.Pada OI kalsium serum dan alkalin fosfatase normal. Kadar 25-hidroksi-vitamin D serum penderita OI sering rendah menunjukkan defisiensi vitamin D sekunder akibat kurangnya paparan terhadap sinar matahari yang sering dialami penderita OI. 1-4
PROGNOSIS
Osteogenesis imperfecta merupakan kondisi kronis yang membatasi tingkat fungsional dan lama hidup penderita. Prognosis penderita OI bervariasi tergantung klinis dan keparahan yang dideritanya. Bayi dengan OI tipe II biasanya meninggal dalam usia bulanan - 1 tahun kehidupan. Sangat jarang seorang anak dengan gambaran radiografi tipe II dan defisiensi pertumbuhan berat dapat hidup sampai usia remaja. Penderita OI tipe III biasanya meninggal karena penyebab pulmonal pada masa anak-anak dini, remaja atau usia 40 tahun-an sedangkan penderita tipe I dan IV dapat hidup dengan usia yang lebih panjang/ lama hidup penuh.
Penderita OI tipe III biasanya sangat tergantung dengan kursi roda. Dengan rehabilitasi medis yang agresif mereka dapat memiliki ketrampilan transfer dan melakukan ambulasi sehari-hari di rumah. Penderita OI tipe IV biasanya dapat memiliki ketrampilan ambulasi di masyarakat juga tak tergantung dengan sekitarnya. 1-5
DAFTAR PUSTAKA
1. Marini JC. Osteogenesis imperfecta. Dalam: Behrman RE, Kliegman RM, Jenson HB,eds. Nelson textbook of pediatrics, edisi ke-17. Philadelphia: Saunders, 2004, 2336-8
2. Beary JF, Chines A, dkk. Clinical features and diagnosis of osteogenesis imperfecta. review version 18.3: September 2010. Di dapat dari
www.Uptodate.com
3. Root AW, Diamond Jr FB. Disorders of calcium metabolism in the child and adolescent. Dalam: Sperling MA, eds. Pediatric endocrinology, edisi ke-2. Philadelphia: Saunders, 2002, 657-85.
4. Nussbaum RL, McInnes RR, Willard HF. The molecular and biochemical basis of genetic disease. Dalam: Thompson and thompson genetic in medicine, edisi ke-6. Philadelphia: Saunders, 2004, 229-346.
5. Chevrel G. Osteogenesis imperfecta. Didapat dari:
www.orpha.net/data/patho/GB/uk-OI.pdf
6. Smith R. Severe osteogenesis imperfecta: new therapeutic options? BMJ
2001;322:63-4. 16.
7. Dimeglio LA, Ford L, McClintock C, Peacock M. A comparison of oral and intravenous biphosphonates therapy for children with osteogenesis imperfecta. J Pediatr Endocrinol Metab 2005;18(1):43-5317.
8. Glorieux FH,. Experience With Bisphosphonates in Osteogenesis Imperfecta.
Pediatrics
2007;119;S163-S165
9. Cole DE. Psychosocial aspects of osteogenesis imperfecta: a n update. Am J Med Genet . 1993 ; 45:207.
10.Antoniazzi F, Mottes M, Fraschini P, et al. Osteogenesis imperfecta: practical treatment guidelines. Paediatr Drugs . 2000; 2:465.
CRTAP AND LEPRE 1 MUTATIONS IN RECESSIVE OSTEOGENESIS IMPERFECTA
PENDAHULUAN
Penderita Osteogeneis Imperfecta (OI) memiliki tulang yang rapuh sehingga meningkatkan resiko terjadinya fraktur. OI diturunkan secara autosomal dominan dan disebabkan oleh mutasi heterogenisitas pada gen COLIA1 dan COLIA2 (MIM #120150 dan #120160). Mutasi ini menyebabkan penurunan produksi secara kuantitatif kolagen tipe 1 yang berstruktur normal sehingga menyebabkan fenotip OI tipe yang paling ringan. Mutasi yang menyebabkan perubahan struktural pada semua rantai prekolagen tipe 1 dapat mempengaruhi asosiasi rantai, formasi tripel helix, sekresi dan atau formasi fibril dan secara keseluruhan menghasilkan fenotip yang lebih berat, termasuk perinatal lethal OI phenotype. Secara biokimia, abnormalitas struktur ini ditandai dengan peningkatan modifikasi post-translasi rantai prekolagen tipe 1.
Perubahan proses post-translasi prekolagen tipe 1 tanpa adanya mutasi juga dapat menyebabkan OI. Berdasarkan kejadian tersebut, mutasi pada CRTAP (MIM #605497) dan LEPRE1 (MIM #610339) yang mengkode protein prolyl 3-hydroxylase-1 (P3H1) dan protein leprecan dapat menyebabkan OI tipe resesif. CRTAP, atau cartilage associated protein, membentuk molecular kompleks di reticulum endoplasmik kasar dengan P3H1 dan cyclophilin B (MIM #123841, CYPB atau peptidylprolyly isomerase B, PPIB) dalam rasio 1:1:1. Baik pada tikus dan manusia, dengan adanya mutasi yang menyebabkan mutasi fungsi dari CRTAP yang menyebabkan hilangnya 3-hydroxylase dari target residu proline single di posisi 986 dalam domain triple helix atau residu 1164 yang diukur dari methionine insiator di rantai proα(1)I. Prolyl 3-hydroxylase merupakan salah satu
stabilitas, dan sekresi prekolagen. Prolyl 4-hydroxylase penting untuk stabilitas suhu dari triple helix sementara hidroksilasi lysyl dan glikosilasi hydroxylysyl berkontribusi terhadap stabilitas crosslink extraseluler antara molekul. OI pada manusia, homozigot atau compound heterozygot mutasi CRTAP dan LEPRE1 berhubungan dengan overmodifikasi post-translasi rantai kolagen tipe 1 yang disintesis oleh kultur fibroblast secara in vitro, yang menyebabkan retardasi mobilitas dalam elektroforesis. Walaupun semua molekul pada kelainan resesif ini overmodifikasi, dan pada kelainan dominan hanya 2/4-3/4 tergantung dari letak gen yang mengalami mutasi, sulit untuk membedakan kolagen overmodifikasi dikarenakan mutasi struktural kolagen tipe 1 atau mutasi pada kompleks prolyl 3-hydroksilasi.
Untuk mengidentifikasi mutasi yang mengganggu prolyl 3-hydroksilasi dan disaat yang bersamaan mengkaji kontribusi mutasi CRTAP, LEPRE1 dan PPIB (yang mengkode CYPB) pada patogenensis OI, kami menskrining DNA dari 78 pasien, yang diidentifikasi baik in utero atau pada saat lahir. Pada penelitian kohort ini, didapatkan 3 tambahan kejadian dimana OI disebabkan oleh mutasi CRTAP termasuk missense mutation yang dilaporkan pertama kali. Kami juga mengidentifikasi 16 kejadian dimana OI disebabkan oleh mutasi LEPRE1. 9 dari 11 mutasi LEPRE 1 adalah baru. Tidak didapatkan mutasi pada gen PPIB.
METODOLOGI
Subjek penelitian
Dipilih 78 subjek untuk skrining berdasarkan laporan hubungan darah, yang berasal dari komunitas dimana didapatkan peningkatan frekuensi konsanguinitas, adanya saudara kandung yang menderita OI atau adanya kegagalan dalam mengidentifikasi mutasi gen COLIA1 dan COLIA2 walaupun kultur sel menghasilkan protein yang abnormal. Beberapa subjek dipilih berdasarkan fenotip klinis dan radiografik mirip dengan kasus mutasi positif yang pernah dilaporkan. Dengan persetujuan Institutional review board, darah, fibroblast atau jaringan diambil dari subjek terpilih dan DNA disiapkan dengan protokol standar.
PCR dan sequence analysis
7 exon CRTAP, 15 exon LEPRE1 dan 5 exon PPIB, termasuk region intronik sekitar, diamplifikasi dari DNA genom dengan PCR dan dianalisis dengan fluorescent dye-terminator sequencing. Hasil dianalisis menggunakan software Sequncher 4.6 atau Mutation Surveyor. Sekuens subject dibandingkan dengan sekuen gen ensemble ENSG00000117385, ENSG00000170275 dan ENSG00000166794 untuk LEPRE1, CRTAP dan PPIB berturut-turut. SNPs diidentifikasi menggunakan kartu gen (www.genecards.org). untuk CRTAP dan LEPRE1, kami menggunakan penomoran kode sekuens konvensional dimana A dari inisiator kodon methionine merupakan nukleotida pertama (+1), dan inisiator metionin adalah asam amino +1. Hal ini menyebabkan poin referensi untuk kode sekuens LEPRE1 berbeda dengan yang digunakan Cabral et al dimana nukleotida pertama dari sekuens cDNA dinomori +1, dimana berdasarkan A pada kodon inisiator metionin bernomor -41.
RT-PCR
RNA total diekstraksi dari fibroblast menggunakan trizol dan sintesis cDNA dilakukan dengan Superscript III reverse trancriptase kit (Invitrogen). Real time quantitative PCR dilakukan menggunakan versi Lightcycler 1.1. Konsentrasi cDNA template ditentukan menggunakan Ribogreen dan untuk masing-masing q-PCR menggunakan konsentrasi ekuivalen. Fluoresen ditangkap pada tiap akhir siklus ekstensi (lebih dari 45 siklus). Poin penyilangan ditentukan oleh derivative kedua algoritme intrinsic dari software Lightcycler dan dinormalisasi menjadi ekspresi gen konstitutif (β2-microglobulin).
Spektrometri massa
3-hydroksilasi dari prolin pada posisi 986 dari triple helix di rantai α1(I) kolagen tipe 1 ditentukan dari kolagen yang disiapkan dari jaringan tulang atau kultur sel kulit fibroblast atau osteoblast tulang. CB peptide atau pepsinized collagens diisolasi dengan SDS-PAGE dan tryptic digest untuk MS/MS. Electrospray MS
menggunakan LCQ Deca XP ion-trap mass spectrometer dengan in-line liquid chromatography.menggunakan kolom kapiler C8 (300mm x 150mm) eluted 4.5 ml/mnt. Mobile gradient dibuat dari buffer A (0,1% formic acid ditambah 2% buffer B dalam air milliQ) dan buffer B (0,1% formic acid dalam 3:1 acetonitrile:n-propanol v/v). Electrospray Ionization Source (ESI) dibawah tekanan atmosfer digunakan untuk emngenalkan sampel pada spectrometer massa. Voltase spray adalah 3kV dan temperaratur inlet kapiler 1600C.
HASIL
Dari 78 individu, didapatkan 3 proband dengan mutasi CRTAP dan 16 proband dengan mutasi gen LEPRE1. 2 dari proband merupakan heterozygous sedangkan sisanya homozygous (table S1). Temuan ini menggambarkan adanya kosanguinitas orangtua pada setidaknya 8 dari 19 probands. Semua probands memiliki bentuk OI berat, baik tipe II (perinatal lethal OI) atau tipe III (severly deforming OI). 7 meninggal 1 tahun pertama setelah lahir, 5 didiagnosa OI berat in utero dan kehamilannya diterminasi, 5 proband dapat melewati 1 tahun pertama kehidupan dengan usia paling tua adalah 16 tahun, dan 1 proband masih hidup dengan usia 5 bulan.
Temuan radiologis pada proband dengan mutasi CRTAP dan LEPRE1 ditunjukkan pada gambar 1 dan 2. Pada saat foto diambil, proband 2 berusia 12 bulan, wanita, kaukasian yang merupakan compound heterozygous mutasi CRTAP (c.278_293dup and c.822_826delAATACinsT). Kedua mutasi memperkirakan frameshift dan downstream terminasi kodon prematur. Saat lahir, dia memiliki fraktur multiple tulang iga, klavikula, humerus, radius, mandibular dan ulna dengan sclerosis, poor definition dan pelebaran metafise tulang panjang (gambar 1). Saat usia 12 bulan, dapat hidup tanpa alat bantu nafas. Proband 7 didapatkan homozygous untuk LEPRE1 nonsense kodon, c.392C>A, p.Ser131X. saat lahir didapatkan fraktur multiple tulang panjang dan iga. (gambar 2A-D), ekstremitas pendek, penurunan tonus dan distress nafas. Secara radiografis, didapatkan osteopenia, rhizomelia, brachycephaly, penurunan ketebalan calvaria, dan tulang iga yang tipis dengan fraktur multiple.
Gambaran radiologis pasien yang bertahan lebih dari 1 tahun pertama kehidupan yang mengalami mutasi CRTAP dan LEPRE1 ditunjukkan pada gambar 3 dan 4
berurutan. Proband 3 merupakan homozygot dari missense mutation CRTAP, c.200T>C(p.leu67Pro). proband ini merupakan keturunan iran yang menikah dengan sepupu pertama. Pada saat studi ini diakukan, pasien ini berusia 9 tahun. Pasien ini mengalami fraktur multipel saat lahir dan menunjukkan keterlambatan pertumbuhan segera setelah lahir. Sejak lahir hingga usia 7,5 tahun telah tercatat mengalami 16 kali fraktur (gambar 3), tetapi 18 bulan terakhir tidak didapatkan. Pada saat usia 7,5 tahun, TB 71 cm (z= -12,87, P50 untuk usia 10,5 bulan) dan BB 11 kg (z= -7,91, P50 untuk usia 18 bulan). Fungsi parunya normal dan sesuai untuk anak seukurannya. Pada anak ini juga didapatkan skoliosis yang signifikan dan bengkok pada tulang panjangnya. Secara radiografis, tulang ini terlihat gracile dan besar, bulbous “popcorn” epiphyses (gambar 5A). Pasien ini tidak dapat berdiri atau berjalan, untuk mobilitas menggunakan wheel bed, dan secara intelektual tergolong normal. Kehamilan kedua pada keluarga ini diterminasi pada usia kehamilan 19 bulan dikarenakan dari USG menunjukkan OI.hasil tes dari kehamilan ini tidak menunjukkan adanya mutasi pada kolagen tipe 1, tetapi menunjukkan homozygositas missense mutasi yang sama dari CRTAP pada proband 3 (c.200T>C; p.Leu67Pro). kedua orangtuanya merupakan heterozygout untuk mutasi tersebut dan varian ini tidak ditemukan pada 100 orang kelompok kontrol. Kadar CRTAP mRNA normal pada fibroblast pasien (tabel 1). Residu asam amino pada CRTAP is evolutionary conserved diantara sequen vertebrata, kecuali beberapa spesies ikan.
Proband 10 merupakan contoh lain yang usia hidupnya lama pada penelitian kohort ini. Kasus ini sebelumnya dilaporkan oleh Cabral et al (2007) sebagai proband 5 dan mutasi yang dicatat oleh Cabral et al (c.1656C>A; p.Tyr552X) juga dikonfirmasi positif pada penelitian ini. Orang tuanya juga masih merupakan sepupu dan memiliki kakek nenek yang sama. Selama kehamilan pertama, didapatkan fraktur in utero dan pada saat lahir. Pada saat dievaluasi usia 6 tahun, anak ini tampak kecil, mobilitasnya menggunakan kursi roda dan memiliki deformitas di semmua ekstremitas serta skoliosis yang signifikan. Pada saat usia 11 tahun, temuan klinis masih sama, tetapi pada temuan radiologisnya didapatkan epifisis tulang panjang yang berbentuk bulbus dan besar dengan regio ireguler (popcorn epiphyses) (gambar 4A-C, 5B).
Dua contoh proband yang usia hidupnya lama menunjukkan suatu striking bulbous “popcorn” epiphyses (gambar 5) yang mungkin merupakan karakteristik
yang dapat membedakan oi tipe dominan dan resesif. Walaupun demikian, dari kasus yang digambarkan diatas, fenotip proband dengan mutasi di CRTAP dan LEPRE1 sulit untuk dibedakan. Kesimpulan ini digambarkan pada gambar 1-4 yang menunjukkan fenotip klinis proband 1 dengan mutasi CRTAP dan proband 4, 5, 14, 16 dan 17 dengan mutasi LEPRE1.
Diantara mutasi CRTAP dan LEPRE1 yang diidentifikasi pada peneitian ini (gambar 6), 7 proband memiliki mutasi frameshift, dimana semuanya mengakibatkan terminasi downstream kodon dan nonsense mediated decay (NMD). Tujuh proband lainnya mengalami mutasi yang mengacaukan splice donor sites dan diprediksi menyebabkan splicing abnormal. Semua proband kecuali 2 (proband 18 dan 19), missplicing menyebabkan terminasi prematur dan NMD. Ini termasuk proband 11, 12, 13, 14 yang semua homozygot mutasi LEPRE1 pada keturunan west Africa, dan proband 15 yang merupakan compound heterozygote dan mutasi nonsense. Empat proband lainnya memiliki mutasi nonsense dimana proband ini diprediksi mengarah ke NMD, sementara mutasi pada proband 9 (c.2041C>T) dalam 50 nukleotida terakhir dari exon penultimate, yang dapat terhindar dari NMD. Proband 3 yang didiskusikan diatas merupakan satu-satunya yang mengalami mutasi missense pada LEPRE1 atau CRTAP. Sehingga, pada mayoritas kasus resesif OI berat, hilangnya stabilitas mRNA mewakili konsekuensi primer dari mutasi.
Untuk mengkonfirmasi efek dari mutasi genomik pada stabilitas transkrip, RT-PCR dilakukan setelah RNA dari kultur sel tersedia. Dari semua kasus dimana frameshift, nonsense atau splice site mutation menyebabkan terminasi kodon prematur, didapatkan reduksi signifikan dari kuantitas transkrip LEPRE1 atau CRTAP dibanding kelompok kontrol yang konsisten dengan efek prediktif NMD (tabel 1). Pada sel dari proband 3, yang homozygot untuk mutasi missense pada CRTAP, tidak didapatkan reduksi jumlah transkrip. Sel dari proband 9, yang homozygot untuk mutasi nonsense dalam 50 pasangan basa terakhir dari exon penultimate LEPRE1, mengandung separuh dari jumlah normal transkrip, yang bisa disimpulkan adanya sejumlah signifikan transkrip yang terhindar dari NMD. Proband 18 dan 19 masing-masing homozygot untuk subtitusi pasangan tunggal basa yang unik pada splice donor site pada permulaan intron 6 dari LEPRE1 (c.1170+2T>A dan c.1170+5G>C). Kedua mutasi ini menghasilkan kuantitas normal transkrip mRNA (tabel 1), karena satu-satunya hasil splice yang jelas dari
sel dari kedua proband adalah melewati in frame dari exon 6 yang menghasilkan delesi 90 nukleotida.
Untuk mengevaluasi efek biokimia dari mutasi, kadar 3-hydroxylase dianalisa menggunakan spectrometri massa dari kolagen tipe 1 yang berasal dari fibroblast-conditioned media, dan tulang. Dari semua sampel yang diperiksa didapatkan penurunan kadar 3-hydroxylase prolin pada posisi 986 dari triplehelix rantai α(I) kolagen tipe 1 yang berasal dari kultur fibroblast seperti pada table 1. Kolagen fibroblast yang berasal dari proband 3, homozygote untuk missense mutasi CRTAP, dan memiliki fenotip paling ringan dibandingkan proband yang jumlah mRNAnya diprediksi hampir tidak ada, menghidroksilasi 79% dari taget residu prolin. Sel dari proband 11, yang memiliki splice site mutasi LEPRE1 yang menghasilkan beberapa produk, memiliki 4% 3-hydroxyproline dalam kolagen fibroblast tetapi 55% 3-hydroxyproline pada kolagen tipe 1 dari bone derived cell (gambar 7). Sebaliknya, proband 1, yang merupakan homozygote dari frameshift mutasi CRTAP, memiliki 4% 3-hydroxyproline dalam kolagen fibroblast dan 2% dalam tulang dibandingkan dengan kelompok control (gambar 7).
DISKUSI
Berdasarkan temuan fenotip dalam penelitian ini, hilangnya fungsi secara komplit atau hampir komplit baik CRTAP maupun LEPRE1 memiliki efek terhadap derajat OI, terutama tipe II dan III. Didapatkan 3 indentifikasi baru alel CRTAP dan 9 identifikasi baru alel LEPRE1 yang memperluas spectrum alel mutant yang ditemukan hingga saat ini (gambar 6). Proband 11, 12, 13 dan 14 homozygot, dan proband 15 heterozygot untuk mutasi pada splice donor site di intron 5 LEPRE1 yang disebut sebagai “West African Allele”. Proband 2 merupakan compound heterozygote mutasi CRTAP dan menurunkan 16 alel duplikasi pasangan basa kepada infant 3 seperti dijelaskan pada Barnes et al.
Missense mutasi CRTAP, c.200T>C(p.Leu67Pro), pada proband 3 menjelaskan gangguan pada sekuens asam amino dari CRTAP dapat menyebabkan OI, dan pada kasus ini mirip walaupun lebih ringan dibandingkan kehilangan komplit dari protein. Penelitian lebih lanjut dibutuhkan untuk menetukan bagaimana penggantian asam amino ini mengganggu fungsi CRTAP.
Adanya hilang fungsi alel LEPRE1 yang unik (c.232delC) pada 3 populasi Irish Traveller dimana keluarganya tidak memiliki hubungan kerabat, memiliki implikasi klinis dan epidemiologis penting pada komunitas ini. Populasi ini memiliki konsanguinitas signifikan dan peningkatan kondisi penyakit genetik dan metabolic yang diturunkan secara autosomal resesif, termasuk OI. Penelitian sebelumnya menunjukkan bahwa OI pada komunitas ini tidak berhubungan dengan 2 gen kolagen tipe 1, COLIA1 dan COLIA2. Peningkatan prevalensi OI pada komunitas ini disebabkan karena tingginya frekuensi karier untuk mutasi LEPRE1 sehingga tes untuk karier dapat diimplementasikan secara efektif. Proband 4, salah satu anggta komunitas Irish Travellers yang memiliki usia hidup lebih panjang dari perkiraan (pada saat penelitian berusia 5 tahun), yang memiliki intelegensi normal dan mobilisasi menggunakan kursi roda, merupakan hasil terapi yang didapat sejak lahir menggunakan bisphosphonates (IV pamidronate, 12 mg/kg/tahun dibagi menjadi dosis tiap 3 bulan) yang sering digunakan sebagai terapi pada anak dengan OI berat. Atau merupakan gambaran latar belakang genetic yang berbeda dimana homozygout mutasi LEPRE1 diekspresikan. Kami berspekulasi bahwa terapi dini dengan obat ini dapat menurunkan perluasan terjadinya bentuk struktur bulbous epiphyses (popcorn epiphyses), mengingat tidak tampaknya gambaran ini pada anak usia 5 tahun yang mendapatkan terapi tetapi pada anak usia 6 tahun yang tidak mendapat terapi, gambaran ini muncul.
Mutasi pada CRTAP dan LEPRE1 masing-masing dapat menyebabkan gambaran klinis yang luas, dari mulai paling berat dan seringnya lethal seperti OI tipe II/III sampai dengan yang ringan (OI tipe VII) yang terlihat pada mutasi CRTAP yang masih dapat memproduksi beberapa protein normal. Konsanguinitas orang tua merupakan feature yang membantu dalam membedakan mereka yang dengan mutasi resesif CRTAP atau LEPRE1 dari mutasi kolagen tipe 1 yang dimana parental mozaicism dapat menyebabkan rekurensi yang menyerupai inheritan resesif. Diantara bayi dengan mutasi CRTAP atau LEPRE1 yang menyisakan sedikit fungsional mRNA, presentasi klinis sulit untuk dibedakan. Bayi0bayi ini sering terdiagnosis in utero dengan penampakan mirip dengan bentuk lethal OI yang dihasilkan dari heterozygote mutasi kolagen tipe 1 dan mungkin mengalami fraktur multiple dari tulang panjang dan tulang iga saat lahir, berkurangnya mineralisasi calvaria, dan penurunan pembentukan tulang. Cabral et al (2007) mengatakan bayi dengan
mutasi LEPRE1 berat berbeda dengan mutasi kolagen tipe 1 dimana pada bayi dengan mutasi LEPRE1 memiliki sclera putih dibandingkan biru, wajah yang bulat dibandingkan segitiga, dan dada barrel-shaped dibandingkan narrow. Berdasarkan temuan kami, masih tidak jelas apakah temuan ini ditemukan secara konsisten pada semua kasus dengan mutasi LEPRE1. Masih diperlukan sampel yang lebih banyak lagi pada bentuk mutasi ini untuk dapat memastikan adanya karakteristik yang khas. Didapatkan gamnbaran popcorn epiphyses pada pasien yang memiliki homozygote missense mutasi dari CRTAP (proband 3, gambar 5A) dan pasien yang memiliki homozygote null mutation P3H1 (proband 10, gambar 5B). epifise abnormal ini menggambarkan dysplasia kartilago pada growth-plate sebagai akibat adanya defek pada formasi tulang yang disebabkan oleh mutasi prolyl 3-hidroxilation complex. Spekulasi ini konsisten dengan kolagen tipe II pada kartilago yang juga dimodifikasi oleh prolyl 3-hidroxilation, dan cartilage CRTAP knock out mice yang tidak menunjukkan adanya prolyl 3-hidroxilation.
Null mutation pada CRTAP dan LEPRE1 mungkin tidak memiliki hasil yang dapat dibandingkan. Contohnya, sementara proband 1 dengan mutasi CRTAP memiliki 2-4% kadar 3-hydroxyroline, pasien lain yaitu proband 11 dengan mutasi LEPRE1 memiliki sekitar 50% 3-hydroxyproline dari kultur osteoblast dan hanya 4% dari kultur dermal fibroblast rantai α1(I). hal ini menunjukkan kemungkinan adanya kompensasi hilangnya fungsi dari P3H1 pada sel tulang, ke,umgkinan oleh P3H2 atau P3H3. Dari penelitian ini tidak dapat dieksklusi adanya data dari fibroblast kemungkinan suatu artefak atau mutasi LEPRE1 menyebabkan keluaran hasil splicing yang berbeda antara di tulang dan fibroblast.
Mekanisme yang pasti dari mutasi CRTAP dan LEPRE1 dalam menyebabkan OI semakin tidak jelas. Populasi produk protein dari LEPRE1 dapat berinteraksi dengan protein CRTAP untuk membentuk kompleks dengan cyclophilin B. protein ini mungkin merupakan “true” secreted protein leprecan yang mengandung 2 situs glycosoglycan tambahan. Fungsi leprecan dan lokalisasinya pada matriks ekstraseluler masih diteliti. Identifikasi sel dengan null mutation dapat menyediakan alat untuk membedah fungsi elemen yang berbeda dari protein kompleks tersebut. Pathogenesis molekulernya kemungkinan lebih kompleks dibandingkan sekedar kekurangan 3-hydroxylation tipe 1 dan mungkin kolagen lain.
Penelitian ini menunjukkan mutasi CRTAP dan LEPRE1 berkontribusi signifikan terhadap kohort individual OI dengan fenotip berat dan resesif. Pada populasi West Africa, bentuk berat dari OI disebabkan mutasi LEPRE1. Pada populasi ini angka karier mencapai 1/100 – 1/280 dengan kemungkinan menghasilkan keturunan dengan OI sebesar 1/40.000 kelahiran. Kemungkinan OI lethal sebesar 1/50.000 – 1/60.000 kelahiran menunjukkan proporsi signifikan dari populasi ini dapat disebabkan oleh allele tunggal saja dibandingkan parental mosaicsm.
Dari penelitian ini timbul pertanyaan bagaimana sebaiknya mendiagnosis OI. Dari 14 subjek penelitian didapatkan sel yang mensintesa rantai prokolagen tipe 1 yang abnormal dan mengubah mobilitas elektroforesis. Tetapi sulit untuk menentukan apakah semua rantai memiliki mobilotas abnormal, sehingga, sementara studi protein mengidentifikasi abnormalitas sel dan mutasi dari CRTAP, LEPRE1 dan gen prokolagen tipe 1, tes terhadap mutasi ini penting untuk mengetahui pola inheritan, perkiraan resiko rekurensi dan menyediakan fasilitas tes untuk parental mosaicsm pada gen kolagen. Data mutasi dapat mendasari penelitian tentang genotip-fenotip, termasuk menetukan apakah “popcorn epiphyses” merupakan tanda klinis khusus untuk menentukan prognosis individu dengan mutasi CRTAP atau LEPRE1. Sampai semua situs splicing bisa diidentifikasi, mengerti bagaiman prognosis dari adanya mutasi baru hanya bisa berdasarkan gambaran klinis dan kerja RNA di sel. Implementasi dari high density arrays akan memfasilitasi adanya delesi dan duplikasi subtle dan dapat menjadi tambahan penting untuk diagnosis berdasarkan sequencing. Untuk mengerti petogenesis molekuler, selain dibutuhkan sampel darah untuk isolasi DNA juga dibutuhkan kultur sel dari jaringan target.
TELAAH KRITIS JURNAL
Penelitian ini bertujuan untuk memberikan gambaran tentang factor penyebab OI tipe resesif dimana pada penelitian sebelumnya didapatkan gambaran klinis OI tanpa adanya mutasi pada COLIA 1 dan COLIA 2 Tujuan dari penelitian tersebut sudah sangat jelas didasarkan pada rumusan masalah yang ada
Teknik pengambilan sampel sudah dijelaskan dengan rinci, termasuk adanya ”inform concent” untuk individu yang dijadikan sampel. Jumlah sampel sudah jelas, karakteristik sampel jelas.
Metode untuk menganalisa data sudah dijelaskan secara rinci. Dari data-data yang telah dianalisis dapat digunakan untuk menjawab rumusan masalah, data-data tersebut sudah dapat dijadikan sumber informasi.
Hasil penelitian sesuai dengan data yang didapat, sudah dapat memberikan gambaran/jawaban atas permasalahan yang ada.
Kesimpulan sudah diambil berdasarkan hasil penelitian.
Hasil dari penelitian ini dapat digunakan kembali untuk penelitian-penelitian selanjutnya.