STUDI KOMPUTASI METODE AB INITIO DFT DALAM KAJIAN STRUKTURAL DAN SIFAT ELEKTRONIK SENYAWA KALSIUM BOROHIDRID-DIAMONIA SEBAGAI
PENYIMPAN HIDROGEN
Muhammad Arsyik Kurniawan S.
Program Studi Kimia FMIPA Universitas Islam Indonesia Jalan Kaliurang KM 14,5, Sleman, Yogyakarta, 55584
E-mail: [email protected]
ABSTRAK
Telah dilakukan kajian teoritis tentang struktur dan sifat elektronik dari senyawa Ca(BH4)2∙2NH3 sebagai salah satu material yang berpotensi digunakan untuk menyimpan dan penghasil hidrogen melalui proses dehidrogenasi. Perhitungan teoritis energi, Density
of States dan celah pita elektron dari struktur kompleks Ca(BH4)2∙2NH3 dilakukan dengan
Density Functional Theory (DFT) dan fungsi energi perubahan dan korelasi menggunakan
metode Generalized Gradient Approximation (GGA).
Berdasarkan analisis struktur dan sifat elektronik kristal kompleks Ca(BH4)2∙2NH3 diperoleh atom–atom hidrogen yang berpotensi sebagai sumber molekul hidrogen yaitu hidrogen (Hδ-) dari BH4 dan hidrogen (Hδ+) dari NH3 dalam bentuk ikatan dihidrogen N– H∙∙∙H–B. Hasil perhitungan simulasi didapatkan besar celah energi band gap sebesar 5,68 eV, yang menyatakan material ini sebagai insulator. Dari data analisis lebih lanjut terhadap studi pelepasan molekul H2 vs NH3 sebesar 2,30 eV vs 1,52 eV, mengindikasikan material Ca(BH4)2∙2NH3 lebih rendah untuk melepaskan molekul H2 dibanding NH3, data ini sesuai dengan hasil pengamatan eksperimen.
Kata kunci: penyimpan hidrogen, density functional theory, ikatan dihidrogen.
ABSTRACT
Study of structure and electronic properties of solid Ca(BH4)2∙2NH3 as a hydrogen storage and production material has been performed. The energy, density of states, and band structure of solid Ca(BH4)2∙2NH3 have calculated within framework of density functional theory (DFT) and generalized gradient approximation (GGA) for the exchange-correlation energy functional.
Structure analysis and electronic properties of Ca(BH4)2∙2NH3 indicated that the source of hydrogen molecule was verified by interaction from hydrogen (Hδ-) from and hydrogen (Hδ+) from NH3 in dihydrogen bonding site B–H∙∙∙H–N. The calculated band structures implied an indirect wide band gap of 5.68 eV, indicated that this material is insulator. The simulation result gets that removal energy of H2 vs NH3 is 1.52 eV vs 2.30 eV, indicated dissociation of H2 is less favorable than NH3 molecule, this theoritical study has a good agreement with experiment observation.
Studi Komputasi Metode Ab Initio Dft Dalam Kajian Struktural dan Sifat Elektronik
Pendahuluan
Minyak bumi yang melimpah sebagian besar digunakan untuk memproduksi bensin atau bahan bakar lainnya, tingginya eksplorasi minyak bumi memungkinkan cadangan minyak bumi akan mencapai masa limit sekaligus meningkatnya pemanasan global akibat produksi karbon dioksida (CO2) yang tinggi. Tuntutan energi global yang melonjak bersama – sama dengan meningkatnya kesadaran akan habisnya sumber daya bahan bakar fosil ini mendorong para peneliti maupun ilmuwan melakukan upaya pencarian sumber energi baru seperti hidrogen (Anonim, 2007).
Keuntungan lain dari gas hidrogen adalah keberadaannya yang melimpah di alam, serta kemungkinannya untuk digunakan dan diproduksi kembali
(renewable), berbeda dengan bahan
bakan hidrokarbon jenis minyak bumi yang sangat melimpah tetapi tidak bisa diproduksi kembali dalam waktu singkat
(unrenewable) sehingga memungkinkan
untuk habis (Irani, 2002).
Dalam konsep energi yang dapat diperbaharui (renewable energy), saat ini teknologi mengarah kepada pemanfaatan material zat padat yang digunakan sebagai bahan penyimpan gas hidrogen yang mudah diproduksi, diantaranya adalah molekul kompleks amonia boran
(NH3-BH3) sebagai material penyimpan hidrogen (Baitalow et al., 2002) yang mampu menyimpan dan menghasilkan gas hidrogen melalui reaksi dehidrogenasi, senyawa amonia-boran (NH3∙BH3) mengandung 19,6% hidrogen telah dipertimbangkan sebagai bahan yang menjanjikan sebagai material penyimpan hidrogen. Namun untuk melepaskan semua hidrogen dari amonia-boran membutuhkan suhu yang sangat tinggi sekitar 1000 oC (Frueh et al., 2011).
Perkembangan eksperimen telah dilakukan untuk meningkatkan kemampuan penyimpanan dan menghasilkan gas hidrogen yang lebih banyak dengan reaksi dehidrogenasi yang lebih baik, diantarannya dengan mereaksikan amonia-boran dengan hidrida logam alkali maupun alkali-tanah.
Campuran amonia-boran dengan hidrida logam alkali yang telah dikembangkan seperti Mg(BH4)2.2NH3 (Soloveichik et al., 2008), Ca(NH2BH3)2 -NH3 (Chua et al., 2009). Namun saat ini muncul masalah lainnya dimana kombinasi amonia-boran dengan hidrida logam justru menghasilkan pelepasan amonia (NH3) daripada gas hidrogen (H2) seperti yang dialami oleh Chu et al.
(2010) pada senyawa Ca(BH4)2∙2NH3, hal ini sangat tidak diinginkan karena selain mempengaruhi efisiensi hasil H2
juga menyebabkan gas H2 yang terkontaminasi amonia (NH3).
Reaksi decomposisi senyawa Ca(BH4)2∙2NH3 seperti pada persamaan reaksi dibawah ini,
Persamaan reaksi 1
Persamaan reaksi 2
Ca(BH4)22NH3 1/4Ca(BH4)2 1/4Ca3(BN2)2 BN
190oC
6H2
Pada reaksi 1 dilakukan didalam sistem reaktor terbuka, sedangkan reaksi 2 pada sistem tertutup (closed vessel).
Pembahasan tentang tentang struktur dan sifat elektronik sangat dibutuhkan dalam memahami sifat decomposisi material dan juga untuk pengembangan studi lebih lanjut mengenai sifat penyimpan hidrogen. Pada artikel ini, kajian struktur dan sifat elektronik material Ca(BH4)2∙2NH3 dipelajari dengan menggunakan tehnik
Projector Augmented-Wave (PAW) dari
metode Density Functional Theory
(DFT), sehingga diharapkan dari kajian ini memberikan gambaran dan pemahaman tentang ikatan dan sifat dekomposisi dari Ca(BH4)2∙2NH3.
Tujuan Penelitian
Penelitian ini dilakukan diantaranya bertujuan untuk hal-hal sebagai berikut :
1. Penggunaan tehnik Projector
Augmented-Wave untuk pemodelan
zat padat (solid state) dalam hal ini material Ca(BH4)2∙2NH3.
2. Mempelajari struktur dan sifat elektronik material Ca(BH4)2∙2NH3
Metode Penelitian Perangkat Keras
Perangkat keras yang digunakan dalam penelitian ini menggunakan komputer dengan spesifikasi: CPU
Processor Intel Core2Quad 2.66 GHz
dengan Physical Memory 4Gb, kapasitas
Storage 200 Gb, serta GPU.
Perangkat Lunak
Perangkat lunak atau Software yang digunakan diantaranya:
1)Program ABINIT, (Gonze et al.
2009) , untuk perhitungan optimasi geometri.
2)Program AtomPAW, (Holzwarth et al. 2001), sebagai generator basis set 3)Program Vesta, (Momma and Izumi,
2011), digunakan untuk visualisasi struktur elektronik.
4)Program GnuPlot, digunakan untuk analisis dan ploting data statistik fungsi gelombang.
Bahan Kajian
Bahan kajian modeling struktur kristal Ca(BH4)2∙2NH3 diperoleh dari spektra
Studi Komputasi Metode Ab Initio Dft Dalam Kajian Struktural dan Sifat Elektronik instrumen XRD dari publikasi
eksperimen Chu et al. (2010).
Rancangan Penelitian
Pembuatan Basis Set Plane-wave
Basis set adalah representasi fungsi matematika yang digunakan sebagai deskriptor fungsi gelombang elektron untuk disertakan dalam perhitungan. Basis set yang digunakan adalah plane-wave Projector Augmented-Wave (PAW) yang berbentuk persamaan
yang dikembangkan oleh Blöchl (1994), indeks j merupakan indeks tiap elektron, koefisien k dipengaruhi oleh ukuran kisi nyata (real lattice) dan koefisien G dipengaruhi oleh ukuran kisi kebalikannya (reciprocal lattice). Secara garis besar basis set plane-wave bergantung pada ukuran kisi sistemnya
Studi Konvergensi Energi Kinetik
Basis set plane wave secara khusus diterapkan dalam pemodelan yang menggunakan tehnik pendekatan
supercell untuk sistem yang tak terhingga
jumlahnya seperti kisi kristal. Parameter khusus seperti energi kinetik cutoff yang digunakan sebagai pembatas jumlah plane wave yang akan digunakan. Seperti pada pers. (2) koefisien ekspansi
dari pers. (1) akan berkurang secara
eksponensial dengan meningkatnya energi kinetik dari fungsi plane wave,
Kualitas basis set plane wave hanya bergantung terhadap ukuran kisi dan nilai energi kinetik cutoff. Fungsi gelombang yang digunakan harus dapat menghitung nilai energi di setiap titik ruang didalam kisi kristal yang terintegrasi dalam zona Brillouin
(Ashcroft dan Mermin, 1976), titik-titik dalam ruang kisi ini disebut k-point.
Studi Struktur dan Elektronik kristal Ca(BH4)2∙2NH3
Metode DFT (Hohenberg dan Kohn, 1964) memperhitungkan bahwa energi sistem merupakan besaran kerapatan sistem sebagai nilai total dari kerapatan semua fungsi gelombang dalam sistem tersebut.
Dengan pendekatan yang dikembangkan oleh Kohn dan Sham (1965), nilai minimum dari fungsi total energi merupakan energi GS (ground-state) sistem, maka kerapatan elektronik yang memiliki nilai minimum ini merupakan energi eksak keadaan dasar dari sistem. Persamaan energi total sistem merupakan jumlah dari energi potensial, kinetik dan energi interaksi pertukaran dan korelasi antar elektron.
Energi pertukaran dan korelasi (xc) dari suku ketiga pers 3 diatas menggunakan metode pendekatan gradien tergeneralisir (GGA) dari Perdew
et al. (1996). Optimasi dilakukan dengan
kondisi toleransi perbedaan gaya hingga mencapai 0,025 eV/Å dan toleransi untuk konvergensi geometri tegangan kisi sebesar 0,05 GPa hingga didapatkan struktur terelaksasikan baik posisi atom, bentuk kisi dan volume kisi, kemudian kajian dilanjutkan dengan mempelajari bentuk struktur dan sifat elektronik dari kristal Ca(BH4)2∙2NH3.
Pembahasan
Pembuatan Basis set Plane-Wave
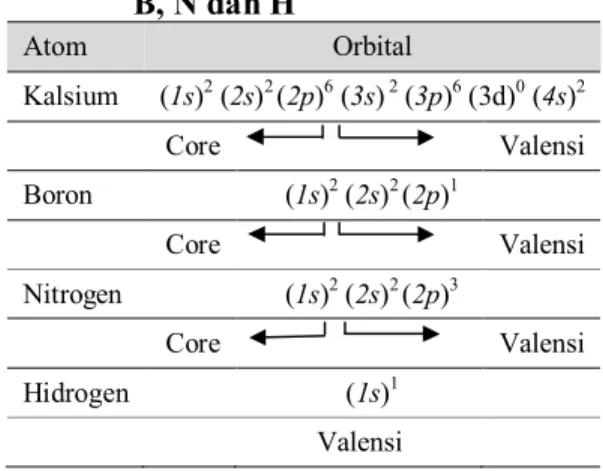
Dalam sistem zat padat dimana atom tidak bisa bergerak secara leluasa seperti pada fasa cair maupun gas, sehingga dapat diasumsikan bahwa dalam zat padat tidak semua elektron ikut serta dalam interaksi dengan atom lain karena sifat rigid sistem padat. Dalam penyusunan basis set fungsi gelombang elektron harus mempertimbangkan keterlibatan seluruh atau sebagian orbital dalam suatu atom untuk berinteraksi dengan atom lain dalam bentuk ikatan, seperti yang disajikan dalam Tabel 1.
Tabel 1. Orbital core-valensi atom Ca, B, N dan H Atom Orbital Kalsium (1s)2 (2s)2 (2p)6 (3s) 2 (3p)6 (3d)0 (4s)2 Core Valensi Boron (1s)2 (2s)2 (2p)1 Core Valensi Nitrogen (1s)2 (2s)2 (2p)3 Core Valensi Hidrogen (1s)1 Valensi
Batas radius optimum orbital core untuk mendapatkan orbital valensi sebagai basis set plane wave dari atom Ca, B, N, dan H masing-masing sebesar 2,9; 1,2; 0,9; dan 0,55Bohr. Penggunaan orbital luar (valensi) akan meringankan proses perhitungan komputasi, dan menghemat waktu untuk simulasi.
Studi Konvergensi Terhadap Energi Kinetik Cutoff
Studi konvergensi energi kinetik merupakan cara untuk mengetahui basis set (fungsi gelombang) telah memenuhi kriteria baik dalam memodelkan sistem, yang berarti dengan studi ini bertujuan mencari energi ground-state terendah sistem dengan parameter energi kinetik. Energi kinetik cutoff merupakan batas perbedaan energi total sistem (Etot) hasil iterasi perhitungan hingga didapat perbedaan energi sistem satu dengan berikutnya sebesar 0,001 Ha.
Studi Komputasi Metode Ab Initio Dft Dalam Kajian Struktural dan Sifat Elektronik
Gambar 1. Struktur kisi kristal Ca(BH4)2∙2NH3
Hasil grafik gambar 1 diperoleh selisih energi total sebesar 0,001 Ha terjadi pada energi kinetik sebesar ±25,7 Ha atau 700 eV, yang berarti dengan energi kinetik sebesar 700 eV telah didapat struktur Ca(BH4)2∙2NH3 yang teroptimasi.
Studi Struktur dan Elektronik kristal Ca(BH4)2∙2NH3
Studi stuktur Ca(BH4)2∙2NH3 bertujuan mendapatkan model terbaik sesuai dengan eksperimen. Cara yang dilakukan adalah dengan optimasi geometri terhadap strukur yang didapat dari eksperimen menggunakan basis set yang telah dibuat dan parameter optimasi antara lain, energi kinetik cutoff 700 eV, skema titik k-point 3x3x3.
Optimasi menggunakan metode
ab initio DFT di dalam program ABINIT
dan energi perubahan dan korelasi (Exc) elektron menggunakan pendekatan gradien tergeneralisir (GGA) dari Perdew
et al. (1996).
Gambar 2. Struktur kisi kristal Ca(BH4)2∙2NH3
Representasi hasil optimasi geometri terhadap kisi kristal Ca(BH4)2∙2NH3 diperlihatkan dalam Gambar 2 dan 3.
Gambar 3. Struktur 3D kristal Ca(BH4)2∙2NH3
Hasil optimasi didapatkan bentuk geometri kristal Ca(BH4)2∙2NH3 seperti pada Gambar 2, dimana dalam satu kisi kristalnya berisi 76 atom yang tersusun dengan space group ortorombik (Pbcn:60). Perhitungan komputasi menggunakan satu kisi kristal Ca(BH4)2∙2NH3 dianggap sudah mewakili struktur kristal Ca(BH4)2∙2NH3 dalam keadaan meruah (bulk) seperti pada Gambar 3 menggunakan teknik
Ca B N H
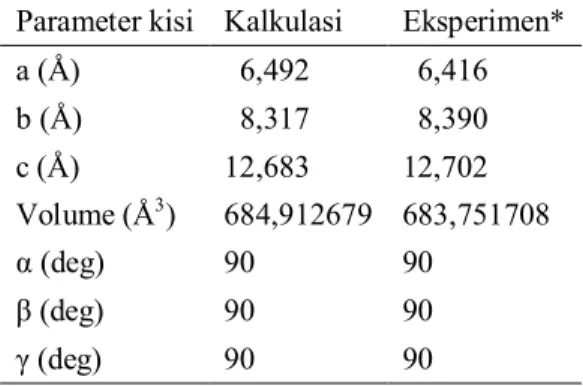
supercell. Parameter kisi hasil optimasi diperlihatkan pada Tabel 2.
Tabel 2. Parameter kisi hasil kalkulasi vs eksperimen
Parameter kisi Kalkulasi Eksperimen*
a (Å) 06,492 06,416 b (Å) 08,317 08,390 c (Å) 12,683 12,702 Volume (Å3) 684,912679 683,751708 α (deg) 90 90 β (deg) 90 90 γ (deg) 90 90
*Sumber: Chu et al. (2010)
Optimasi struktur kisi yang disajikan pada Tabel 2 memperlihatkan bahwa setiap parameter kisi menghasilkan data yang tidak jauh berbeda dengan hasil eksperimen, hal ini menjadi poin penting dalam optimasi struktur kristal Ca(BH4)2∙2NH3 untuk menghasilkan data kisi kristal yang serupa dan dapat dibandingkan dengan eksperimen. Hal ini diperjelas
dengan geometri struktur posisi atom di dalam kisi yang juga tidak mengalami banyak perubahan seperti yang ditampilkan dalam Tabel 3.
Data hasil optimasi geometri atom-atom ditampilkan sebagai perbandingan koordinat struktur geometri senyawa Ca(BH4)2∙2NH3 hasil kalkulasi dengan eksperimen seperti pada Tabel 3.
Dalam Tabel 3 menampilkan 10 atom yang menjadi perwakilan dari 76 atom kisi kristal Ca(BH4)2∙2NH3, dimana koordinat atom dari Ca(BH4)2∙2NH3 menghasilkan koordinat struktur yang mirip.
Analisis struktur kristal Ca(BH4)2∙2NH3 dilanjutkan dengan menganalisis bentuk struktural, sifat
Tabel 3. Perbandingan koordinat hasil kalkulasi vs eksperimen
Koordinat kalkulasi (x y z) Koordinat eksperimen (x y z)* Ca 06,134 11,669 05,992 06,062 11,831 06,000 B 09,346 08,085 03,656 09,191 08,098 03,535 N 09,216 11,514 09,629 09,247 11,651 09,473 H1(B) 09,849 10,367 03,828 09,723 10,355 03,729 H2(B) 07,610 07,762 02,137 07,501 07,827 01,984 H3(B) 08,690 07,273 05,752 08,503 07,259 05,592 H4(B) 11,278 07,022 02,899 11,093 07,012 02,776 H5(N) 08,744 10,320 11,086 08,916 10,379 10,920 H6(N) 10,959 10,916 09,013 11,019 11,209 08,773 H7(N) 09,526 13,229 10,475 09,460 13,375 10,357 *Sumber: Chu et al. (2010)
Studi Komputasi Metode Ab Initio Dft Dalam Kajian Struktural dan Sifat Elektronik elektronik dan juga sifat dehidrogenasi.
Hal ini dilakukan agar dapat mempelajari hasil pengamatan eksperimen melalui model komputasi.
Analisis struktural, sifat elektronik
dan sifat dehidrogenasi kristal
Ca(BH4)2∙2NH3
Metode komputasi Ab Initio DFT adalah metode yang sangat baik
digunakan untuk penentuan struktur dan sifat dari molekul yang bersifat statis atau kurang mengalami perubahan dalam strukturnya (Leach, 2001). Analisis ini memberikan pemahaman mengenai interaksi, panjang ikatan maupun sudut yang terdapat di dalam kristal Ca(BH4)2∙2NH3.
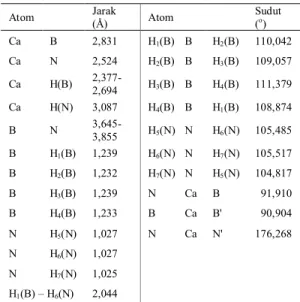
- Struktural kristal Ca(BH4)2∙2NH3 Struktur kompleks sistem Ca(BH4)2∙2NH3 berbentuk oktahedral dengan ion Ca2+ sebagai atom pusat yang dikelilingi oleh 4 molekul pada sisi bidang dan 2 molekul NH3 pada sisi atas- bawah. Seperti yang ditampilkan pada Gambar 4.
Pada Gambar 4. diperlihatkan bahwa jarak antara Ca-B sebesar 2,831 Å, mirip dengan Ca(BH4) (dCa-B = 2,88 Å) dari penelitian Majzoub dan Rönnebro (2009), mengindikasikan bahwa sistem kristal Ca(BH4)2∙2NH3 mirip dengan
sistem Ca(BH4). Jarak Ca-N sebesar 2,524 Å, mirip dengan Ca(NH2)2 (dCa-N = 2,442 – 2,573 Å) dari penelitian Senker et al., (1998) mengindikasikan bahwa sistem ini juga mirip dengan sistem Ca(NH2)2.
Struktur dari Gambar 4. memiliki bentuk tetrahedral dengan panjang ikatan dB-H = 1,233 Å dan besar sudut H-B-H = 109,41˚ – 110,04˚, panjang B-H ini sedikit lebih panjang dibanding pada Ca(BH4)2 (dB-H = 1,220 Å) dari penelitian Majzoub dan Rönnebro (2009). Dan untuk molekul NH3 memiliki
Gambar 4. Panjang ikatan dalam kristal Ca(BH4)2∙2NH3 2,524 2,831 H1(B) 3,855 H6(N) 2,044 H3(B) H1(B) H2(B) 1,239 2,524 3,087 1,027 2,524 1,239 1,027 H3(B) H7(N) 2,377 H5(N)
bentuk piramida dengan jarak dN-H = 1,025 – 1,027 Å dengan sudut H-N-H sebesar 104,81˚ – 105,51˚, panjang ikatan N-H ini lebih panjang dibanding pada fasa gas (dN-H = 1,017 Å), untuk sudut H– N–H sebesar 104,81˚ – 105,51˚, besar sudut ini lebih kecil dibandingkan sudut pada fasa gas NH3 (107,80o) (Wells, 1984). Data lengkap disajikan dalam Tabel 4.
Data Tabel 4. memperlihatkan atom H dari memiliki jarak lebih dekat dengan atom pusat Ca2+ mengindikasikan atom H(B) memiliki muatan parsial negatif (δ-), sehingga lebih menyukai untuk interaksi dengan atom pusat Ca2+ yang memiliki muatan positif (+).
Tabel 4. Panjang dan sudut ikatan pada kristal Ca(BH4)2∙2NH3
Atom Jarak (Å) Atom Sudut (o) Ca B 2,831 H1(B) B H2(B) 110,042 Ca N 2,524 H2(B) B H3(B) 109,057 Ca H(B) 2,377-2,694 H3(B) B H4(B) 111,379 Ca H(N) 3,087 H4(B) B H1(B) 108,874 B N 3,645-3,855 H5(N) N H6(N) 105,485 B H1(B) 1,239 H6(N) N H7(N) 105,517 B H2(B) 1,232 H7(N) N H5(N) 104,817 B H3(B) 1,239 N Ca B 091,910 B H4(B) 1,233 B Ca B' 090,904 N H5(N) 1,027 N Ca N' 176,268 N H6(N) 1,027 N H7(N) 1,025 H1(B) – H6(N) 2,044
Jarak ikatan N-H ini lebih panjang dari NH3 pada fasa gas (1,027:1,017)
mengindikasikan adanya interaksi atom N dengan Ca, dimana struktur NH3 bersifat basa lewis yang mendonorkan pasangan elektron bebas untuk berinteraksi dengan kation Ca2+ yang bersifat asam lewis, sehingga menyebabkan kerapatan elektron pada N lebih besar dan kerapatan ikatan N-H menjadi lebih rendah, dan menyebabkan ikatan N-H menjadi memanjang.
Pada Gambar 4. juga didapatkan interaksi dua atom H yang saling berdekatan, yakni antara H6(N) dengan H1(B), data jarak interaksi terdekatnya adalah sebesar 2,044 Å, interaksi ini adalah interaksi dihidrogen antara dari molekul NH3 dengan dari molekul yang berpotensi sebagai sumber molekul hidrogen (H2). Interaksi antar dua hidrogen ini mirip dengan ikatan hidrogen tapi bedanya dengan ikatan dihidrogen adalah interaksi dimana atom hidrogen biasanya tidak akan saling berdekatan dengan atom hidrogen lainnya dengan jarak lebih kecil dari 2,4 Å, ciri karakteristik ikatan dihidrogen adalah jarak antar atom hidrogen mendekati 1,8 Å (Custelcean dan Jackson, 2001).
- Sifat elektronik sistem kristal Ca(BH4)2∙2NH3
Analisis terhadap sifat elektron di dalam sistem dapat memberikan gambaran sifat sistem kimia. Kerapatan
Studi Komputasi Metode Ab Initio Dft Dalam Kajian Struktural dan Sifat Elektronik keadaan orbital atom atau biasa disebut
Density of State (DOS), merupakan
analisis terhadap jumlah keberadaan per interval energi dari setiap level energi yang dapat terisi oleh elektron (Atkins, 2006).
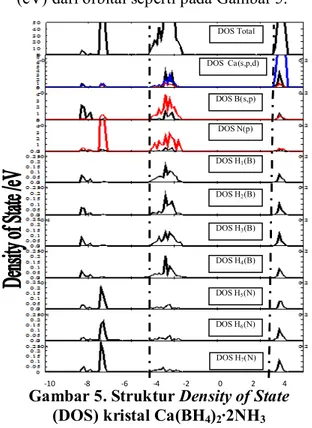
Analisis DOS digunakan untuk menganalisis keterlibatan ikatan kimia antar spesies kimia dari sistem kristal Ca(BH4)2∙2NH3. DOS memiliki bentuk besar intensitas total kerapatan (eV) dari tiap orbital atom berbanding besar energi (eV) dari orbital seperti pada Gambar 5.
Gambar 5. Struktur Density of State
(DOS) kristal Ca(BH4)2∙2NH3 Data DOS kristal Ca(BH4)2∙2NH3 terbagi menjadi 3 daerah, daerah 1 merupakan daerah DOS pita valensi dengan energi rendah yakni dibawah -4,35 eV, daerah 2 merupakan daerah DOS pita valensi energi tinggi (-4,35 eV
sampai -2,17 eV) dan daerah 3 adalah DOS pita konduksi (3,51 eV).
Daerah 1 di sekitar 8,70 sampai -7,89 eV merupakan kontribusi dari kerapatan orbital s atom B dan H dari molekul yang menandakan adanya interaksi kuat hibridisasi orbital sp dari B–H dan di atas -7,48 eV sampai -6,93 eV merupakan kontribusi dari orbital p
atom N dan s dari atom H dari molekul yang mengindikasikan adanya interaksi kovalen hibridisasi orbital sp
yang kuat antara N dan H.
Daerah 2 di sekitar -4,35 eV sampai -2,17 eV merupakan dominasi kontribusi orbital p atom B dan s atom H dari molekul , ditambah kontribusi yang kecil dari orbital p dari atom N, serta kontribusi kecil orbital s dan p dari atom Ca. Daerah pita valensi (daerah 1 dan 2) ini memperlihatkan DOS dari orbital hibridisasi kombinasi dari orbital s-B, p-B dan s-H yang membentuk orbital hibrid sp3, sedangkan kebutuhan 1 elektron yang digunakan untuk membentuk sp3 diperoleh dari atom Ca terdekat, hal inilah yang memberikan alasan tentang arah kecendrungan atom H pada yang lebih dekat dengan atom Ca dibanding B. Dan daerah 3 di atas 3,51 eV merupakan pita konduksi dari orbital d atom Ca. Atom B dan N
-10 -8 -6 -4 -2 0 2 4 DOS H7(N) DOS Ca(s,p,d) DOS Total DOS B(s,p) DOS N(p) DOS H1(B) DOS H2(B) DOS H3(B) DOS H4(B) DOS H5(N) DOS H6(N)
memberikan kontribusi kecil terhadap pita konduksi. Dalam grafik DOS pada daerah
pita valensi paling rendah di sekitar -7,89 eV yang merupakan kontribusi DOS atom H(B) dari molekul dan sangat berdekatan dengan DOS atom H(N) dari molekul di daerah 7,48 eV, menguatkan indikasi interaksi ikatan dihidrogen dapat terjadi dari atom H(B) pada molekul dan H(N) pada molekul . Hal ini dikuatkan dengan
analisis struktur pita elektron dari kristal Ca(BH4)2∙2NH3 yang diperlihatkan dalam grafik pita energi pada Gambar 6.
Pada Gambar 6. memperlihatkan wilayah pita elektron H(B) dan H(N) yang berdekatan dan sangat memungkinkan untuk saling berinteraksi satu sama lain dengan proses eksitasi. Terjadinya eksitasi membutuhkan energi eksternal, sehingga tidak aneh bila sistem
ini membutuhkan energi panas eksternal untuk terjadinya proses dekomposisi dan dehidrogenasi.
Pita elektron valensi energi rendah di sekitar -7,89 eV merupakan kontribusi pita elektron atom H(B) dari molekul dan sangat berdekatan dengan pita elektron atom H(N) dari molekul di daerah 7,48 eV, selisih
celah energinya sekitar 0,41 eV, dekatnya celah ini menguatkan indikasi interaksi elektronik dapat terjadi dari ikatan dihidrogen pada atom H(B) dan H(N).
Sejauh penelitian ini dilakukan belum ada hasil penelitian eksperimen tentang besar dari nilai celah pita energi
band gap dari kristal Ca(BH4)2∙2NH3,
namun dengan membandingkan dengan
Gambar 6. Struktur pita elektron kristal Ca(BH4)2∙2NH3 5.68 eV
2.58 eV
0.41 eV
Orbital 2s B, 1s H(B) Orbital 2p N, 1s H(N)
Studi Komputasi Metode Ab Initio Dft Dalam Kajian Struktural dan Sifat Elektronik hasil secara teoritis dari penelitian lain
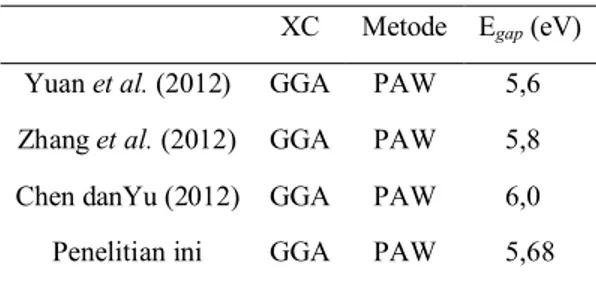
seperti pada Tabel 5. maka dapat terlihat bahwa kristal ini memiliki celah pita energi yang besar (5,68 eV) yang mengindikasikan bahwa sistem ini merupakan kristal non-metal dan bersifat insulator.
Tabel 5. Perbandingan besar band gap
XC Metode Egap (eV) Yuan et al. (2012) GGA PAW 5,60
Zhang et al. (2012) GGA PAW 5,80
Chen danYu (2012) GGA PAW 6,00
Penelitian ini GGA PAW 5,68
Bentuk pola pita elektron kristal Ca(BH4)2∙2NH3 (Gambar 6) yang berbentuk flat menggambarkan sistem yang cenderung ionik (Ashcroft dan Mermin, 1976).
Gambar 7. Distribusi kerapatan elektron
Pada Gambar 7. memperlihatkan bahwa kerapatan potensial atom Ca paling besar. Pada molekul
distribusi elektron lebih besar pada atom H(B) daripada atom B, mengindikasikan muatan H(B) lebih cenderung parsial negatif (δ-) sedangkan pada atom B sebagai parsial positifnya (δ+).
Distribusi kerapatan elektron pada molekul menunjukkan kerapatan yang besar pada di sekitar atom N tapi tidak pada atom H(N), mengindikasikan muatan N lebih parsial negatif (δ-) sedangkan pada atom H(N) sebagai parsial positifnya (δ+). Posisi atom N dan H(B) yang mengarah ke atom Ca mengindikasikan interaksi muatan negatif dari N dan H(B) dengan Ca yang memiliki potensial inti positif (δ+) dan interaksi yang terjadi juga akibat distorsi yang diberikan oleh orbital d atom Ca.
Muatan negatif yang dimiliki H(B) merupakan kontribusi ionik dari Ca2+, hal ini terlihat dari muatan Ca yang mengalami penurunan menjadi +1,53. Kejelasan besarnya nilai kerapatan tiap spesies ditampilkan dalam Tabel 6.
Pada Tabel 6. memperlihatkan distribusi kerapatan elektron lebih dominan terdapat pada atom H(B) dibandingkan atom B pada molekul
, sehingga memberikan gambaran bahwa atom H(B) lebih bermuatan negatif ( ) dibanding atom B.
Tabel 6. Nilai kerapatan elektron dan muatan atom
Elemen ρ Muatan atom
Ca 0,8154 +1,53 B 0,1094 +1,80 H(B) 0,3125 -0,70 N 0,4972 -1,05 H(N) 0,3414 +0,35 Pair e- N 0,5995
Berbeda dengan molekul dimana kerapatan elektron atom N lebih besar ( ) dibandingkan pada atom H(N), terutama lebih dominan pada pasangan elektron bebasnya, sehingga atom H(N) lebih bermuatan positif ( ). Hal ini akan mendukung adanya interaksi dari H(B) dan H(N) sebagai interaksi dihidrogen karena masing-masing memiliki muatan parsial yang berbeda.
- Sifat dehidrogenasi Ca(BH4)2∙2NH3 Dengan maksud mempelajari lebih lanjut sifat dehidrogenasi maupun dekomposisi material Ca(BH4)2∙2NH3. Perhitungan energi ikat hidrogen (ΔE) dilakukan dengan persamaan 3 terhadap hidrogen yang teridentifikasi ikatan dihidrogen seperti pada Gambar 4.
(4)
dimana adalah energi total kristal Ca(BH4)2∙2NH3, adalah energi molekul H2 yang terisolasi,
adalah energi total kristal Ca(BH4)2∙2NH3 setelah atom H terlepas, dan menyatakan jumlah atom hidrogen yang terlepas.
Data Tabel 7 memperlihatkan bahwa energi ikat H(B) lebih rendah dibandingkan energi ikat H(N) (2,363:
3,429) yang mengidentifikasikan bahwa
hidrogen dari atom boron lebih diprioritaskan untuk terjadi disosiasi
terlebih dahulu dibandingkan hidrogen dari nitrogen.
Tabel 7. Perbandingan energi ikat hidrogen
Posisi atom ΔE1 (eV)
H-B 2,363
H-N 3,429
H-B & H-N (2H) 2,304
Ca(BH4)2∙NH3--NH3 1,516
Studi dekomposisi material dengan pelepasan molekul NH3 dilakukan untuk mempelajari interaksi NH3 dengan material Ca(BH4)2∙2NH3. Energi pelepasan molekul , sebagai energi ikat terhadap kristal Ca(BH4)2∙2NH3 didapatkan sebesar 1,516 eV. Hal ini menunjukkan energi ikat NH3 yang lebih rendah dibanding energi ikat 2H, disini dapat disimpulkan bahwa pelepasan dua hidrogen menjadi H2 kurang dominan dibandingkan NH3. Fenomena dekomposisi hasil perhitungan ini didapat dan sesuai dengan fakta eksperimental seperti yang telah dilakukan oleh Chu et al., (2010)
Kesimpulan.
Model senyawa Ca(BH4)2∙2NH3 terbaik telah berhasil didapat dengan menggunakan tehnik Projector
Augmented-Wave (PAW) dari metode
Density Functional Theory (DFT) yang
merupakan tehnik yang lebih cocok untuk material zat padat. Hasil model yang
Studi Komputasi Metode Ab Initio Dft Dalam Kajian Struktural dan Sifat Elektronik mirip dengan eksperimen dapat
digunakan untuk mempelajari bentuk struktur dan sifat elektronik senyawa Ca(BH4)2∙2NH3, termasuk menjelaskan sifat penyimpanan hidrogen didalamnya.
Pustaka
Anonim, 2007. Hydrogen Storage Materials. Mater. Matters 2, 3–7.
Ashcroft, N.W., Mermin, N.D., 1976. Solid state physics. Harcourt College Publishers, New York; London.
Atkins, P.W., 2006. Atkins’ Physical chemistry, 8th ed. ed. Oxford University Press, Oxford ; New York.
Baitalow, F., Baumann, J., Wolf, G., Jaenicke-Rößler, K., Leitner, G., 2002. Thermal decomposition of B–N–H compounds investigated by using combined thermoanalytical methods. Thermochim. Acta 391, 159–168. doi:10.1016/S0040-6031(02)00173-9
Blöchl, P.E., 1994. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979.
doi:10.1103/PhysRevB.50.17953
Chen, X., Yu, X., 2012. Electronic Structure and Initial Dehydrogenation Mechanism of M(BH4)2·2NH3 (M = Mg, Ca, and Zn): A First-Principles Investigation. J. Phys. Chem. C
116, 11900–11906.
doi:10.1021/jp301986k
Chua, Y.S., Wu, G., Xiong, Z., He, T., Chen, P., 2009. Calcium Amidoborane Ammoniate— Synthesis, Structure, and
Hydrogen Storage Properties. Chem. Mater. 21, 4899–4904. doi:10.1021/cm9020222
Chu, H., Wu, G., Xiong, Z., Guo, J., He, T., Chen, P., 2010. Structure and Hydrogen Storage Properties of
Calcium Borohydride
Diammoniate. Chem. Mater. 22, 6021–6028.
doi:10.1021/cm1023234
Custelcean, R., Jackson, J.E., 2001. Dihydrogen Bonding: Structures, Energetics, and Dynamics. Chem. Rev. 101, 1963–1980. doi:10.1021/cr000021b
Frueh, S., Kellett, R., Mallery, C., Molter, T., Willis, W.S., King’ondu, C., Suib, S.L., 2011. Pyrolytic Decomposition of Ammonia Borane to Boron Nitride. Inorg. Chem. 50, 783– 792. doi:10.1021/ic101020k
Gonze, X., Amadon, B., Anglade, P.-M., Beuken, J.-M., Bottin, F., Boulanger, P., Bruneval, F., Caliste, D., Caracas, R., Côté, M., Deutsch, T., Genovese, L., Ghosez, P., Giantomassi, M., Goedecker, S., Hamann, D.R., Hermet, P., Jollet, F., Jomard, G., Leroux, S., Mancini, M., Mazevet, S., Oliveira, M.J.T., Onida, G., Pouillon, Y., Rangel, T., Rignanese, G.-M., Sangalli, D., Shaltaf, R., Torrent, M., Verstraete, M.J., Zerah, G., Zwanziger, J.W., 2009. ABINIT: First-principles approach to material and nanosystem properties. Comput. Phys. Commun. 180, 2582–2615. doi:10.1016/j.cpc.2009.07.007
Hohenberg, P., Kohn, W., 1964. Inhomogeneous Electron Gas. Phys. Rev. 136, B864–B871. doi:10.1103/PhysRev.136.B864
Holzwarth, N.A.W., Tackett, A.R., Matthews, G.E., 2001. A Projector Augmented Wave (PAW) code for electronic structure calculations, Part I: atompaw for generating atom-centered functions. Comput. Phys. Commun. 135, 329–347.
doi:10.1016/S0010-4655(00)00244-7
Irani, R.S., 2002. Hydrogen storage: high-pressure gas contaiment 27, 680–684.
Kohn, W., Sham, L.J., 1965. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 140, A1133–A1138. doi:10.1103/PhysRev.140.A1133
Leach, A.R., 2001. Molecular modelling: principles and applications. Prentice Hall, Harlow [etc.].
Majzoub, E.H., Rönnebro, E., 2009. Crystal Structures of Calcium Borohydride: Theory and Experiment. J. Phys. Chem. C
113, 3352–3358.
doi:10.1021/jp8064322
Momma, K., Izumi, F., 2011. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 44, 1272– 1276.
doi:10.1107/S0021889811038970
Perdew, J.P., Burke, K., Ernzerhof, M., 1996. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865–3868. doi:10.1103/PhysRevLett.77.3865
Senker, J., Jacobs, H., Müller, M., Press, W., Müller, P., Mayer, H.M.,
Ibberson, R.M., 1998. Reorientational Dynamics of Amide Ions in Isotypic Phases of Strontium and Calcium Amide. 1. Neutron Diffraction Experiments. J. Phys. Chem. B 102, 931–940. doi:10.1021/jp972907y
Soloveichik, G., Her, J.-H., Stephens, P.W., Gao, Y., Rijssenbeek, J., Andrus, M., Zhao, J.-C., 2008. Ammine Magnesium Borohydride Complex as a New Material for Hydrogen Storage: Structure and Properties of Mg(BH4)2·2NH3. Inorg. Chem. 47, 4290–4298. doi:10.1021/ic7023633
Wells, A.F., 1984. Structural inorganic chemistry, 5th ed. ed. Clarendon Press ; Oxford University Press, Oxford [Oxfordshire] : New York.
Yuan, P.-F., Wang, F., Sun, Q., Jia, Y., Guo, Z.-X., 2012. Structural, energetic and thermodynamic analyses of Ca(BH4)2·2NH3 from first principles calculations. J. Solid State Chem. 185, 206– 212.
doi:10.1016/j.jssc.2011.11.009
Zhang, G., Yang, J., Fu, H., Zheng, J., Li, Y., Li, X., 2012. Structural and electronic properties of the hydrogen storage compound Ca(BH4)2·2NH3 from first-principles. Comput. Mater. Sci.
54, 345–349.
doi:10.1016/j.commatsci.2011.10. 037