UNIVERSITAS INDONESIA
LAPORAN PRAKTEK KERJA PROFESI APOTEKER
DI PT. CLINISINDO LABORATORIES
JL. ULUJAMI RAYA NO 12 JAKARTA SELATAN
PERIODE 12 MARET – 30 APRIL 2012
LAPORAN PRAKTEK KERJA PROFESI APOTEKER
LIANNE CYNTHIA CAROLINA LIE, S.Farm.
1106047070
ANGKATAN LXXIV
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM PROGRAM PROFESI APOTEKER - DEPARTEMEN FARMASI
DEPOK JUNI 2012
ii
UNIVERSITAS INDONESIA
LAPORAN PRAKTEK KERJA PROFESI APOTEKER
DI PT. CLINISINDO LABORATORIES
JL. ULUJAMI RAYA NO 12 JAKARTA SELATAN
PERIODE 12 MARET – 30 APRIL 2012
LAPORAN PRAKTEK KERJA PROFESI APOTEKER
Diajukan sebagai salah satu syarat untuk memperoleh gelar Apoteker
LIANNE CYNTHIA CAROLINA LIE, S.Farm.
1106047070
ANGKATAN LXXIV
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM PROGRAM PROFESI APOTEKER - DEPARTEMEN FARMASI
DEPOK JUNI 2012
iii
Laporan Praktek Kerja Profesi Apoteker ini diajukan oleh:
Nama : Lianne Cynthia Carolina Lie, S.Farm.
NPM : 1106047070
Program Studi : Apoteker – Departemen Farmasi FMIPA UI Judul Laporan : Laporan Praktek Kerja Profesi Apoteker di PT.
Clinisindo Laboratories Jl. Ulujami Raya No. 12 Jakarta Selatan Periode 12 Maret – 30 April 2012
Telah berhasil dipertahankan di hadapan Dewan Penguji dan diterima sebagai bagian persyaratan yang diperlukan untuk memperoleh gelar Apoteker pada Program Studi Apoteker – Departemen Farmasi, Fakultas Matematika dan Ilmu Pengetahuan Alam, Universitas Indonesia.
DEWAN PENGUJI
Pembimbing I : Budi Prasaja, S.Si., MM., Apt. ( )
Pembimbing II : Dr. Harmita, Apt.pt. ( )
Penguji I : ( )
Penguji II : ( )
Penguji III : ( )
Ditetapkan di : Depok Tanggal :
iv
Puji syukur penulis panjatkan kepada Tuhan Yang Maha Esa, karena atas penyertaan, kasih, berkat, dan anugerah-Nya, penulis dapat menyelesaikan penyusunan laporan ini. Laporan Praktek Kerja Profesi Apoteker ini diajukan sebagai salah satu syarat untuk memperoleh gelar Apoteker pada Program Profesi Apoteker Fakultas Matematika dan Ilmu Pengetahuan Alam Universitas Indonesia.
Penulis menyadari bahwa, tanpa bantuan serta bimbingan dari berbagai pihak dalam penyusunan laporan ini, sangatlah sulit bagi penulis untuk menyelesaikan laporan ini. Oleh karena itu, pada kesempatan kali ini, penulis ingin menyampaikan rasa terima kasih kepada semua pihak yang telah membantu selama praktek kerja dan penyusunan laporan ini, antara lain:
(1) Bapak Budi Prasaja, S.Si., MM., Apt., selaku Manajer Teknis PT. Clinisindo Laboratories dan Pembimbing PKPA atas kesempatan yang diberikan, bimbingan, saran, serta bantuan yang diberikan selama PKPA dan penyusunan laporan ini .
(2) Bapak Dr. Harmita, Apt., selaku Ketua Program Profesi Apoteker Departemen Farmasi FMIPA UI dan Pembimbing PKPA atas bimbingan, saran serta bantuan yang diberikan selama penyusunan laporan ini.
(3) Ibu Prof. Dr. Yahdiana Harahap, MS., Apt., selaku Ketua Departemen Farmasi FMIPA UI dan Pembimbing Akademis atas kesempatan yang diberikan.
(4) Kak Windy Lusthom, S.Si., Apt., selaku Asisten Manajer Teknis atas pengarahan dan bantuan selama pelaksanaan Praktek Kerja Profesi Apoteker. (5) Kak Hardiyanti, S.Si., Apt., selaku Manajer Mutu atas pengarahan dan
bantuan yang diberikan selama PKPA.
(6) Kak Theresia Sinandang, S.Si., Apt., selaku Supervisor Laboratorium atas pengarahan dan bantuan yang diberikan.
(7) Lia Yumi Yusvita, S.Farm., Apt., Evan, Dedek, serta seluruh karyawan PT. Clinisindo Laboratories lainnya yang telah banyak memberikan pengarahan dan bantuan selama PKPA.
v
(8) Seluruh staf pengajar Program Profesi Apoteker Departemen Farmasi Fakultas Matematika dan Ilmu Pengetahuan Alam Universitas Indonesia atas segala bimbingannya selama masa perkuliahan.
(9) Keluargaku terkasih, Mama, Alm. Papa, Kakak-kakak, dan Adikku, yang tak henti-hentinya memberikan dukungan moril dan materil, doa, penghiburan, dan motivasi selama studi di Farmasi.
(10) Teman-teman seperjuangan Apoteker UI angkatan 74, dan keluarga kecilku di Farmasi angkatan 2005-2008. Terima kasih untuk segala dukungan, bantuan, saran, dan semangat yang telah diberikan kepada penulis.
(11) Semua pihak yang tidak dapat disebutkan satu per satu, yang telah memberikan dukungannya selama PKPA dan penulisan laporan ini.
Penulis menyadari dalam penyusunan laporan ini masih jauh dari sempurna. Oleh karena itu, penulis memohon maaf bila terdapat kesalahan dalam penulisan laporan ini. Semoga laporan ini dapat bermanfaat bagi semua pihak yang membutuhkan.
Penulis 2012
Halaman
HALAMAN JUDUL ... ii
HALAMAN PENGESAHAN ... iii
KATA PENGANTAR ... iv
DAFTAR ISI ... vi
DAFTAR GAMBAR ... vii
DAFTAR TABEL ... viii
DAFTAR LAMPIRAN ... ix
BAB 1 PENDAHULUAN ... 1
1.1 Latar Belakang ... 1
1.2 Tujuan ... 2
BAB 2 TINJAUAN UMUM ... 3
2.1 Sejarah Organisasi ... 3
2.2 Visi dan Misi ... 4
2.3 Struktur Organisasi ... 5
2.4 Spesifikasi Jabatan ... 7
2.5 Bangunan dan Fasilitas ... 14
2.6 Peralatan ... 15
2.7 Dokumentasi ... 16
2.8 Pengolahan Limbah ... 17
BAB 3 TINJAUAN KHUSUS ... 18
3.1 Pendahuluan Uji Bioavailabilitas dan Bioekivalensi ... 18
3.2 Alur Uji Bioavailabilitas dan Bioekivalensi ... 19
3.3 Kriteria Uji Bioekivalensi ... 21
3.4 Desain dan Pelaksanaan Studi Bioekivalensi ... 25
3.5 Laporan Hasil Uji Bioekivalensi ... 29
BAB 4 PEMBAHASAN ... 32
BAB 5 KESIMPULAN DAN SARAN ... 35
5.1 Kesimpulan ... 35
5.2 Saran ... 35
vii Universitas Indonesia
Gambar Halaman
2.1 Struktur organisasi PT. Clinisindo Laboratories ... 5
3.1 Alur tata cara permohonan uji bioekivalensi ... 20
3.2 Tahapan pelaksanaan studi BA/BE ... 21
Tabel Halaman 3.1 Perbandingan jumlah subyek terhadap koefisien variasi intrasubyek ... 27
ix Universitas Indonesia
Lampiran Halaman 1. Daftar obat copy yang mengandung zat aktif wajib uji bioekivalensi ... 37
1.1 Latar Belakang
Produk obat copy harus memiliki standar mutu, efikasi, dan keamanan yang sama dengan produk obat originator. Bukti atas efikasi dan keamanan dari produk obat copy merupakan salah satu syarat yang diperlukan untuk mendapatkan izin edar. Obat copy harus ekivalen secara terapetik dengan obat originator, sehingga dapat menggantikan obat originator. Uji bioekivalensi antara produk obat copy dengan produk obat originator merupakan salah satu cara untuk menunjukkan ekivalensi terapetik tanpa perlu melakukan uji preklinik dan uji klinik yang melibatkan banyak subyek dan memerlukan waktu yang lama (World Health Organization, 2006; Medicines Control Council, 2003; Tamboli, Todkar, Zope, & Sayyad, 2010).
Agar dapat menghasilkan efek terapetik yang optimal, sejumlah obat harus dapat mencapai reseptornya pada konsentrasi efektif selama jangka waktu tertentu. Pada uji bioekivalensi keamanan dan efikasi dari obat uji (obat copy) diprediksi berdasarkan pada pengukuran konsentrasi sistemik obat tersebut dibandingkan terhadap konsentrasi sistemik obat originator dengan dosis yang sama. Berdasarkan asumsi, pada subyek yang sama konsentrasi obat yang sama dalam plasma akan menghasilkan konsentrasi obat yang sama pada loka aksi, sehingga akan menghasilkan efek terapetik yang sama pula (World Health Organization, 2006; Medicines Control Council, 2003).
Uji bioekivalensi merupakan bukti tidak langsung atas keamanan dan efikasi dari produk obat copy, karena itu uji ini perlu dilakukan dengan cara yang benar. Pengujian yang dilakukan di laboratorium harus mengikuti prinsip Good Laboratory Practice (GLP) dan karena melibatkan subyek manusia uji bioekivalensi juga harus dilakukan dengan menerapkan Cara Uji Klinik yang Baik (CUKB) atau Good Clinical Practices (GCP). Dengan demikian hasil uji bioekivalensi yang diperoleh dapat dipercaya dan akurat, serta hak, integritas, dan kerahasiaan dari subyek uji klinik pun terlindungi (World Health Organization, 2006; Badan POM RI, 2004).
Universitas Indonesia
Suatu lembaga pengujian yang melakukan uji bioekivalensi harus memenuhi semua persyaratan tersebut. Selain itu, uji bioekivalensi juga menuntut adanya independensi dari lembaga pengujian agar hasil uji yang diperoleh terbebas dari konflik kepentingan dan intervensi pihak lain.
1.2 Tujuan
Praktek Kerja Profesi Apoteker (PKPA) yang dilaksanakan di PT. Clinisindo Laboratories ini bertujuan untuk:
1.2.1 Mengetahui penerapan Cara Uji Klinik Yang Baik (CUKB)/Good Clinical Practice (GCP) dan Good Laboratory Practice yang dilakukan PT. Clinisindo Laboratories.
1.2.2 Mengetahui kegiatan yang dilakukan oleh PT. Clinisindo Laboratories. 1.2.3 Mengetahui peranan apoteker dalam laboratorium pengujian
2.1 Sejarah Organisasi
PT. Clinisindo Laboratories adalah lembaga penelitian independen (Independent Contract Research Organization) yang didirikan pada tahun 2004. PT. Clinisindo Laboratories bergerak dalam bidang pengujian dan pengembangan metode analisis bioavailabilitas/bioekivalensi (BA/BE).
PT. Clinisindo Laboratories didirikan pada tanggal 20 September 2004, dengan akte pendirian perusahaan Perseroan Terbatas nomor 31, dimana pemegang saham menyerahkan sepenuhnya operasional dari PT. Clinisindo Laboratories kepada direktur yang sepenuhnya terlepas dari tugas/tanggung jawab lainnya. Tujuannya adalah untuk menghindari terjadinya pertentangan kepentingan dalam pelaksanaan pengujian di Laboratorium PT. Clinisindo, sehingga seluruh keputusan, hasil pengujian dan laporan pengujian dilakukan secara profesional dan independen tanpa ada intervensi dari pemegang saham.
Laboratorium ini didirikan dengan tujuan untuk memenuhi kebutuhan pengujian bioekivalensi dan pengembangan metode analisis berdasarkan standar Good Laboratory Practice (GLP) dan Good Clinical Practice (GCP) serta standar lain yang berlaku. Guna menghindari benturan kepentingan dalam proses kegiatan laboratorium maka PT. Clinisindo menerapkan asas independensi seperti penerapan sistem musyawarah mufakat dan berimbang dalam pengambilan keputusan tanpa adanya dominasi dan tekanan dari pihak manapun, serta tersedianya sumber daya keuangan yang dalam pengelolaannya bebas dari benturan berbagai pihak yang berkepentingan.
PT. Clinisindo Laboratories berlokasi di Jl. Ulujami Raya No.12, Pesanggrahan Jakarta Selatan, sekitar 30 km dari Jakarta Pusat. Memiliki luas area sekitar 500 m2 dan dilengkapi dengan fasilitas klinik, fasilitas analitik, dan fasilitas kantor.
Pegawai yang bekerja di PT. Clinisindo Laboratories berjumlah 22 orang (termasuk pegawai kontrak), yang terdiri dari peneliti utama, peneliti pembantu, ahli farmakokinetika dan statistik, apoteker, dokter, analis kesehatan, perawat,
Universitas Indonesia
analis kimia, dan petugas kebersihan. Personel PT. Clinisindo Laboratories diseleksi dari para profesional yang berpengalaman di bidang analisis dan studi bioekivalen. Peneliti utama memegang seluruh tanggung jawab atas kegiatan klinik pada studi, termasuk aspek-aspek klinis pada desain penelitian, pemberian produk selama penelitian, menghubungi pemerintah lokal dan komisi etik, dan menandatangani protokol dan laporan.
PT. Clinisindo Laboratories mempunyai sistem dokumen mutu seperti Quality Manual (QM), Standard Operation Procedure (SOP) dan Standard Operating Instruction (SOI). Personel quality assurance (QA) bebas dari campur tangan bagian klinik dan analitik pada penelitian. Terdapat program audit internal untuk inspeksi diri yang diatur dan diorganisasikan oleh manajer mutu (QA) sebagai sistem penjaminan mutu (QA system). Audit internal dilakukan minimal 1 kali setahun untuk semua divisi.
Dalam melakukan penelitiannya PT. Clinisindo Laboratories juga bekerja sama dengan pihak ketiga dalam pemeriksaan kesehatan subyek dan penanganan limbah.
2.2 Visi dan Misi
PT. Clinisindo Laboratories memiliki visi untuk “Menjadi Laboratorium Pengujian BA/BE yang Kompeten, Berkualitas, dan Diakui secara Nasional, Regional, dan Internasional”. Untuk mencapai visi itu PT. Clinisindo Laboratories memiliki misi, yaitu sebagai berikut:
2.2.1 Memberikan pelayanan pengujian BA/BE dan pengembangan metode analisis yang berkualitas dengan semangat ilmiah sesuai persyaratan ISO/IEC 17025, Good Clinical Practice (GCP) dan Good Laboratory Practice (GLP).
2.2.2 Mencapai kepuasan pelanggan dengan memberikan laporan hasil pengujian yang akurat, tepat waktu dan sesuai dengan persyaratan pelanggan serta standar pengujian ter-up date dan/atau standar nasional, regional maupun internasional dengan tetap menjaga kerahasiaan informasi dan hak kepemilikan pelanggan.
2.2.3 Menerapkan dan meningkatkan efektivitas sistem manajemen mutu secara berkelanjutan dengan menetapkan sasaran mutu dan mengevaluasinya tiap tahun dalam rapat tinjauan manajemen.
2.2.4 Meningkatkan kemampuan sumber daya manusia agar semua karyawan jelas dengan tugas dan tanggung jawabnya serta terus menerus mengevaluasi kemampuan tersebut.
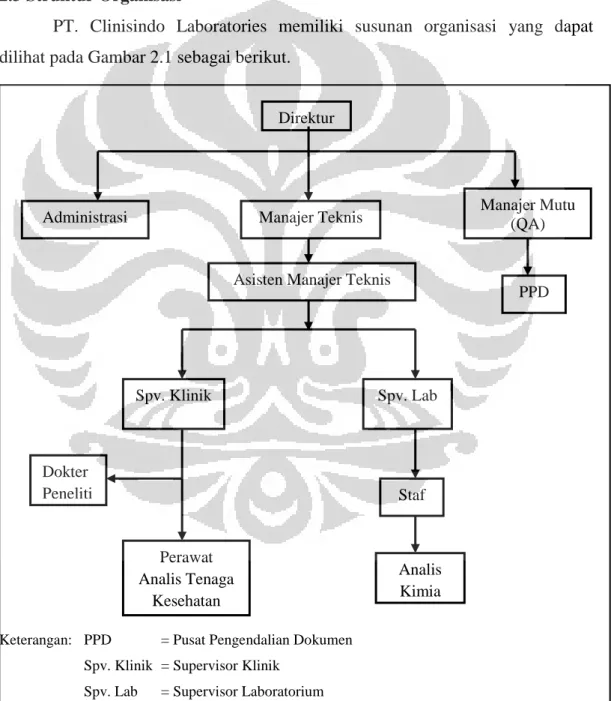
2.3 Struktur Organisasi
PT. Clinisindo Laboratories memiliki susunan organisasi yang dapat dilihat pada Gambar 2.1 sebagai berikut.
Keterangan: PPD = Pusat Pengendalian Dokumen Spv. Klinik = Supervisor Klinik
Spv. Lab = Supervisor Laboratorium
Gambar 2.1 Struktur organisasi PT. Clinisindo Laboratories Direktur
Administrasi Manajer Teknis Manajer Mutu (QA)
PPD Spv. Klinik Spv. Lab Dokter Peneliti Perawat Analis Tenaga Kesehatan Staf Analis Kimia Asisten Manajer Teknis
Universitas Indonesia
Berdasarkan struktur organisasi tersebut, personil inti laboratorium PT. Clinisindo Laboratories terdiri atas:
2.3.1 Direktur
Direktur merupakan manajemen puncak yang mempunyai tanggung jawab penuh terhadap semua kegiatan laboratorium serta memimpin organisasi untuk mencapai tingkat prestasi yang terbaik. Direktur memiliki wewenang untuk membuat keputusan terhadap kebijakan maupun sumber daya laboratorium untuk mencapai mutu data pengujian yang sesuai dengan kebutuhan dan kepuasan customer.
Direktur bertugas untuk menetapkan dan memelihara kebijakan mutu dan sasaran mutu laboratorium serta mempromosikannya ke seluruh organisasi untuk meningkatkan kesadaran, motivasi dan pelibatan. Direktur juga menjamin proses komunikasi yang tepat telah ditetapkan, diimplementasikan, dan dipelihara untuk menjamin tercapainya sasaran mutu dalam penerapan sistem manajemen mutu yang efektif dan efisien. Komunikasi dilakukan melalui pertemuan antar personil laboratorium dalam rapat tinjauan manajemen yang dipimpin direktur.
2.3.2 Manajer Mutu
Manajer mutu ditunjuk secara resmi oleh direktur. Manajer mutu memiliki kewenangan untuk memberi pengarahan kepada semua manajer lainnya dalam hal penerapan sistem manajemen mutu laboratorium. Manajer mutu bertanggung jawab untuk memberikan masukan serta usulan kepada direktur dalam memelihara dan meningkatkan sistem manajemen mutu di laboratorium. Selain itu manajer mutu juga bertanggung jawab untuk memastikan bahwa segala aspek dari mutu dilaksanakan sesuai dengan peraturan dan prosedur mutu laboratorium.
2.3.3 Manajer Teknis
Manajer teknis bertanggung jawab atas semua aspek operasional teknis dan ketersediaan sumber daya yang diperlukan untuk memastikan bahwa mutu yang dipersyaratkan dalam kegiatan laboratorium tercapai dan sesuai dengan kebutuhan dan kepuasan dari pelanggan. Dalam pengujian bioekivalensi manajer teknis bertanggung jawab atas pengurusan ethical clearence dan Persetujuan
Pelaksanaan Uji Bioekivalensi (PPUB), pemilihan/seleksi dari calon subyek, pemberian produk yang akan diuji kepada subyek, pencarian dan pengembangan metode analisis, validasi metode, pengolahan data secara statistik, serta pembuatan laporan pengujian.
2.4. Spesifikasi Jabatan
Spesifikasi jabatan asli masing-masing karyawan disimpan oleh manajer mutu dan salinan spesifikasi jabatan diberikan kepada karyawan yang bersangkutan. Perubahan atas spesifikasi jabatan harus diajukan oleh manajer mutu ke direktur.
2.4.1 Bagian Laboratorium
2.4.1.1 Manajer Teknis/Asisten Manajer Teknis
Manajer teknis/asisten manajer teknis adalah seorang apoteker yang sudah memiliki pengalaman bekerja di laboratorium. Tugas dan tanggung jawab yang dimiliki oleh seorang manajer teknis/asisten manajer teknis adalah sebagai berikut:
a. Mengelola seluruh aspek kegiatan pengujian untuk memastikan bahwa setiap pengujian sudah mengikuti pedoman Good Clinical Practice (GCP) dan Good Laboratory Practice (GLP).
b. Bersama dengan bagian administrasi menerima permintaan pelanggan, melakukan tinjauan kontrak dan membuat penawaran harga dan membuat perjanjian kontrak apabila harga sudah disepakati.
c. Bertanggung jawab dalam pengurusan ethical clearence dari Komisi Etik dan menjalin kerjasama dengan pelanggan/sponsor dalam pengurusan Persetujuan Pelaksanaan Uji Bioekivalensi (PPUB) dari Badan POM.
d. Mengkoordinasikan penerapan jaminan mutu dan pengendalian mutu (QA/QC) untuk semua jenis pengujian yang dilakukan oleh laboratorium.
e. Memonitor supervisor laboratorium dalam melaksanakan pengembangan dan validasi metode analisis.
f. Bertanggung jawab terhadap pengujian ulang (reanalyze) terhadap retained sample, apabila diperlukan.
Universitas Indonesia
g. Merencanakan, mengorganisasikan dan mengevaluasi partisipasi uji profisiensi atau uji banding laboratorium.
h. Bertanggung jawab terhadap pelaporan hasil pengujian dan penandatanganan sertifikat pengujian.
i. Menyusun budget, rencana investasi serta target tahunan sesuai dengan target dari manajemen.
j. Menyusun analisis kebutuhan pelatihan dan program pelatihan yang diperlukan dalam upaya untuk memastikan bahwa setiap personil yang ada mempunyai kompetensi untuk melaksanakan tugas sesuai uraian kerjanya.
k. Memberi penilaian terhadap karyawan di bawahnya dan mengusulkan perubahan gaji, promosi dan demosi.
l. Memberikan delegasi kepada supervisor laboratorium/klinik, apabila berhalangan.
2.4.1.2 Supervisor Laboratorium
Supervisor laboratorium adalah seorang apoteker yang memiliki pengalaman di bidang analisis dan instrumentasi. Supervisor laboratorium memiliki tugas dan tanggung jawab sebagai berikut:
a. Melaksanakan proses pengembangan metode analisis berdasarkan kompendial, literatur yang relevan, atau pengembangan sendiri.
b. Membuat protokol validasi sebelum melakukan validasi metode.
c. Memastikan validasi metode dilaksanakan berdasarkan protokol yang dibuat, mencatat bila terdapat penyimpangan terhadap protokol.
d. Membuat laporan hasil validasi metode.
e. Menyusun dokumen instruksi yang digunakan di dalam laboratorium.
f. Memantau pelaksanaan proses kerja yang dilakukan staf dan analis sesuai dengan prosedur yang sudah ditentukan.
g. Memberikan penjelasan dan training kepada staf dan analis sebelum memulai suatu pengujian.
h. Bertanggung jawab dalam pemeliharaan dan program kalibrasi instrumen dan alat-alat di laboratorium.
i. Bersama manajer teknis memberikan pelatihan dan evaluasi terhadap staf dan analis di bawahnya.
j. Memastikan sistem dokumentasi laboratorium berjalan dengan baik, termasuk proses pemindahan dan pengolahan data sudah dilakukan dengan benar.
k. Mengatur agar semua peralatan dan pereaksi yang dibutuhkan tersedia dalam jumlah yang cukup dan digunakan sebagaimana mestinya.
l. Mengatur tugas staf dan analis secara efisien dan efektif.
m. Mengatur pelaksanaan replika pengujian dalam rangka jaminan mutu.
n. Menunjuk staf atau analis yang menjadi tanggung jawabnya, apabila berhalangan.
2.4.1.3 Staf Analisis
Staf analisis adalah seorang apoteker atau minimal sarjana farmasi/kimia yang memiliki pengalaman di bidang analisis dan instrumentasi. Staf analisis memiliki tugas dan tanggung jawab sebagai berikut:
a. Melaksanakan proses pengembangan metode analisis berdasarkan kompedial, literatur yang relevan atau pengembangan sendiri.
b. Bersama supervisor laboratorium membuat protokol validasi sebelum melakukan validasi metode.
c. Melakukan validasi metode berdasarkan protokol yang dibuat, mencatat dan melaporkan bila terdapat penyimpangan terhadap protokol.
d. Membuat laporan hasil validasi metode.
e. Menyusun dokumen instruksi yang digunakan di dalam laboratorium.
f. Melakukan pengujian sampel dengan metode analisis yang sudah tervalidasi. g. Bekerjasama dan memonitor proses kerja dari analis.
h. Bertanggung jawab dalam pengoperasian dan pemeliharaan instrumen analisis (HPLC dan LC-MS/MS).
i. Melaporkan kepada supervisor laboratorium kebutuhan peralatan dan peraksi yang digunakan dalam analisis.
j. Bertanggung jawab terhadap kebersihan dan ketertiban di tempat kerja.
k. Bersama supervisor laboratorium memberikan pelatihan dan evaluasi terhadap analis di bawahnya.
Universitas Indonesia
2.4.1.4 Analis
Analis adalah seorang lulusan Sekolah Menengah Analis Kimia atau Akademi Analis Kimia/D3 Kimia yang bekerja di laboratorium bioekivalensi dan memiliki tugas dan tanggung jawab sebagai berikut:
a. Membantu supervisor dan staf dalam melakukan proses pengujian.
b. Membuat larutan pereaksi (reagen) yang menunjang dan diperlukan dalam pengujian.
c. Melakukan preparasi sampel.
d. Melakukan pencatatan berkala pada formulir yang sudah disediakan, seperti monitoring suhu ruangan, suhu lemari es dan freezer, dan lainnya.
e. Mencatat setiap penimbangan, pemasukan material (bahan kimia/pereaksi), pembuatan pereaksi dan pemakaian alat ke dalam logbook yang sudah disediakan.
f. Melaporkan kepada supervisor laboratorium bila ada kebutuhan pereaksi dan alat gelas.
g. Bertanggung jawab terhadap kebersihan dan ketertiban di tempat kerja.
2.4.2 Bagian Klinik 2.4.2.1 Supervisor Klinik
Supervisor Klinik adalah seorang apoteker yang memiliki tugas dan tanggung jawab sebagai berikut:
a. Bersama dengan manajer teknis melakukan proses perekrutan subyek mulai dari skrining sampai proses pengambilan sampel.
b. Bersama dengan dokter peneliti membuat desain case report form
c. Memberikan penjelasan dan training singkat kepada perawat/analis kesehatan sebelum proses pengambilan sampel dilaksanakan.
d. Bertanggung jawab terhadap standardisasi kondisi subyek.
e. Memantau proses pengambilan sampel selama pengujian sesuai protokol. f. Memonitor proses dokumentasi klinis berupa data/rekaman ataupun dokumen
yang berhubungan dengan subyek.
g. Mengatur kebutuhan peralatan yang diperlukan dalam pengambilan sampel (jarum suntik, kapas dan lain-lain).
h. Menyusun prosedur dan instruksi kerja yang digunakan di dalam pengujian bioekivalensi.
i. Mengolah data yang diberikan oleh Supervisor Laboratorium dan melakukan perhitungan sesuai dengan prinsip farmakokinetika dan melakukan analisis secara statistik.
2.4.2.2 Staf Klinik
Staf Klinik minimal adalah seorang D3 farmasi/kimia/S1 kesehatan. Staf klinik bertugas dan bertanggung jawab untuk:
a. Membantu supervisor klinik dalam melakukan studi klinik, seperti: proses perekrutan subyek dan sampling (pemberian obat, standardisasi kondisi subyek, (termasuk makanan, snack, minuman), proses monitoring, pencatatan efek samping, kepatuhan subyek, pengambilan sampel serta kegiatan lainnya. b. Membantu supervisor klinik dalam proses dokumentasi klinik berupa
data/rekaman ataupun dokumen yang berhubungan dengan subyek
c. Bertanggung jawab bahwa alat-alat medis yang digunakan berfungsi dengan baik atau terkalibrasi
d. Bertanggung jawab mengatur kebutuhan peralatan yang diperlukan dalam pengambilan sampel (jarum suntik, kapas dan lain-lain)
2.4.2.3 Dokter Peneliti
Dokter peneliti adalah seorang dokter yang bertanggung jawab untuk: a. Melakukan penjelasan kepada subyek (informed consent) sebelum mereka ikut
serta dalam penelitian.
b. Memberikan penjelasan kepada subyek terhadap hasil pemeriksaan. c. Bersama dengan supervisor klinik merancang desain case report form.
d. Bertanggung jawab dalam pengisian dan melakukan koreksi terhadap case report form.
e. Melakukan skrining terhadap subyek, meliputi evaluasi hasil pemeriksaan laboratorium, fisik dan lain-lain sebagai bahan pertimbangan apakah subyek diperbolehkan ikut serta dalam pengujian.
Universitas Indonesia
2.4.2.4 Perawat/Analis Kesehatan
Perawat/analis kesehatan adalah seorang perawat yang minimal merupakan lulusan D3 Keperawatan atau Sekolah Menengah Analis Kesehatan. Perawat/analis kesehatan memiliki tugas dan tanggung jawab sebagai berikut: a. Melaksanakan proses pengambilan sampel darah maupun urin sesuai dengan
prosedur dan protokol yang sudah ditetapkan dan melakukan pencatatan setiap selesai pengambilan sampel.
b. Memberikan pelayanan yang diperlukan kepada subyek selama proses pengambilan sampel.
c. Membantu dokter dalam memonitor dan menangani setiap kejadian yang tidak diinginkan (adverse event) dan melaporkan ke dokter atau supervisor klinik untuk diambil tindakan lebih lanjut.
2.4.3 Bagian Quality Assurance (QA)/Mutu 2.4.3.1 Manajer Mutu
Manajer Mutu adalah seorang apoteker yang memiliki tugas dan tanggung jawab untuk:
a. Mengelola Tim ISO/IEC17025:2005 dengan tanggung jawab untuk membuat usulan dalam memprakarsai prosedur mutu untuk melaksanakan pengelolaan sistem mutu perusahaan agar sesuai dengan standar ISO/IEC17025:2005; melaksanakan peraturan atau prosedur mutu yang berlaku pada bagiannya; mengadakan pelatihan kepada bawahan masing-masing dalam penerapan sistem manajemen mutu; memantau dan memastikan apakah hasil pekerjaan sesuai standar dan sistem manajemen mutu yang sudah ditetapkan di bagiannya masing-masing dan melakukan tindakan koreksi dan pencegahan apabila ada kekurangan; serta memberi masukkan kepada pimpinan perusahaan tentang status penerapan sistem manajemen mutu dan memberi usulan perbaikan peningkatan kepada manajemen.
b. Merencanakan, mengkoordinir, mengevaluasi penyusunan dan melakukan kaji ulang/tinjauan manajemen mutu untuk menentukan kesesuaian, kecukupan dan efektifitas penerapan sistem manajemen mutu laboratorium sehingga mencapai sasaran yang telah ditetapkan.
c. Mengelola Tim Audit Mutu Internal dengan tanggung jawab untuk membentuk tim auditor untuk mengelola kegiatan audit mutu internal sesuai prosedur yang berlaku; mengarahkan program audit internal secara keseluruhan untuk melihat keefektifan pelaksanaan dan pengendalian sistem mutu perusahaan; memastikan kegiatan audit internal dilaksanakan sesuai standar dan jadwal yang ditetapkan; memastikan tindakan perbaikan dilaksanakan secara efektif dan jadwal waktu yang disetujui; memastikan para auditor mutu cukup terlatih dalam melaksanakan tugas auditnya
d. Memastikan Pusat Pengendali Dokumen (PPD) sudah menjalankan tugasnya sesuai pengarahan yang tertuang pada prosedur yang berlaku.
e. Mengelola Kegiatan Tindakan Perbaikan dan Pencegahan dengan cara memastikan semua keluhan dari pelanggan atau wakilnya ditangani dengan efektif oleh manajemen yang terkait; memastikan kekurangan mutu atas setiap titik pelaksanaan pekerjaan ditangani secara efektif dan dilakukan tindakan perbaikan dan pencegahan oleh bagian yang terkait; menyetujui kesempurnaan tindakan perbaikan dan pencegahan yang dilaksanakan oleh pihak terkait dari aspek mutu; memacu strategi dan rencana peningkatan mutu agar dilaksanakan oleh pihak terkait dari aspek mutu; serta memacu strategi dan rencana peningkatan mutu agar dilaksanakan oleh pihak terkait.
f. Melakukan review laporan akhir suatu studi dan memastikan bahwa laporan yang sudah dibuat sesuai dengan data mentah.
2.4.3.2 Pusat Pengendali Dokumen (PPD)
Pusat Pengendali Dokumen (PPD) dikelola oleh minimal seorang lulusan Akademi Sekretaris/D3 Ekonomi. Tugas dan tanggung jawab yang dimiliki oleh staf PPD adalah:
a. Mengontrol keluar masuknya dokumen, arsip, surat, memo dan dokumen-dokumen lain.
b. Mengatur dan mengontrol sistem pendistribusian dan pengarsipan dokumen. c. Ikut menjaga dan mengamankan dokumen-dokumen yang bersifat rahasia dan
Universitas Indonesia
d. Menyiapkan rapat-rapat rutin intern serta ekstern yang diselenggarakan dan membuat serta mendistribusikan notulensi hasil rapat yang diselenggarakan.
2.4.3.3 Administrasi
Staf administrasi adalah seorang lulusan D3/SMF/SMAK yang mempunyai tugas dan tanggung jawab sebagai berikut:
a. Bertanggung jawab untuk mencari pemasok kebutuhan laboratorium sesuai dengan spesifikasi yang sudah ditentukan, meminta penawaran dan pemesanan barang.
b. Memelihara sistem administrasi pembelian laboratorium termasuk pembuatan Purchase Order (PO), pengarsipan surat jalan dan invoice.
c. Memelihara data Approval Pemasok dan melakukan up-date secara rutin. d. Bekerjasama dengan user untuk melakukan kualifikasi pemasok sebelum
memasukkan ke dalam Daftar Approval Pemasok. e. Melakukan evaluasi pemasok secara berkala.
f. Bersama manajer teknis melakukan kaji ulang permintaan dan kontrak dari pelanggan sesuai dengan bagian tugasnya dan membuat penawaran kepada pelanggan.
2.5 Bangunan dan Fasilitas 2.5.1 Fasilitas Klinik
Fasilitas klinik PT. Clinisindo Laboratories memiliki luas wilayah sekitar 216 m2 yang dilengkapi dengan fasilitas seperti:
a. Wilayah untuk registrasi dan skrining subyek.
b. Wilayah pelayanan subyek (Subject Service Area) terdiri dari 2 kamar subyek (ward/sleeping area) yang memiliki 26 tempat tidur yang terpisah untuk subyek pria dan wanita, ruang rekreasi yang dilengkapi dengan televisi dan fasilitas internet, dilengkapi dengan air conditioner untuk kenyamanan subyek, ruang istirahat (toilet dan mushola), dan kantin.
c. Ruang sampling untuk melakukan proses sampling dan mengumpulkan sampel (darah atau urin) dari subyek. Terdapat pass box khusus untuk mentransfer sampel dari ruang sampling ke ruang preparasi.
d. Fasilitas yang memadai untuk perawatan subyek yang memerlukan penanganan emergensi atau penanganan medis lainnya.
e. Tersedia emergency trolley untuk digunakan pada ruang emergensi.
2.5.2 Fasilitas Analitik
Fasilitas analitik PT. Clinisindo Laboratories memiliki luas wilayah sekitar 152 m2 yang dilengkapi dengan fasilitas seperti:
a. Ruang preparasi untuk memproses sampel (seperti proses pemisahan plasma dan proses ekstraksi).
b. Ruang timbang dengan spesifikasi suhu 20-28oC dan kelembaban < 60%. c. Ruang instrumen dengan spesifikasi suhu 20-28oC dan kelembaban < 60%. d. Ruang penyimpanan untuk bahan-bahan kimia dan reagen
e. Lemari pendingin (refrigerator) dan frezzer bersuhu -20 dan -70oC yang ditujukan sebagai tempat penyimpanan bahan standar dan sampel biologis.
2.5.3 Fasilitas Kantor
Fasilitas kantor PT. Clinisindo Laboratories memiliki luas wilayah sekitar 132 m2 yang dilengkapi dengan ruangan administrasi, ruangan manajer, dan ruang rapat.
2.6 Peralatan
Instrumen utama yang digunakan untuk menganalisis sampel adalah HPLC (High Performance Liquid Chromatography) dan LC-MS/MS (kromatografi cair yang dilengkapi dengan detektor massa). Peralatan laboratorium lainnya yang digunakan dalam analisis adalah timbangan analitik, pH meter, solid phase extraction (SPE), freezer (-20 dan –70oC), alat uji disolusi, water purified system, evaporator, vortex mixer, lemari asam, dan sentrifus.
2.6.1 Kualifikasi, Kalibrasi dan Perawatan
Kegiatan kualifikasi dan kalibrasi diimplementasikan pada semua instrumen dan peralatan yang digunakan untuk memproses sampel, analisis, penyimpanan yang menyangkut (mencakup) massa, volume, suhu, kelembaban,
Universitas Indonesia
dan kecepatan. Kualifikasi dilakukan oleh supplier, termasuk kualifikasi instalasi (IQ), kualifikasi operasional (OQ), dan kualifikasi performance (PQ).
Kegiatan kalibrasi baik internal maupun eksternal dilakukan secara teratur dan terjadwal dengan baik. Frekuensi kalibrasi ditentukan berdasarkan frekuensi penggunaan, kondisi lingkungan, umur instrumen atau peralatan, akurasi dari instrumen atau peralatan, dan rekomendasi menurut buku manual. Hasil kalibrasi diberikan sebagai laporan kalibrasi dan diberi label “Terkalibrasi/Calibrated”. Adanya penyimpangan pada kalibrasi harus dilaporkan dan di follow up. Karena kalibrasi harus dilakukan secara periodik, maka label juga harus mencantumkan tanggal dilakukannya rekalibrasi. Selama waktu pemeriksaan dan perbaikan instrumen tidak boleh digunakan dan diberi label “out of service”.
Perawatan juga direncanakan untuk setiap instrumen dan peralatan utama berdasarkan Prosedur Kontrol Peralatan. Penggunaaan peralatan dicatat pada logbook.
2.7 Dokumentasi
Berdasarkan Prosedur Kontrol Dokumen, kontrol dokumen mencakup pemformatan, penomoran, penerbitan, pendistribusian, sirkulasi, pengisian, pengubahan dokumen dan pemusnahan dokumen diklasifikasikan sebagai controlled document (dokumen terkendali).
Dokumen terkendali adalah semua dokumen yang berkaitan dengan sistem manajemen mutu dari ISO/IEC 17025 dan GCP seperti Quality manual, Standard Operating Procedures (SOP), Standard Operating Instruction (SOI) dan juga pencatatan yang terkait dengan kegiatan laboratorium. Kontrol dokumen merupakan tanggung jawab dari Central Document Controller (PPD) yang berada di bawah pengawasan manajer QA.
Seluruh data mentah asli (seperti perhitungan, kromatogram, dan lainnya) didokumentasikan dengan tujuan agar dapat terlacak dengan menggunakan nomor sampel, peralatan yang digunakan, tanggal dan waktu analisis, dan nama dari analis (staf yang melakukan analisis).
2.8 Pengolahan Limbah
PT. Clinisindo Laboratories menetapkan sistem pemisahan limbah saat pembuangan berdasarkan kode warna kantung. Analis dibantu petugas kebersihan bertanggung jawab melakukan pembuangan limbah ke dalam wadah yang sesuai. Penanganan limbah kimia dan biologi selanjutnya diserahkan pada pihak ketiga.
Limbah yang dihasilkan PT. Clinisindo Laboratories dibagi menjadi 3 macam, yaitu:
a. Limbah medis/biologis
Limbah medis merupakan limbah yang dihasilkan selama kegiatan sampling klinik maupun bioanalisis, seperti darah, plasma, urin, retained sample, jarum suntik, kapas terkena darah, micropore, kasa, perban, dan sarung tangan satu kali pakai. Limbah-limbah biologis/medis ini dimasukkan ke dalam kantung plastik berwarna kuning dan diberi lambang biohazard. Khusus untuk limbah benda tajam, seperti jarum suntik dibuang ke dalam needle box berwarna kuning.
b. Limbah kimia
Limbah kimia adalah bahan-bahan kimia baik cair maupun padat yang dihasilkan dari kegiatan bioanalisis dan analisis laboratorium. Limbah cair organik dibuang ke dalam wadah khusus (tong/jerigen) bertutup rapat dan diberi label “Limbah Cair Organik”. Khusus untuk asam-asam dapat langsung dibuang ke pembuangan air setelah dinetralkan terlebih dahulu.
c. Limbah umum
Limbah umum seperti alat tulis kantor, kemasan pembungkus, kardus, makanan sisa, dan lainnya dibuang ke dalam kantung plastik hitam.
18 Universitas Indonesia
TINJAUAN KHUSUS
3.1 Pendahuluan Uji Bioavailabilitas dan Bioekivalensi (Badan POM RI, 2004)
Badan Pengawas Obat dan Makanan (BPOM) mempunyai kewajiban untuk menilai semua produk obat sebelum dipasarkan, memberikan izin pemasaran, dan selanjutnya melakukan pengawasan terhadap produk obat tersebut setelah dipasarkan untuk memberikan jaminan kepada masyarakat bahwa produk obat tersebut memenuhi standar efikasi, keamanan dan mutu yang dibutuhkan. Produk obat yang mengandung zat aktif berupa zat kimia baru (new chemical entity/NCE) perlu dinilai efikasi, keamanan dan mutunya secara lengkap. NCE ini yang dipatenkan oleh pabrik penemunya disebut juga sebagai obat inovator. Sedangkan untuk produk obat yang merupakan produk copy hanya dibutuhkan standar mutu yang antara lain berupa bioekivalensi dengan produk obat inovator sebagai produk pembanding (reference product) yang merupakan baku mutu.
Bioavailabilitas (ketersediaan hayati) adalah persentase dan kecepatan zat aktif dalam suatu produk obat yang mencapai/tersedia dalam sirkulasi sistemik dalam bentuk utuh/aktif setelah pemberian produk obat tersebut, diukur dari kadarnya dalam darah terhadap waktu atau dari ekskresinya dalam urin. Bila dibandingkan dengan sediaan intravena yang bioavailabilitasnya 100% disebut sebagai bioavailabilitas absolut dan bila dibandingkan dengan sediaan bukan intravena dinamakan bioavailabilitas relatif.
Dua produk obat yang mengandung zat aktif yang sama dalam jumlah yang sama dan bentuk sediaan yang sama disebut ekivalensi farmasetik. Sedangkan bila keduanya mengandung zat aktif yang sama tetapi berbeda dalam bentuk kimia (garam, ester, dan lainnya) atau bentuk sediaan atau kekuatan, maka disebut sebagai alternatif farmasetik. Disebut bioekivalen jika keduanya mempunyai ekivalensi farmasetik atau merupakan alternatif farmasetik dan pada pemberian dengan dosis molar yang sama akan menghasilkan bioavailabilitas yang sebanding sehingga efeknya akan sama, dalam hal efikasi maupun keamanan. Jika bioavailabilitasnya yang tidak memenuhi kriteria bioekivalen maka kedua produk obat tersebut disebut bioinekivalen.
Dua produk obat mempunyai ekivalensi terapeutik jika keduanya mempunyai ekivalensi farmasetik atau merupakan alternatif farmasetik dan pada pemberian dengan dosis molar yang sama akan menghasilkan efikasi klinik dan keamanan yang sebanding. Dengan demikian, ekivalensi/inekivalensi terapeutik seharusnya ditunjukkan dengan uji klinik. Akan tetapi, untuk produk obat yang bekerja sistemik, uji klinik mempunyai kendala yaitu pada penyakit ringan tidak terlihat sedangkan pada penyakit berat tidak etis. Selain itu, endpoint yang diukur seringkali kurang akurat sehingga variabilitasnya besar sekali, sehingga dibutuhkan jumlah sampel yang besar. Oleh karena itu, sebagai alternatif dilakukan uji bioekivalensi yang endpoint-nya sangat akurat (yakni kadar obat dalam plasma) sehingga variabilitasnya rendah, dan dengan demikian sampel yang dibutuhkan jauh lebih kecil. Jika terdapat perbedaan yang bermakna secara klinik dalam bioavailabilitasnya maka kedua produk obat tersebut dinyatakan inekivalen secara terapeutik (inekivalensi terapeutik).
Produk obat pembanding (reference product) adalah produk obat inovator yang telah diberi izin pemasaran di Indonesia berdasarkan penilaian dossier lengkap yang membuktikan efikasi, keamanan dan mutu. Jika produk obat inovator tidak dipasarkan di Indonesia atau tidak lagi dikenali yang mana karena sudah terlalu lama beredar di pasar, maka dapat digunakan produk obat inovator dari primary market (negara dimana produsennya menganggap bahwa efikasi, keamanan dan kualitas produknya terdokumentasi paling baik) atau menggunakan produk yang merupakan market leader yang telah diberi izin pemasaran di Indonesia dan telah lolos penilaian efikasi, keamanan dan mutu. Produk obat pembanding yang akan digunakan harus disetujui oleh Badan POM.
3.2 Alur Uji Bioavailabilitas dan Bioekivalensi (Badan POM RI, 2001; Badan POM RI, 2004)
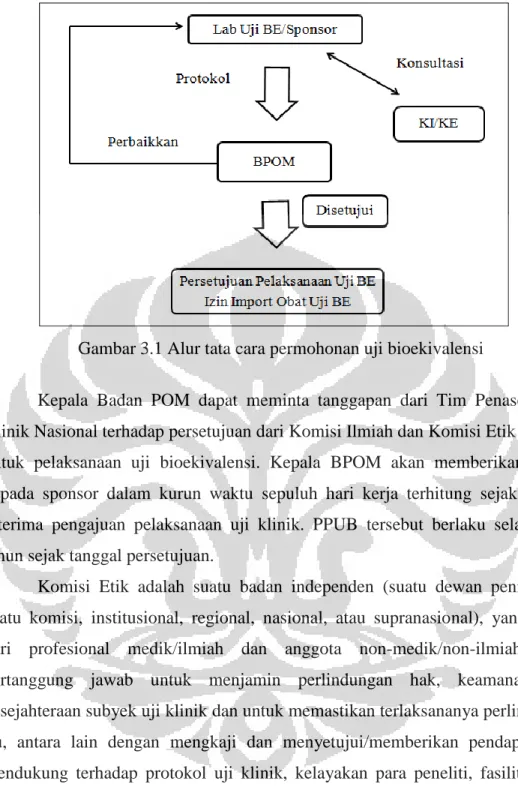
Persetujuan Pelaksanaan Uji Bioekivalensi (PPUB) adalah surat persetujuan uji bioekivalensi yang dikeluarkan oleh Kepala Badan POM. Pengajuan pelaksanaan uji bioekivalensi dilakukan oleh sponsor atau yang bertindak sebagai sponsor kepada Kepala Badan POM. Alur tata cara permohonan uji bioekivalensi dapat dilihat pada Gambar 3.1 sebagai berikut.
Universitas Indonesia
Gambar 3.1 Alur tata cara permohonan uji bioekivalensi
Kepala Badan POM dapat meminta tanggapan dari Tim Penasehat Uji Klinik Nasional terhadap persetujuan dari Komisi Ilmiah dan Komisi Etik Institusi untuk pelaksanaan uji bioekivalensi. Kepala BPOM akan memberikan PPUB kepada sponsor dalam kurun waktu sepuluh hari kerja terhitung sejak tanggal diterima pengajuan pelaksanaan uji klinik. PPUB tersebut berlaku selama dua tahun sejak tanggal persetujuan.
Komisi Etik adalah suatu badan independen (suatu dewan penilai atau suatu komisi, institusional, regional, nasional, atau supranasional), yang terdiri dari profesional medik/ilmiah dan anggota non-medik/non-ilmiah, yang bertanggung jawab untuk menjamin perlindungan hak, keamanan, dan kesejahteraan subyek uji klinik dan untuk memastikan terlaksananya perlindungan itu, antara lain dengan mengkaji dan menyetujui/memberikan pendapat yang mendukung terhadap protokol uji klinik, kelayakan para peneliti, fasilitas, cara dan bahan yang digunakan untuk memperoleh dan mendokumentasikan persetujuan setelah penjelasan dari subyek uji klinik tersebut. Komisi Ilmiah adalah suatu badan independen yang terdiri dari para tenaga kesehatan yang bertanggung jawab melakukan kajian aspek ilmiah termasuk manfaat yang diharapkan terhadap dokumen uji klinik.
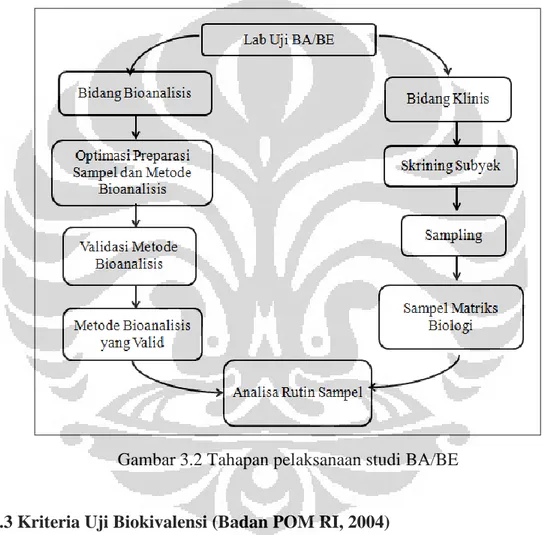
Laboratorium bioavailabilitas dan bioekivalensi (BA/BE) terbagi menjadi dua bidang, yaitu bidang klinis dan bagian bioanalisis. Bidang klinis bertanggung
jawab atas skrining subjek hingga proses sampling terlaksana, sedangkan bidang bioanalisis memiliki tanggung jawab untuk melakukan pengembangan metode analisis sampel. Bidang bioanalisis melakukan optimasi metode preparasi sampel hingga optimasi metode analisis. Setelah itu akan dilakukan validasi metode bioanalisis untuk mendapatkan metode bioanalisis yang valid yang kemudian akan digunakan untuk analisa sampel rutin. Metode analisa yang valid diperlukan untuk menjamin keabsahan hasil uji yang diperoleh. Bagan tahapan pelaksanaan studi BA/BE dapat dilihat pada Gambar 3.2 sebagai berikut.
Gambar 3.2 Tahapan pelaksanaan studi BA/BE
3.3 Kriteria Uji Biokivalensi (Badan POM RI, 2004)
Tidak semua obat copy perlu dilakukan uji bioekivalensi sebelum dipasarkan. Ada beberapa obat yang tidak memerlukan uji bioekivalensi secara in vivo, tetapi cukup dilakukan uji bioekivalensi in vitro saja yaitu dengan Uji Disolusi Terbanding (UDT). Selain itu ada juga sediaan obat copy yang tidak perlu diuji ekivalensinya, misalnya sediaan intravena yang bioavailabilitasnya mencapai 100%.
Universitas Indonesia
3.3.1 Kriteria produk obat yang memerlukan uji bioekivalensi in vivo
1. Produk obat oral lepas cepat yang bekerja sistemik dan memenuhi satu atau lebih kriteria berikut ini:
a. Obat-obat untuk kondisi yang serius yang memerlukan respon terapi yang pasti (critical use drugs). Contohnya antituberkulosis, antiretroviral, antimalaria, antibakteri, antihipertensi, antiangina, obat gagal jantung, antiepilepsi, dan antiasma.
b. Batas keamanan/indeks terapi sempit, ditandai dengan kurva dosis-respons yang curam. Misalnya digoksin, antiaritmia, antikoagulan, obat-obat sitostatik, litium, fenitoin, siklosporin, sulfonilurea, dan teofilin.
c. Terbukti terdapat masalah bioavailabilitas atau bioinekivalensi dengan obat yang bersangkutan atau obat-obat dengan struktur kimia atau formulasi yang mirip (tidak berhubungan dengan masalah disolusi), misal absorpsinya bervariasi atau tidak lengkap; eliminasi presistemik yang tinggi (>70%); farmakokinetik nonlinear; dan sifat-sifat fisiokimia yang tidak menguntungkan (misalnya kelarutan rendah, permeabilitas rendah, tidak stabil, dan lainnya). d. Eksipien dan proses pembuatannya diketahui mempengaruhi bioekivalensi. 2. Produk obat non-oral dan non-parenteral yang didesain untuk bekerja sistemik,
seperti sediaan transdermal, supositoria, permen karet nikotin, gel testosteron dan kontraseptif bawah kulit.
3. Produk obat lepas lambat atau termodifikasi yang bekerja sistemik.
4. Produk kombinasi tetap untuk bekerja sistemik, yang paling sedikit salah satu zat aktifnya memerlukan studi in vivo.
5. Produk obat bukan larutan untuk penggunaan nonsistemik (oral, nasal, okular, dermal, rektal, vaginal) dan dimaksudkan untuk bekerja lokal (tidak untuk diabsorpsi sistemik). Bioekivalensi dari produk-produk tersebut harus ditunjukkan dengan studi klinik atau farmakodinamik, dermatofarmakokinetik komparatif dan/atau studi in vivo. Pada kasus-kasus tertentu, pengukuran kadar obat dalam darah masih diperlukan dengan alasan keamanan untuk melihat adanya absorpsi yang tidak diinginkan.
Daftar obat copy yang mengandung zat aktif wajib uji bioekivalensi dapat dilihat pada Lampiran 1.
3.3.2 Produk obat yang cukup dilakukan uji ekivalensi in vitro (uji disolusi terbanding/UDT)
1. Produk obat yang tidak memerlukan studi in vivo. 2. Produk obat copy yang hanya berbeda kekuatan
Jika studi ekivalensi telah dilakukan sedikitnya pada salah satu kekuatan (biasanya kekuatan yang tertinggi, kecuali untuk alasan keamanan dipilih kekuatan yang lebih rendah); uji disolusi terbanding dapat diterima untuk kekuatan yang lebih rendah berdasarkan perbandingan profil disolusi, dengan ketentuan sebagai berikut:
a. Tablet lepas cepat
Produk obat copy dengan kekuatan berbeda, yang dibuat oleh pabrik obat yang sama di tempat produksi yang sama, jika semua kekuatan mempunyai proporsi zat aktif dan inaktif yang persis sama atau untuk zat aktif yang sangat poten (sampai 10 mg per satuan dosis), zat inaktifnya sama banyak untuk semua kekuatan; studi ekivalensi telah dilakukan sedikitnya pada salah satu kekuatan (biasanya kekuatan yang tertinggi, kecuali untuk alasan keamanan dipilih kekuatan yang lebih rendah); dan/atau profil disolusinya mirip antar kekuatan.
b. Kapsul berisi butir-butir lepas lambat
Jika kekuatannya berbeda hanya dalam jumlah butir yang mengandung zat aktif, maka perbandingan profil disolusi dengan satu kondisi uji yang direkomendasi sudah cukup.
c. Tablet lepas lambat
Jika produk uji dalam bentuk sediaan yang sama tetapi berbeda kekuatan, dan mempunyai proporsi zat aktif dan inaktif yang persis sama atau untuk zat aktif yang sangat poten (sampai 10 mg per satuan dosis) zat inaktifnya sama banyak, dan mempunyai mekanisme pelepasan obat yang sama, kekuatan yang lebih rendah tidak memerlukan studi in vivo jika menunjukkan profil disolusi yang mirip dalam 3 pH yang berbeda (antara pH 1,2 dan 7,5) dengan metode uji yang direkomendasi.
3. Berdasarkan sistem klasifikasi biofarmasetik (Biopharmaceutic Classification System/BCS) dari zat aktif, serta karakteristik disolusi, dan profil disolusi dari
Universitas Indonesia
produk obat. Berlaku untuk produk obat oral lepas cepat, tetapi tidak berlaku untuk produk obat oral lepas cepat yang disebutkan dalam butir 1.
a. Zat aktif memiliki kelarutan dalam air yang tinggi dan permeabilitas dalam usus yang tinggi (BCS kelas 1), serta merupakan produk obat memiliki disolusi yang sangat cepat, atau produk obat yang memiliki disolusi yang cepat dan profil disolusinya mirip dengan produk pembanding.
b. Zat aktif memiliki kelarutan dalam air yang tinggi tetapi permeabilitas dalam usus yang rendah (BCS kelas 3), serta merupakan produk obat memiliki disolusi yang sangat cepat dan produk obat tidak mengandung zat inaktif yang diketahui mengubah motilitas dan/atau permeabilitas saluran cerna.
c. Zat aktif memiliki permeabilitas dalam usus yang tinggi tetapi kelarutan dalam air yang rendah (kelarutan dalam air tinggi hanya pada pH 6,8; BCS kelas 2 asam lemah), serta merupakan produk obat memiliki disolusi yang cepat pada pH 6,8, dan produk obat yang memiliki profil disolusi yang mirip dengan produk pembanding (juga berlaku jika disolusi < 10% pada salah satu pH).
3.3.3 Produk obat yang tidak memerlukan uji ekivalensi
1. Produk obat copy intravena (larutan dalam air) yang mengandung zat aktif yang sama dalam kadar molar yang sama dengan pembanding.
2. Produk obat copy parenteral lain (intramuskular, subkutan) sebagai larutan dalam air dan mengandung zat aktif yang sama dalam kadar molar yang sama dan eksipien yang sama atau mirip (similar) dalam kadar yang sebanding seperti dalam produk pembanding. Eksipien tertentu (misalnya pengawet, antioksidan) boleh berbeda asalkan perubahan eksipien ini diperkirakan tidak mempengaruhi keamanan dan/atau efikasi obat.
3. Produk obat copy berupa larutan oral (sirup, eliksir, tingtur atau bentuk larutan lain bukan suspensi), yang mengandung zat aktif dalam kadar molar yang sama dengan produk pembanding, dan hanya mengandung eksipien yang diketahui tidak mempunyai efek terhadap transit atau permeabilitas dalam saluran cerna. 4. Produk obat copy berupa bubuk untuk dilarutkan dan larutannya memenuhi
kriteria 1, 2, atau 3 tersebut diatas. 5. Produk obat copy berupa gas.
6. Produk obat mata atau telinga sebagai larutan dalam air. 7. Produk obat topikal sebagai larutan dalam air.
8. Produk obat copy berupa larutan untuk aerosol atau inhalasi nebulizer atau semprot hidung, yang digunakan dengan atau tanpa alat yang praktis sama,
3.4 Desain dan Pelaksanaan Studi Bioekivalensi (Badan POM RI, 2004) Studi bioekivalensi (BE) adalah studi bioavailabilitas (BA) komparatif yang dirancang untuk menunjukkan bioekivalensi antara produk uji (suatu produk obat copy) dengan produk obat inovator/pembandingnya. Caranya dengan membandingkan profil kadar obat dalam darah atau urin antara produk-produk obat yang dibandingkan pada subyek manusia. Desain dan pelaksanaan studi BE harus mengikuti Pedoman Cara Uji Klinik yang Baik (CUKB), termasuk harus lolos Kaji Etik dan mendapatkan PPUB. Protokol studi BA/BE harus lolos kaji etik dan mendapatkan PPUB terlebih dahulu sebelum studi dapat dimulai karena studi BA/BE menggunakan subyek manusia (suatu uji klinik).
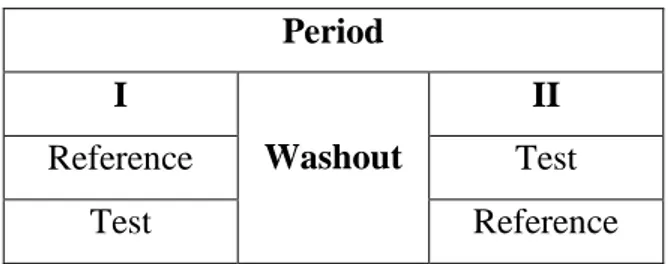
Studi biasanya dilakukan pada subyek yang sama (dengan desain menyilang/cross over) untuk menghilangkan variasi biologik antar subyek (karena setiap subyek menjadi kontrolnya sendiri), hal ini sangat memperkecil jumlah subyek yang dibutuhkan. Desain 2-way crossover (desain menyilang dua arah) adalah desain studi 2 periode untuk pemberian 2 produk obat pada setiap subyek. Pemberian produk obat yang pertama harus dilakukan secara acak agar efek urutan (order effect) maupun efek waktu (period effect) seimbang.
Kedua perlakuan dipisahkan oleh periode washout yang cukup untuk eliminasi produk obat yang pertama diberikan (biasanya lebih dari lima kali waktu paruh obat, atau lebih lama jika mempunyai metabolit aktif dengan waktu paruh yang lebih panjang). Jika obat mempunyai kecepatan eliminasi yang sangat bervariasi antar subyek, periode washout yang lebih lama diperlukan untuk memperhitungkan kecepatan eliminasi yang lebih rendah pada beberapa subyek. Oleh karena itu untuk obat dengan waktu paruh eliminasi yang panjang (> 24 jam), dapat dipertimbangkan penggunaan desain 2 kelompok paralel. Bentuk desain 2-way crossover dapat dilihat pada Gambar 3.3 sebagai berikut.
Universitas Indonesia Period I Washout II Reference Test Test Reference
Gambar 3.3 Desain 2-way crossover
3.4.1 Kriteria seleksi subyek
Kriteria inklusi dan eksklusi subyek harus dinyatakan dengan jelas di dalam protokol, yaitu:
a. Sukarelawan sehat (untuk mengurangi variasi antar subyek).
b. Sedapat mungkin pria dan wanita (jika wanita pertimbangkan risiko pada wanita usia subur).
c. Umur antara 18-55 tahun.
d. Berat badan dalam kisaran normal (BMI 18-25).
e. Kriteria sehat berdasarkan uji laboratorium klinis yang baku (hematologi rutin, fungsi hati, fungsi ginjal, gula darah, dan urinalisis), riwayat penyakit, dan pemeriksaan fisik.
f. Pemeriksaan khusus mungkin harus dilakukan sebelum, selama dan setelah studi selesai, bergantung pada kelas terapi dan profil keamanan obat yang diteliti. Misalnya untuk obat dari kelas fluorokuinolon yang diketahui dapat memperpanjang interval QT, harus dilakukan pemeriksaan EKG.
g. Sebaiknya bukan perokok. Jika perokok sedang (kurang dari 10 batang sehari) diikutsertakan, harus disebutkan dan efeknya pada hasil studi harus didiskusikan.
h. Tidak mempunyai riwayat ketergantungan pada alkohol atau penyalahgunaan obat.
i. Tidak kontraindikasi atau hipersensitif terhadap obat yang diuji.
j. Digunakan penderita dengan indikasi yang sesuai untuk obat yang terlalu toksik untuk diberikan kepada sukarelawan sehat (misalnya obat sitostatik atau obat antiaritmia).
k. Uji serologis terhadap Hepatitis B (HBsAg), Hepatitis C (anti-HCV) dan HIV (anti-HIV).
3.4.2 Jumlah subyek
Jumlah subyek yang dibutuhkan dihitung berdasarkan parameter bioavailabilitas yang utama, yakni AUC (Area Under Curve) atau area dibawah kurva kadar obat dalam darah terhadap waktu, yang menunjukkan jumlah obat yang masuk peredaran darah sistemik.
Untuk desain menyilang 2-way crossover, jumlah subyek yang dibutuhkan ditentukan oleh perbedaan nilai rata-rata AUC antara produk uji (test/T) dan produk pembanding (reference/R) yang sesuai dengan kriteria bioekivalen, yakni rasio nilai rata-rata geometrik (AUC)T/(AUC)R = 1,00 dengan 90% CI sebesar 0,80-1,25. Selain itu faktor yang juga berpengaruh adalah koefisien variasi (coefficient of variation/CV) intrasubyek dari AUC obat yang diteliti yang diperkirakan dari percobaan pendahuluan, dari studi sebelumnya atau dari data terpublikasi.
Dengan ketentuan tersebut diatas, maka jumlah subyek tergantung dari koefisien variasi (CV) intrasubyek sebagai berikut (umumnya, CV intrasubyek kurang dari 20%) seperti yang dapat dilihat pada Tabel 3.1 sebagai berikut :
CV intrasubyek (%)* Jumlah subyek
15,0 12 17,5 16 20,0 20 22,5 24 25,0 28 27,5 34 30,0 40
* CV2 = varians residual pada ANOVA untuk desain menyilang 2-way
Tabel 3.1 Perbandingan jumlah subyek terhadap koefisien variasi intrasubyek. Jumlah subyek minimal adalah 12 orang dan umumnya yang digunakan adalah 18-24 orang. Kemungkinan drop-out dan withdrawal juga harus diperhitungkan. Ada dua cara (cara yang dipilih harus disebutkan dalam protokol) yaitu tambahkan sejumlah tertentu subyek (satu atau dua untuk setiap urutan)
Universitas Indonesia
kepada jumlah subyek yang telah dihitung, atau tambahkan sejumlah tertentu subyek ke dalam studi.
Hanya jika ada subyek yang drop-out maka sampel darah subyek tambahan tersebut diukur kadar obatnya. Withdrawal yang terjadi setelah kadar obatnya diukur, maka hasilnya harus dilaporkan. Jika jumlah subyek ternyata kurang karena variasi yang diperkirakan ternyata lebih besar, maka jumlah subyek dapat ditambah dengan tidak kurang dari setengah jumlah subyek awalnya. Hasil dapat digabung asal digunakan protokol yang sama dan produk obat uji dari batch yang sama.
3.4.3 Standardisasi kondisi studi
Kondisi studi harus dibakukan (untuk mengurangi variabilitas berbagai faktor yang terlibat kecuali produk yang diuji), yaitu:
a. Lama puasa pada malam sebelum pemberian produk minimal 10 jam.
b. Jika obat harus diberikan bersama makanan untuk mengurangi efek samping saluran cerna, maka studi BE harus dilakukan bersama makanan standar. c. Volume air yang diminum bersama produk harus konstan (antara 150-200 ml)
karena dapat mempengaruhi pengosongan lambung.
d. Semua makanan dan minuman yang dikonsumsi setelah pemberian produk harus dibakukan komposisi dan waktu pemberiannya selama periode pengambilan sampel darah, seperti air boleh diminum kapan saja kecuali 1 jam sebelum dan 2 jam sesudah pemberian produk, dan makanan standar diberikan tidak kurang dari 4 jam setelah pemberian produk.
e. Subyek tidak boleh makan obat lain apapun (termasuk obat bebas dan obat tradisional) selama beberapa waktu sebelum penelitian (minimal 1 minggu) dan selama penelitian. Dalam keadaan darurat, penggunaan obat apapun harus dilaporkan (dosis dan waktu penggunaan).
f. Subyek tidak boleh mengkonsumsi makanan dan minuman yang dapat berinteraksi dengan fungsi sirkulasi, saluran cerna, hati atau ginjal (misalnya merokok, minum alkohol, kopi, teh, kola, coklat atau jus buah) selama 24 jam sebelum penelitian dan selama periode pengambilan sampel darah.
g. posisi tubuh dan aktivitas fisik juga harus distandardisasi sepanjang hari penelitian karena akan mempengaruhi motilitas dan aliran darah saluran cerna.
3.4.4 Pengambilan sampel
Sampel yang digunakan biasanya adalah sampel darah, meskipun sampel urin juga dapat digunakan. Sampel darah harus diambil pada waktu-waktu tertentu sehingga dapat menggambarkan fase-fase absorpsi, distribusi, dan eliminasi obat. Kebanyakan obat memerlukan 12-18 sampel darah, yang terdiri dari:
a. 1 sampel sebelum obat/pada waktu nol (t0) b. 2-3 sampel sebelum kadar maksimal (Cmax) c. 4-6 sampel sekitar Cmax
d. 5-8 sampel setelah Cmax, sampai sedikitnya 3 atau lebih waktu paruh eliminasi obat dalam plasma (> 3 x t1/2).
Obat atau obat yang metabolit aktifnya mempunyai waktu paruh eliminasi (t1/2) yang panjang (lebih dari 24 jam), sampel darah harus diambil sampai sedikitnya 72 jam jika variabilitas intra-subyek kecil, atau lebih lama jika variabilitas intra-subyek besar.
Sampel urin hanya digunakan jika kadar obat dalam darah terlalu kecil untuk dapat dideteksi dan eliminasi obat dalam bentuk utuh melalui ginjal cukup besar (>40%). Urin dikumpulkan di tempat studi secara periodik sampai sedikitnya 3 x waktu paruh eliminasi obat (3 x t1/2). Waktu sampling untuk studi selama 24 jam biasanya 0-2, 2-4, 4-8, 8-12 dan 12-24 jam. Volume urin setiap interval waktu tersebut harus diukur dan dilaporkan. Kemudian dibuat kurva jumlah obat kumulatif yang diekskresi dalam urin terhadap waktu.
3.5 Laporan Hasil Uji Bioekivalensi
Produk uji (test/T) dan produk pembanding (reference/R) dikatakan bioekivalen jika:
a. Rasio nilai rata-rata geometrik (AUC)T/(AUC)R = 1,00 dengan 90% Cl = 80-125%, untuk obat-obat dengan indeks terapi yang sempit, interval ini mungkin perlu dipersempit (90-111%). Interval yang lebih lebar mungkin dapat diterima jika didasari pertimbangan klinik yang jelas.
Universitas Indonesia
b. Rasio nilai rata-rata geometrik (Cmax)T/(Cmax)R = 1,00 dengan 90% CI = 80-125%. Cmax lebih bervariasi dibanding AUC, maka interval yang lebih lebar mungkin cocok. Interval ini harus ditetapkan sebelumnya, misal 75-133% atau 70-143%, dan harus diberikan alasan dengan mempertimbangkan efikasi dan keamanannya..
c. Perbandingan tmax dilakukan hanya jika ada klaim yang relevan secara klinik mengenai pelepasan atau kerja yang cepat atau adanya tanda-tanda yang berhubungan dengan efek samping obat.
Nilai confidence interval (CI) tidak boleh dibulatkan, jadi untuk CI 80- 125% nilainya harus minimal 80,00 dan tidak lebih dari 125,00. Jika bioavailabilitas produk uji lebih besar dibandingkan produk pembandingnya (suprabioavailabilitas), maka harus dilakukan reformulasi. Studi bioekivalensi harus dilakukan lagi dengan produk reformulasi tersebut. Laporan studi BE harus mencantumkan:
a. Nama dan afiliasi serta tandatangan para peneliti, tempat studi, dan waktu pelaksanaan studi.
b. Dokumentasi bahwa pelaksanaan studi sesuai dengan prinsip Cara Uji Klinik yang Baik (CUKB), termasuk surat persetujuan Komisi Etik setempat, dan informed consent yang ditandatangani oleh setiap subyek penelitian.
c. Nama, nomor batch dan komposisi produk obat uji; spesifikasi obat jadi dalam bentuk sertifikat analisis dan hasil uji disolusi terbanding; pernyataan sponsor bahwa produk obat uji identik dengan produk yang didaftarkan untuk izin pemasaran.
d. Nama, nomor batch dan tanggal kadaluarsa produk pembanding.
e. Validasi metode pengukuran kadar obat dalam plasma/urin, mencakup seluruh kisaran kadar yang diukur dalam spesimen.
f. Data kadar obat dalam plasma/urin terhadap waktu dari masing-masing subyek disertai statistik deskriptifnya (rata-rata, median, SD, minimum dan maksimum).
g. Kurva kadar obat dalam plasma/urin terhadap waktu dari masing-masing subyek, dalam skala biasa (arithmetic) maupun skala logaritmik (ln).
h. Nilai parameter bioavailabilitas dari masing-masing subyek disertai statistik deskriptifnya
i. Data yang dibuang disertai alasannya
j. Data dari subyek yang dropout dan mengundurkan diri.
k. Analisis statistik yang cukup rinci agar dapat diulang jika perlu dan cara perhitungannya, termasuk 90% CI.
32 Universitas Indonesia
PEMBAHASAN
Biaya kesehatan yang semakin lama semakin tinggi menuntut adanya substitusi obat originator/inovator (paten) dengan obat copy generik. Obat copy yang dimaksud adalah produk obat yang mempunyai ekivalensi farmasetik atau merupakan alternatif farmasetik dengan produk obat inovator atau pembandingnya, dapat dipasarkan dengan nama generik atau dengan nama dagang.
Obat copy generik ini dapat menghemat biaya 40-60% dibandingkan dengan inovator. Hal ini disebabkan karena pembuatan obat copy generik tidak melakukan pengembangan senyawa kimia baru/new chemical entity (NCE) seperti pada obat inovator/paten. Pada obat copy generik hanya dilakukan pengembangan formulasi produk obat yang sudah off patent agar sama dengan inovator, sehingga tidak perlu dilakukan uji pada hewan dan juga uji klinik untuk keamanan dan efektivitasnya. Tetapi perbandingan efek terapetik antara dua produk obat yang mengandung zat aktif yang sama ini harus dibuktikan.
Persyaratan yang diperlukan untuk obat substitusi/obat copy ini adalah harus ekivalen secara terapetik dengan obat inovator. Ekivalen secara terapetik dapat diasumsikan sebagai bioekivalen. Bioekivalensi adalah bila dua produk obat yang dibandingkan mempunyai ekivalensi farmasetik atau alternatif farmasetik, pada pemberian dosis molar yang sama akan menghasilkan bioavailabilitas yang sebanding sehingga diperkirakan efeknya akan sama dalam hal efikasi maupun keamanannya.
Tujuan dari uji ini adalah untuk menjamin efikasi, keamanan, dan mutu obat yang beredar. Adanya uji bioekivalensi menyebabkan meningkatnya riset obat generik, menghasilkan industri generik yang kompetitif, meningkatnya akses obat yang terjangkau, mendorong inovasi, dan meningkatkan peran Indonesia dalam pasar generik global.
Tidak semua obat harus diuji bioekivalensinya. Ada beberapa obat yang tidak memerlukan uji ekivalensi in vivo (bioekivalensi) tetapi cukup dengan uji ekivalensi in vitro saja (Uji Disolusi Terbanding/UDT), ada pula obat yang tidak perlu uji ekivalensi, dan ada obat yang wajib untuk diuji ekivalensi in vivo.
Kriteria untuk uji ekivalensi ini dapat dilihat pada buku Pedoman Uji Bioekivalensi yang dikeluarkan oleh BPOM tahun 2004.
Laboratorium BA/BE harus menerapkan Cara Uji Klinik Yang Baik (CUKB) atau Good Clinical Practices (GCP). Cara Uji Klinik yang Baik (CUKB) adalah suatu standar untuk desain, pelaksanaan, pencapaian, pemantauan, pengauditan, perekaman, analisis, dan pelaporan uji klinik yang memberikan jaminan bahwa data dan hasil yang dilaporkan dapat dipercaya dan akurat, dan bahwa hak, integritas, dan kerahasiaan subyek uji klinik dilindungi.
Pengujian yang dilakukan di laboratorium BA/BE harus mengikuti prinsip Good Laboratory Practice (GLP). Penilaian terhadap Laboratorium BA/BE di Indonesia dilakukan oleh Subdirektorat Standardisasi dan Penilaian Bioavailabilitas/Bioekivalensi Obat yang berada di bawah Direktorat Standardisasi Produk Terapeutik dan PKRT Badan Pengawas Obat dan Makanan (BPOM) Republik Indonesia. Akreditasi terhadap institusi yang memiliki laboratorium BA/BE dilakukan oleh Komite Akreditasi Nasional (KAN).
Di Indonesia terdapat beberapa laboratorium pengujian bioavailabilitas dan bioekivalensi (BA/BE) yang telah terakreditasi oleh Komite Akreditasi Nasional (KAN) yaitu Laboratorium Uji Bioekivalensi Jurusan Farmasi FMIPA UI, Laboratorium Uji Bioekivalensi Fakultas Kedokteran UI, Laboratorium Uji Bioekivalensi Fakultas Farmasi UGM, Laboratorium Uji Bioekivalensi, Farmakologi, dan Toksikologi Fakultas Kedokteran UGM, Laboratorium Pengujian Bioekivalensi Fakultas Farmasi UNAIR, Laboratorium Pengujian Bioekivalensi Sekolah Farmasi ITB, Center for Drug Evaluation Analysis Fakultas Farmasi Universitas Surabaya, PT. Equilab Internasional Indonesia, PT. Clinisindo Laboratories, PT. San-Clin Eq, PT. Pharmametric, PT. Econolab, PT. Omega Medika Farma, dan Laboratorium Bioekivalensi Independen PT. Citra Sintesa Mustika.
PT. Clinisindo Laboratories adalah salah satu laboratorium pengujian bioekivalensi swasta yang didirikan pada tahun 2004. Sebagai suatu badan usaha yang independen PT. Clinisindo Laboratories sepenuhnya terbebas dari tugas dan tanggung jawab lain untuk menghindari terjadinya pertentangan kepentingan sehingga seluruh keputusan hasil pengujian dan laporan hasil pengujian dilakukan
Universitas Indonesia
secara profesional dan independen tanpa ada intervensi dari pihak lain. Semua pengujian yang dilakukan di PT. Clinisindo Laboratories menerapkan prinsip Good Clinical Practice (GCP) dan Good Laboratory Practice (GLP). Pengujian dilakukan dengan menggunakan instrumen dan peralatan yang terkualifikasi dan terkalibrasi dan dilakukan oleh personil yang kompeten.
35 Universitas Indonesia
BAB 5
KESIMPULAN DAN SARAN
5.1 Kesimpulan
5.1.1 PT. Clinisindo Laboratories telah menerapkan setiap aspek Cara Uji Klinik Yang Baik (CUKB)/Good Clinical Practice (GCP) dan Good Laboratory Practice (GLP) dengan baik dalam tiap aspek dan pengujiannya yang meliputi aspek personalia, bangunan dan fasilitas, peralatan, dokumentasi, inspeksi diri, kesehatan dan keselamatan kerja (K3), pengolahan limbah laboratorium, dokumentasi, serta kualifikasi dan validasi.
5.1.2 Kegiatan yang dilakukan PT. Clinisindo Laboratories dibagi menjadi dua bagian yaitu kegiatan bidang klinik dan bagian laboratorium/bioanalisis. Kegiatan di bidang klinik meliputi skrining subjek hingga proses sampling terlaksana, sedangkan bidang laboratorium/bioanalisis memiliki tanggung jawab untuk melakukan pengembangan metode analisis, validasi metode bioanalisis, dan analisis sampel rutin.
5.1.3 Apoteker memegang peranan yang sangat penting dalam laboratorium pengujian bioavailabilitas/bioekivalensi (BA/BE), khususnya di PT. Clinisindo Laboratories, yaitu sebagai manajer teknis, manajer mutu, supervisor klinik, supervisor laboratorium, dan staf analisis. Fungsi apoteker adalah sebagai tenaga profesional yang ikut dalam pengujian dan penentuan bioekivalensi produk obat uji (copy) terhadap obat originator/inovatornya.
5.2 Saran
Penerapan aspek CUKB/GCP dan GLP di PT. Clinisindo Laboratories perlu dipertahankan dan terus ditingkatkan agar dapat menjamin keabsahan hasil pengujian.