SIMULASI DINAMIKA MOLEKUL
PROTEIN 1GB1
MENGGUNAKAN
NOT JUST ANOTHER MOLECULAR
DYNAMICS PROGRAM
(NAMD)
KANIA NUR SAWITRI
DEPARTEMEN FISIKA
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM INSTITUT PERTANIAN BOGOR
PERNYATAAN MENGENAI SKRIPSI DAN
SUMBER INFORMASI SERTA PELIMPAHAN HAK CIPTA*
Dengan ini saya menyatakan bahwa skripsi berjudul Simulasi Dinamika Molekul Protein 1GB1 Menggunakan Not Just Another Molecular Dynamcs
Program (NAMD) adalah benar karya saya dengan arahan dari dosen
pembimbing dan belum diajukan dalam bentuk apa pun kepada perguruan tinggi mana pun. Sumber informasi yang berasal atau dikutip dari karya yang diterbitkan maupun tidak diterbitkan dari penulis lain telah disebutkan dalam teks dan dicantumkan dalam Daftar Pustaka di bagian akhir skripsi ini.
Dengan ini saya melimpahkan hak cipta dari karya tulis saya kepada Institut Pertanian Bogor.
Bogor, Juni 2013
Kania Nur Sawitri
iv
ABSTRAK
KANIA NUR SAWITRI. Simulasi Dinamika Molekul Protein 1GB1 Menggunakan Not Just Another Molecular Dynamcs Program (NAMD).
Dibimbing oleh TONY IBNU SUMARYADA dan SETYANTO TRI WAHYUDI.
Protein 1GB1 membantu Streptomyces griseus menghindari pertahanan
inang melalui sifat pengikatan protein. Tujuan penelitian ini adalah mempelajari mekanisme unfolding dan dinamika molekul protein 1GB1 dengan variasi suhu
berdasarkan beberapa parameter. File koordinat 1GB1 hasil NMR (Nuclear Magnetic Resonance) diunduh melalui protein data bank yang kemudian diolah
melalui beberapa proses simulasi dinamika molekuler yaitu minimisasi, pemanasan, ekuilibrasi dan production run menggunakan metode PBC (Periodic Boundary Condition) dan PME (Particle Mesh Ewald). Simulasi dilakukan pada
temperatur 325K, 350K, 375K, 400K, 450K dan 500K. Kecuali simulasi 500K yang dilakukan selama 1 ns, semua simulasi dilakukan selama 10 ns. Beberapa parameter menunjukkan proses unfolding 1GB1 terjadi pada waktu 900 ps dengan
temperature 500K. Struktur sekunder yang pertama kali mengalami kerusakan adalah alpha-helix yang berubah menjadi turn dan coil.
Kata kunci: simulasi dinamika molekul, VMD, NAMD, protein 1GB1, unfolding
ABSTRACT
KANIA NUR SAWITRI. Molecular Dynamics Simulation of Protein 1GB1 Using
Not Just Another Molecular Dynamics Program (NAMD). Supervised by TONY
IBNU SUMARYADA and SETYANTO TRI WAHYUDI.
Protein 1GB1 helps Streptomyces griseus to avoid host defense by
immunoglobulin binding site properties. The aim of this study are to learn the mechanism of unfolding and molecular dynamics of protein 1GB1 by temperature variance based on some parameters. Coordinate file of 1GB1 that given by NMR (Nuclear Magnetic Resonance) can be downloaded from Protein Data Bank, then it was processed by some steps such as minimization, heating, equilibration and production run using PBC (Periodic Boundary Condition) and PME (Particle Mesh Ewald) methods. Simulation running at 325K, 350K, 375K, 400K, 450K dan 500K. Except simulation 1 ns at 500K, all simulations running for 10 ns. Some parameters shown unfolding process of 1GB1 at 900 ps in 500 K simulation. Secondary structures that broke for the first time is alpha-helix that converted to turn and coil.
Keywords: molecular dynamics simulation, VMD, NAMD, Protein 1GB1,
Skripsi
sebagai salah satu syarat untuk memperoleh gelar Sarjana Sains
pada
Departemen Fisika
SIMULASI DINAMIKA MOLEKUL
PROTEIN 1GB1
MENGGUNAKAN
NOT JUST ANOTHER MOLECULAR
DYNAMICS PROGRAM
(NAMD)
KANIA NUR SAWITRI
DEPARTEMEN FISIKA
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM INSTITUT PERTANIAN BOGOR
Judul Skripsi : Simulasi Dinamika Molekul Protein 1GB1 Menggunakan Not Just
Another Molecular Dynamcs Program (NAMD)
Nama : Kania Nur Sawitri NIM : G74090004
Disetujui oleh
Dr. Tony Ibnu Sumaryada Pembimbing I
Setyanto Tri Wahyudi, M.Si Pembimbing II
Diketahui oleh
Dr. Akhiruddin Maddu Ketua Departemen Fisika
viii
PRAKATA
Alhamdulillahirabbil'alamin. Puji syukur penulis panjatkan kepada Allah SWT, karena berkat rahmat, hidayah dan karunia-Nya penulis dapat menyelesaikan karya ilmiah yang berjudul Simulasi Dinamika Molekul
Streptococcal Protein 1GB1 Menggunakan Not Just Another Molecular Dynamics Program (NAMD) yang dilaksanakan sejak bulan Desember 2012. Shalawat serta
salam semoga tetap tercurahkan kepada Nabi Muhammad SAW.
Ucapan terimakasih penulis sampaikan kepada kesua dosen pembimbing, Dr. Tony Ibnu Sumaryada dan Setyanto Tri Wahyudi, M.Si yang telah meluangkan waktunya untuk membimbing penulis selama penelitian. Terimakasih penulis sampaikan kepada seluruh dosen dan staf Departemen Fisika FMIPA-IPB.
Ucapan terima kasih penulis sampaikan kepada Mamak, Bapak, abang dan adik tersayang yang telah mendoakan, memberikan motivasi dan kasih sayangnya kepada penulis. Begitu juga dengan rekan-rekan mahasiswa/i fisika angkatan 46 dan Pasca sarjana fisika angkatan 7 yang senantiasa memberikan motivasi, saran dan bimbingannya selama ini.
Semoga karya ilmiah ini dapat bermanfaat untuk mengembangkan simulasi dinamika molekul di Departemen Fisika FMIPA-IPB.
Bogor, Juni 2013
DAFTAR ISI
Proses Unfolding Protein 3
Jari-Jari Girasi 3
Root Mean Square Deviation (RMSD) dan Root Mean Square Fluctutation
(RMSF) 3
Energi 3
Solvent Accessible Surface Area (SASA) 4
x
Jari-jari girasi 11
RMSD 12
RMSF 13
SASA 14
Jembatan Garam 16
SIMPULAN DAN SARAN 17
Simpulan 17
Saran 17
DAFTAR PUSTAKA 18
DAFTAR TABEL
Tabel 1 Residu penyusun struktur sekunder 1GB1 2
Tabel 2 Peningkatan jumlah ikatan hidrogen selama simulasi dengan variasi
temperatur 11
DAFTAR GAMBAR
Gambar 1 Perbandingan faktor beta simulasi dengan ekperimen 7 Gambar 2 Energi konformasi selama simulasi dengan variasi temperatur 8 Gambar 3 Energi non ikatan selama simulasi dengan variasi temperatur 9 Gambar 4 Jumlah ikatan hidrogen selama simulasi dengan variasi temperatur 10 Gambar 5 Jari-jari girasi protein 1GB1 dengan variasi temperatur 12 Gambar 6 Kenaikan jari-jari girasi selama simulasi dengan variasi temperatur 12 Gambar 8 Rata-rata RMSD 1GB1 selama simulasi dengan variasi temperatur 13 Gambar 7 RMSD 1GB1 selama simulasi dengan variasi temperatur 13 Gambar 9 selama simulasi tiap residu dengan variasi temperatur. 14 Gambar 10 SASA (a), (b) total (c), (d) backbone (e), (f) non polar (g), (h) polar
selama simulasi dengan variasi temperatur 15 Gambar 11 Kenaikan persentase SASA akibat variasi temperatur 16 Gambar 12 Letak pasangan jembatan garam (a) E56/K10 (b) E27/K31 16 Gambar 13 Energi elektrostatik pasangan jembatan garam (a) E56-K10 (b)
E27-K31 17
DAFTAR LAMPIRAN
Lampiran 1 Perubahan struktur sekunder protein 1GB1 selama simulasi pada (a) 325 K, (b) 350 K, (c) 375 K, (d) 400 K dan (e) 500 K 19 Lampiran 2 Karakteristik residu yang terdapat pada 1GB1 20 Lampiran 3 Pasangan residu penyusun jembatan garam 21 Lampiran 4 Konformasi akhir setiap simulasi (a) 325 K, (b) 350 K, (c) 375 K, (d)
PENDAHULUAN
Latar Belakang
Protein merupakan senyawa organik kompleks dengan bobot molekul tinggi. Protein juga merupakan suatu polimer yang terdiri dari monomer-monomer asam amino yang dihubungkan dengan ikatan peptida. Protein memiliki banyak fungsi diantaranya sebagai enzim, hormon dan antibodi. Di alam, bentuk protein spesifik untuk suatu fungsi. Oleh karena itu agar suatu polipeptida yang baru dibentuk siap menjadi protein yang berfungsi secara biologis dan mampu mengkatalisis suatu reaksi metabolik, menggerakkan sel, atau makromolekul, polipeptida tersebut harus mengalami pelipatan membentuk susunan tiga dimensi tertentu atau konformasi.1
Struktur tiga dimensi protein penting untuk diketahui karena dapat merepresentasikan aktivitas, fungsi, stabilitas, maupun paramater fisika-kimia lainnya. Metode penentuan struktur tiga dimensi protein yang banyak digunakan saat ini adalah kristalografi sinar-X (X-ray crystallography). Kristalografi sinar-X
menggunakan pancaran sinar-X yang ditembakkan mengenai suatu protein yang telah dimurnikan atau memiliki kemurnian tinggi sehingga berbentuk kristal.2 Selain metode Kristalografi sinar-X, metode Nuclear Magnetic Resonance (NMR)
juga dapat memberikan informasi struktural tingkat atom dari protein pada keadaaan unfolded yang penting dalam karakterisasi proses pelipatan protein.3
Simulasi dinamika molekul adalah teknik menghasilkan lintasan atom dari suatu sistem N partikel dengan integrasi numerik persamaan Newton tentang gerak, untuk potensial interatomik spesifik dengan kondisi awal dan kondisi batas tertentu.4 Protein 1GB1 termasuk protein yang memiliki kestabilan termal tinggi. Hal ini dibuktikan dengan proses unfolding beta hairpin yang merupakan salah
satu fragmen dari protein 1GB1 pada suhu 400K. Oleh karena itu diperlukan simulasi dinamika molekul protein 1GB1 pada suhu tinggi.
Perumusan Masalah
Simulasi dinamika molekul untuk struktur lengkap protein 1GB1 masih jarang dilakukan. Sehingga proses unfolding 1GB1 belum dapat dijelaskan secara
detil. Adapun perumusan masalah yang penulis ajukan adalah sebagai berikut: 1. Bagaimana dinamika yang terjadi pada protein 1GB1 selama simulasi? 2. Pada suhu berapa protein 1GB1 mengalami unfolding?
Tujuan Penelitian
Tujuan dari penelitian ini adalah mempelajari mekanisme unfolding dan
2
Manfaat Penelitian
Penelitian ini diharapkan dapat menjelaskan dinamika molekul protein 1GB1 akibat pengaruh suhu. Sehingga penelitian ini dapat menjadi awalan simulasi dinamika molekul di Departemen Fisika IPB.
Ruang Lingkup Penelitian
Ruang lingkup penelitian ini adalah menemukan perubahan beberapa parameter saat terjadi unfolding. Penelitian ini dibatasi oleh waktu simulasi yaitu
selama 10 ns untuk suhu 325K, 350K, 400K dan 450K. Sedangkan untuk suhu 500K penelitian dibatasi selama 1 ns. Simulasi 300K dilakukan hanya untuk validasi. Pembatasan tersebut ditujukan untuk mempercepat computing time.
TINJAUAN PUSTAKA
Protein 1GB1
Protein 1GB1 adalah protein dari kelompok G Streptococcus dengan domain B1. Permukaan multidomain selnya besar dan berfungsi untuk membantu organisme menghindari pertahanan inang melalui sifat pengikatan protein. Protein 1GB1 terdiri dari 56 residu yang membentuk 2 pasang beta-hairpin, 2 coil, 2 turn
dan 1 alpha-helix.5 Jumlah residu penyusun struktur sekunder tersebut dapat
negatif, yang nantinya akan membentuk jembatan garam. Tabel 1 Residu penyusun struktur sekunder 1GB1
3
19 s/d 22 E, A, V, D 36 s/d 41 D, N, G, V, D, G 55 s/d 56 T, E
Proses Unfolding Protein
Mekanisme unfolding dapat dirangkum menjadi empat keadaan utama � →
� → → . F (frayed) adalah keadaan folded dimana strukturnya berjumbai. H
adalah keadaan tanpa struktur sekunder tapi inti hidrophobiknya masih tertutup. S adalah keadaan sebagian inti hidropobik terlarut. U adalah keadaaan unfolded
sempurna.6 Proses unfolding protein dapat dijelaskan melalui beberapa parameter, diantaranya adalah:
Jari-Jari Girasi
Jari-jari girasi � adalah parameter yang menjelaskan konformasi seimbang dari sistem total dan kekompakan protein.
R2g=∑Ni=1(ri-RC)2⁄N (1)
dimana adalah jarak atom ke-i terhadap pusat massa, adalah koordinat pusat massa dan N adalah jumlah atom.7
Root Mean Square Deviation (RMSD) dan Root Mean Square Fluctutation (RMSF)
RMSD didefinisikan sebagai besaran jarak antara dua buah atom.
= √∑� ���= � − � frame ke-i dan �̅ adalah posisi rata-rata residu selama frame T.9
4 menunjukkan interaksi atom-atom yang berikatan kovalen (1 sampai 3 ikatan kovalen). Suku kedua menunjukkan sudut ikatan. Suku elektrostatik dalam bentuk potensial Coulomb ditunjukan ⁄ . Suku ketiga menunjukkan sudut putar dihedral. Dua suku terakhir menunjukkan interaksi non-ikatan (lebih dari 3 ikatan kovalen).10
Interaksi Van der Waals direpresentasikan dalam persamaan potensial Lennard Jones.
suku pertama berfungsi untuk menghitung peningkatan tolakan karena awan elektron dari atom yang tumpang tindih. Suku kedua menunjukkan daya tarik electron lemah diantara semua atom. adalah jarak antar atom.10
Solvent Accessible Surface Area (SASA)
SASA digunakan untuk menghitung luas area permukaan protein yang dapat diakses atau yang terpapar oleh molekul pelarut.11
Jembatan Garam
Jembatan garam adalah interaksi elektrostatik antara kelompok residu polar didalam protein. Kekuatan jembatan garam sama dengan ikatan hidrogen tetapi
bekerja pada jarak yang lebih jauh.12
Ikatan Hidrogen
Ikatan hidrogen menunjukkan tipe interaksi tertentu yang terjadi antara kelompok D, pendonor proton, yang bersifat sangat polar (FH, OH, NH, SH, dll) dengan atom A, akseptor proton, yang bersifat sangat elektronegatif (F,O,N,dll). Bentuk ikatan hidrogen ditunjukkan dengan D-H…A. Jarak antara D dengan A bergantung pada atom-atom yang terlibat12. Ikatan hidrogen termasuk interaksi non kovalen.
Simulasi Dinamika Molekul
5
Simulasi dilakukan menggunakan NVT ensemble (Jumlah, Volume dan
Temperatur tetap). Distribusi Boltzman untuk ensembel NVT kanonik dalam NAMD dihitung menggunakan persamaan Langevin
M v= F r -γv+ √2γkBT
M R(t) (7)
dimana adalah massa, � kecepatan, � gaya, jarak, � koefisien gesek. � konstanta boltzman, temperatur dan � adalah proses acak Gaussian.14
METODE
Metode yang digunakan dalam simulasi ini adalah PBC dan PME. Metode PBC (Periodic Boundary Condition) digunakan agar sistem mendekati kondisi
sebenarnya. Metode PME (Particle Mesh Ewald) menggunakan ekspansi Taylor
untuk menghitung interaksi elektrostatik sistem. Parameter untuk kedua metode tersebut dimasukkan dalam file konfigurasi. File topologi yang digunakan adalah par_all27_prot_na_lipid.inp
Alat
Penelitian ini menggunakan peralatan berupa alat tulis, laptop dengan spesifikasi 2,7GHz, 8 GB RAM dan sistem operasi Linux Ubuntu 12.04. Laptop tersebut dilengkapi dengan beberapa program pendukung. VMD versi 1.9.1 digunakan pada tahap persiapan dan analisa, untuk menampilkan, menganalisis dan menganimasikan molekul15. NAMD versi 2.9 digunakan sebagai simulator dinamika molekul. NAMD bekerja dengan fungsi potensial, parameter, dan format file CHARMM. Data yang dihasilkan oleh NAMD diolah kembali menggunakan VBA Ms.Excel 2007 dan CatDCD versi 4.0. Plot data menggunakan program Gnuplot versi 4.4. Konversi format file gambar menggunakan Gimp 2.6.
Preparasi molekul
Tahap ini dilakukan dengan bantuan program VMD. Data 1GB1.pdb menjadi masukan awal simulasi dan dapat diunduh dari Protein Data Bank
(www.pdb.org). Data tersebut memuat koordinat semua atom residu, faktor struktur kristalografi dan data eksperimen NMR sehingga dapat ditentukan struktur tiga dimensi protein 1GB1. Ada empat langkah preparasi molekul dengan program VMD, yang pertama adalah mengatur data 1GB1.pdb. Jumlah frame
6
Kedua, membuat data 1GB1-psf.pdb dan 1GB1-psf.psf. Data 1GB1.pdb tidak memuat informasi sepesifik yang diperlukan untuk menerapkan medan gaya tertentu ke dalam sistem molekul, sehingga dibutuhkan data 1GB1-psf yang dihasilkan melalui automatic psf builder. Keluaran dari program tersebut adalah
1GB1-psf.pdb dan 1GB1-psf.psf. Ketiga, pelarutan molekul, Protein 1GB1 ditempatkan dalam kotak air berdimensi (80x80x80)�. Air berperan sebagai media pelarut molekul 1GB1. Jumlah air selama simulasi adalah tetap. Keluaran dari tahap ini adalah 1GB1-solv.pdb dan 1GB1-solv.psf.
Keempat, penetralan sistem, molekul yang telah dilarutkan belum netral sepenuhnya, karena masih terdapat ion-ion dari residu-residu polar. Residu polar bermuatan positif dari protein 1GB1 adalah Lysine. Sedangkan residu polar yang
bermuatan negatif adalah Asparagine dan Glutamic Acid. NaCl 0.5 M berperan
sebagai penetral sistem. Larutan akan secara otomatis terus ditambahkan ke sistem hingga sistem mencapai keadaan netral. Keluaran dari tahap ini adalah 1GB1-ion.pdb dan 1GB1-ion.psf.
Simulasi dinamika molekul
Tahap ini menggunakan program NAMD. Masukan awal program berupa file konfigurasi. File konfigurasi berisi parameter apa saja yang digunakan dan berapa nilai parameter tersebut. Selain itu data konfigurasi juga mengontrol bagaimana sistem akan disimulasikan. Protokol untuk kontol yang dipilih adalah Langevin dengan time step sebesar 2 fs. Ada tiga langkah simulasi yang dilakukan.
Pertama adalah minimisasi. Minimasasi diperlukan untuk meminimalkan energi yang dimiliki molekul, sehingga pada kondisi awal molekul tidak terlalu banyak bergerak. Masukan awal minimisasi adalah tiga fixed atoms file masing-masing
berupa data dengan faktor beta diberikan sama dengan 1 untuk protein, atom-atom
backbone dan atom-atom �. Minimisasi dilakukan selama 100 ps.
Kedua adalah Pemanasan dan ekuilibrasi. Suhu awal simulasi adalah 0K sedangkan suhu akhir divariasikan 325K, 350K, 375K, 400K, 450K dan 500K. Suhu dinaikkan setiap 25K dengan iterasi 10.000 kali. Kemudian, sistem diekuilibrasi dengan protokol Langevin agar suhu tetap berada di suhu akhir selama 20 ps. Selama proses ini sistem masih merasakan kekangan berupa konstanta pegas. Ekuilibrasi akhir dilakukan dengan menghilangkan constraint
selama 40 ps. File keluaran dari proses ini adalah 1GB1-heat-equil.dcd.
Ketiga adalah Production run. Pada tahap ini sistem sudah tidak dikontrol
lagi suhunya sehingga molekul bebas bergerak. Tahap ini merupakan tahap inti dari simulasi dinamika molekul protein 1GB1. Production run dilakukan selama
10 ns dimana prosesnya dibagi menjadi 10 sub-proses. Dinamika yang terjadi selama Production run seperti perubahan temperatur, tekanan, energi elektrostatik
dan lainnya direkam dalam log file. Keluaran dari proses ini adalah File
1GB1-md.dcd.
Pengolahan data
7
dengan cara menggabungkan data 1GB1-index.ind kedalam kedua file tadi sehingga didapatkan 1GB1-heat-equil-nobox.dcd dan 1GB1-md-nobox.dcd. Melalui program VMD maka akan didapatkan beberapa data analisis, pertama adalah jumlah ikatan hidrogen antara protein/protein, backbone/backbone dan
protein/air. Khusus untuk perhitungan ikatan hidrogen protein/air, penelitian ini menggunakan data 1GB1-heat-equil.dcd dan 1GB1-md.dcd. Kedua adalah dinamika energi konformasi dan non-ikatan. Ketiga adalah RMSD, yang menunjukkan pergerakan lintasan atom selama simulasi. Keempat, energi elektrostatik tiap jembatan garam . Perhitungan jari-jari girasi, Root Mean Square Fluctuation (RMSF), jumlah residu yang membentuk struktur sekunder, dan Solvent Accessible Surface Area (SASA) juga dilakukan melalui VMD.
HASIL DAN PEMBAHASAN
Validasi
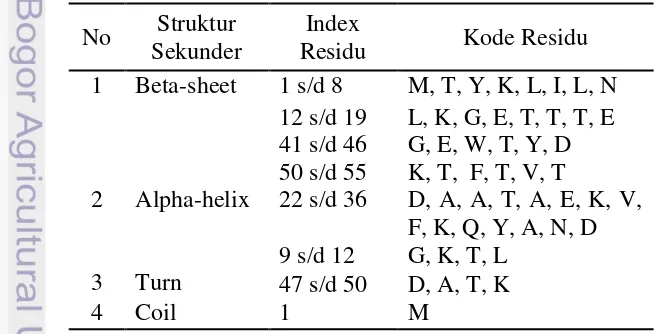
Hasil simulasi perlu divalidasi untuk melihat kesesuaiannya. Parameter yang dapat dibandingkan adalah faktor beta. Faktor beta menunjukkan pergeseran kerapatan elektron pada suatu atom dalam kristal. Pemilihan temperatur 300K sebagai pembanding dikarenakan protein tidak mengalami perubahan konformasi yang banyak berbeda dari struktur awal. Data faktor beta simulasi pada temperatur 300K menunjukkan bahwa hasil simulasi memiliki pola yang sama dengan eksperimen (didapat dari protein data bank) walaupun nilainya berbeda. Oleh
karena itu simulasi ini dapat dilanjutkan pada temperatur berikutnya (325K sampai 500K).
.
Energi konformasi
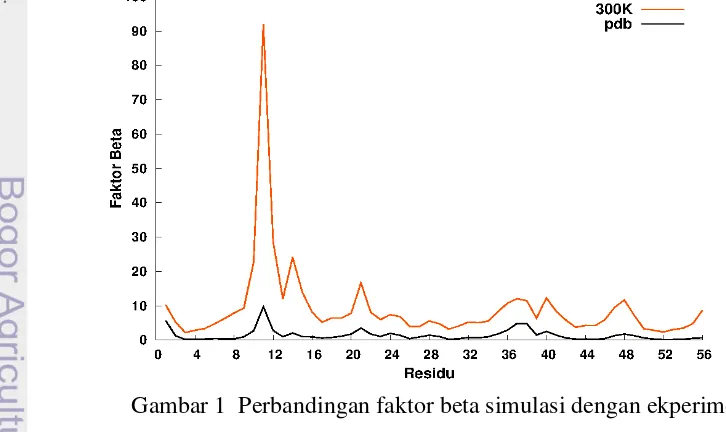
Energi konformasi mencakup energi ikatan, sudut, dihedral dan improper.
8
ke bentuk konformasi yang lain. Semakin tinggi temperatur maka energi konformasi juga akan semakin besar (Gambar 2(a)). Perbedaan energi sebesar 50Kcal/mol dihasilkan dari perbedaan temperatur sebesar 25K. Terlihat pada simulasi temperatur 500K (Gambar 2(b)) energi konformasi mengalami lonjakan pada waktu 900 ps karena pada saat tersebut protein 1GB1 mengalami unfolding
(dijelaskan pada subbab struktur sekunder). Unfolding merupakan keadaan yang
tidak stabil dari sistem, sehingga akan berada pada energi yang tinggi. Energi konformasi kemudian stabil kembali tetapi dengan nilai lebih tinggi dari kondisi sebelum unfolding.
(a) (b)
Energi non-ikatan
Energi non-ikatan sudah mencakup energi elektrostatik dan van der Waals. Energi elektrostatik dihasilkan dari pasangan residu polar, baik yang bermuatan maupun yang netral. Sedangkan energi van der Waals adalah interaksi elektrostatik antara residu polar netral yang teriduksi dipol oleh residu polar bermuatan. Jangkauan energi van der waals sangat rendah sehingga dapat mewakili kekompakan protein. Tidak ada perubahan yang signifikan terhadap energi non-ikatan dengan bertambahnya temperatur (berada di rentang -1000 sampai dengan -500Kcal/mol) (Gambar 3(a)). Hal ini sudah benar karena tidak ada perubahan pH yang dilakukan selama simulasi yang dapat mengubah konsentrasi ion dalam sistem ditambah lagi jumlah residu polar dan bermuatan yang tetap selama simulasi. Energi non ikatan meningkat saat 900ps (Gambar 3(b)) karena sistem menjadi tidak stabil.
9
(a) (b)
Ikatan Hidrogen
Gambar-gambar yang akan ditampilkan pada subbab ini adalah gambar yang telah diperhalus menggunakan moving average untuk setiap 50 frame agar
memudahkan pembacaan data. Ikatan hidrogen yang dihitung adalah ikatan hidrogen dimana jarak donor-akseptor proton masih berada di rentang 3.0Å. Pada Gambar 4(a) dan 4(b) ikatan hidrogen antar backbone cenderung konstan selama
simulasi. Kenaikan suhu menyebakan energi sistem bertambah sehingga vibrasi antar ikatan semakin kuat dan akan menyebabkan putusnya ikatan hidrogen yang termasuk ikatan lemah. Backbone adalah rantai utama peptida tanpa memasukkan
rantai samping atau residunya. Pada proses unfolding terlihat penurunan jumlah
ikatan hidrogen (Gambar 4(b)). Ini dikarenakan jarak beberapa backbone yang
menjauh sehingga memutuskan ikatan hidrogen.
Pada Gambar 4(c) dan 4(d) ikatan hidrogen antar protein juga menunjukkan pola yang sama dengan Gambar 4(a) dan 4(b) hanya jumlahnya lebih banyak. Hal ini sudah benar karena telah memasukkan ikatan hidrogen antar residu dan ikatan hidrogen residu/backbone. Karena pada proses unfolding jumlah
ikatan hidrogen antar protein tetap (Gambar 4(d)), dapat disimpulkan bahwa terjadi peningkatan jumlah ikatan hidrogen antar residu. Jarak residu terjadap
backbone akan tetap walaupun terjadi unfolding, sehingga tidak akan ada
perbedaan jumlah ikatan hidrogen residu/backbone saat unfolding.
Pada Gambar 4(e) dan 4(f) terlihat jelas pengaruh temperatur terhadap jumlah ikatan hidrogen protein/air. Semakin tinggi temperatur maka jumlah ikatan hidrogen protein/air akan semakin berkurang. Terjadi penurunan ikatan hidrogen protein/air yang tajam saat terjadi unfolding. Ini dikarenakan ukuran protein yang
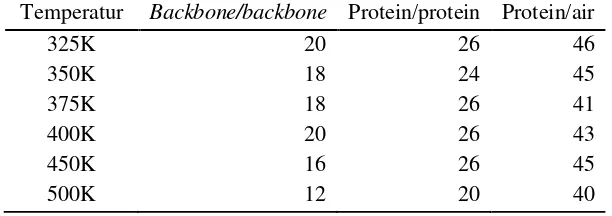
membesar secara drastis (dijelaskan pada subbab jari-jari girasi) dan tiba-tiba sehingga mengacaukan sistem wadah air yang telah dibuat. Selama simulasi terjadi fluktuasi jumlah ikatan hidrogen, dikarenakan sistem yang bervibrasi akibat pemberian panas. Peningkatan jumlah ikatan hidrogen selama simulasi dihitung dari nilai maksimal dikurangi nilai minimal yang pernah ada saat simulasi tersebut (Tabel 2).
10
(a) (b)
(c) (d)
(e) (f)
11
Tabel 2 Peningkatan jumlah ikatan hidrogen selama simulasi dengan variasi temperatur
Temperatur Backbone/backbone Protein/protein Protein/air
325K 20 26 46
Simulasi 325K sampai dengan 450K memiliki jumlah struktur sekunder yang tetap. Sedangkan pada simulasi 500K alpha-helix akan berubah menjadi turn
dan coil. Selama simulasi residu penyusun masing-masing struktur sekunder akan
berubah (Lampiran 1). Fluktuatif penyusun struktur sekunder menunjukkan bahwa struktur-struktur tersebut tersebut berusaha mempertahankan keberadaannya. Semakin tinggi temperatur simulasi maka akan semakin sering terjadi pertukaran penyusun struktur. Residu penyusun asli dari 1GB1 dapat dilihat pada Tabel 1. Saat terjadi proses unfolding struktur sekunder yang pertama kali rusak adalah alpha-helix. Hal ini sudah sesuai dengan penelitian sebelumnya.
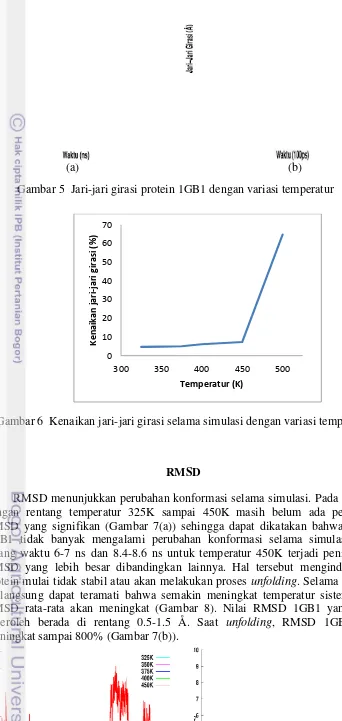
Jari-jari girasi
Jari-jari girasi 1GB1 selama simulasi temperatur 325K sampai 450K belum ada perubahan yang signifikan (Gambar 5(a)) sehingga selama rentang temperatur tersebut protein 1GB1 masih kompak. Jari-jari girasi 1GB1 berada di rentang 10.5-11.25 Å. Jari-jari girasi rata-rata selama simulasi untuk temperatur 325K, 350K, 375K, 400K dan 450K masing-masing adalah 10.73219 Å, 10.77799 Å, 10.70131 Å, 10.78662 Å dan 10.86688 Å. Pada proses unfolding di temperatur
500K terjadi lonjakan jari-jari girasi dari 11 Å sampai 18 Å (Gambar 5(b)). Terlihat pada Gambar 6, semakin meningkat temperatur maka fluktuatif jari-jari girasi juga akan semakin besar. Fluktuasi dapat diartikan sebagai persen kenaikan jari-jari girasi dari nilai minimal ke nilai maksimal yang ada dalam simulasi.
12
(a) (b)
Gambar 6 Kenaikan jari-jari girasi selama simulasi dengan variasi temperatur
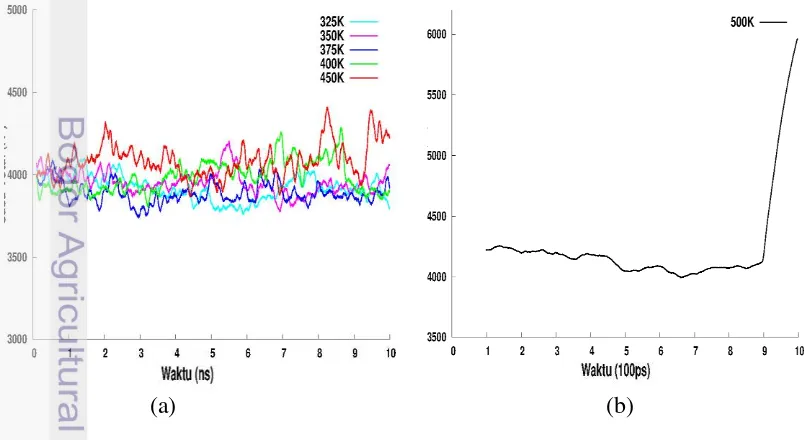
RMSD
RMSD menunjukkan perubahan konformasi selama simulasi. Pada simulasi dengan rentang temperatur 325K sampai 450K masih belum ada perubahan RMSD yang signifikan (Gambar 7(a)) sehingga dapat dikatakan bahwa protein 1GB1 tidak banyak mengalami perubahan konformasi selama simulasi. Pada selang waktu 6-7 ns dan 8.4-8.6 ns untuk temperatur 450K terjadi peningkatan RMSD yang lebih besar dibandingkan lainnya. Hal tersebut mengindikasikan protein mulai tidak stabil atau akan melakukan proses unfolding. Selama simulasi
berlangsung dapat teramati bahwa semakin meningkat temperatur sistem maka RMSD rata-rata akan meningkat (Gambar 8). Nilai RMSD 1GB1 yang dapat
13
(a) (b)
Gambar 8 Rata-rata RMSD 1GB1 selama simulasi dengan variasi temperatur
RMSF
RMSF menunjukkan fleksibilitas residu selama simulasi. Skrip analisis RMSF hanya menghitung untuk atom C-� dari protein 1GB1. Analisis pergerakan atom C- � mewakili pergerakan rata-rata dari suatu residu protein selama simulasi. Ditunjukkan pada gambar 9(a) beberapa residu yang memiliki nilai RMSF tertinggi pada setiap simulasi. Residu tersebut adalah M1, T11, G14, V21, D40, dan E56. Simulasi 500K menghasilkan RMSF yang berbeda dengan yang lainnya. Banyak residu yang tadinya tidak fleksibel menjadi fleksibel. Hal ini dikarenakan terjadinya proses unfolding yang merubah fleksibilitas residu.
14
(a) (b)
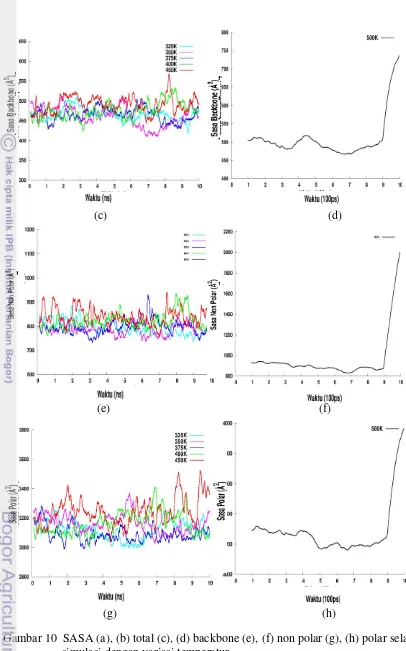
SASA
Sasa juga merupakan salah satu parameter yang menunjukkan kekompakan protein. Grafik yang ditampilkan merupakan data yang sudah diperhalus dengan
moving average untuk setiap 50 frame. Semakin meningkatnya suhu maka
perlahan-lahan nilai total SASA juga akan semakin meningkat (Gambar 10(a) dan (b)). Secara rata-rata nilai total SASA 1GB1 yang diperoleh dari masing-masing simulasi berada pada rentang 3590-4597� . Saat unfolding nilai SASA meningkat
drastis ke angka 6851� . Hal ini dikarenakan terbukanya gugus hidrophobik protein yang ditunjukkan persentase kenaikan SASA non polar tertinggi dibandingkan SASA lainnya (Gambar 11).
(a) (b)
15
(c) (d)
(e) (f)
(g) (h)
16
Gambar 11 Kenaikan persentase SASA akibat variasi temperatur
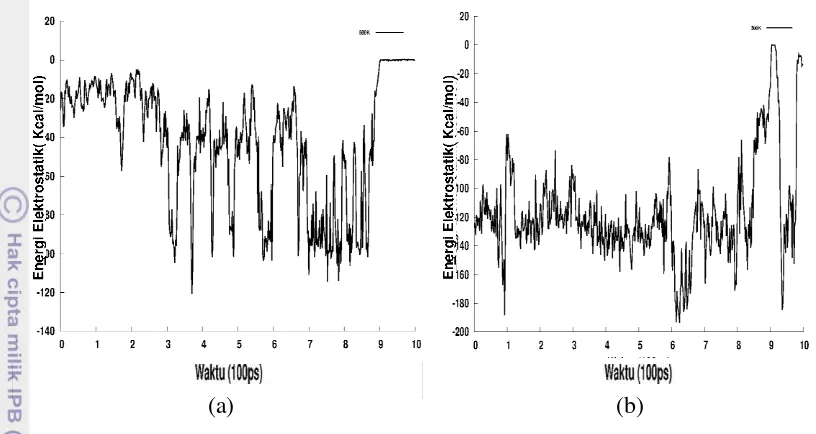
Jembatan Garam
Jembatan garam merupakan interaksi elektrostatik antara − pada residu
polar negatif (Glutamic acid atau Aspartic acid) dengan �+ pada residu polar
netral (Lysine). Pergerakan protein yang fluktuatif menyebabkan interaksi
elektrostatik juga hilang timbul. Secara umum jembatan garam yang ada selama simulasi dirangkum dalam Lampiran 3. Pasangan jembatan garam E15/K4, E27/K41 dan E56/K10 selalu ada di setiap simulasi dengan temperatur berapapun. Seperti telah dijelaskan sebelumnya pada subbab struktur sekunder bahwa struktur
alpha-helix yang pertama kali rusak. Hal tersebut didukung dengan energi
elektrostatik pasangan jembatan garam E56-K10, yang menghubungan antar beta-hairpin (Gambar 12(a)), semakin lama semakin besar amplitudo fluktuatifnya
yang menandakan gejala putusnya interaksi elektrostatik dan akhirnya menuju nilai nol yang menandakan sudah tidak ada interaksi lagi (Gambar 13(a)). Sedangkan pasangan jembatan garam E27-K31 (Gambar 12(b)) terdapat pada struktur alpha-helix. Proses unfolding menyebabkan pasangan residu tersebut
semakin jauh sehingga interaksi elektrostatiknya semakin menuju nol (Gambar 13(b)).
(a) (b)
17
(a) (b)
SIMPULAN DAN SARAN
Simpulan
Proses unfolding protein 1GB1 terjadi pada waktu 900 ps dengan temperatur
500K dimulai dengan rusaknya struktur alpha-helix. Struktur tersebut merupakan
tempat yang didominasi oleh residu hidrophobik. Sehingga nilai SASA residu hidrophobik meningkat 200% dari semula. Keadaan unfolded merupakan keadaan
yang tidak stabil dari protein sehingga memiliki energi yang lebih tinggi daripada keadaan folded. Membukanya protein 1GB1 secara keseluruhan ditunjukkan
dengan meningkatnya jari-jari girasi hingga 60%. Ikatan hidrogen dan jembatan garam berfungsi menstabilkan protein. Tingginya energi yang dimiliki protein akan menyebabkan putusnya ikatan hidrogen yang termasuk ikatan lemah dan melemahnya interaksi elektrostatik antar residu penyusun jembatan garam. RMSD dan RMSF masing-masing menunjukkan simpangan rata-rata setiap konformasi protein dan residu selama simulasi. Peningkatan suhu menyebabkan konformasi protein semakin berbeda dari aslinya (ditunjukkan dengan nilai RMSD yang meningkat). Proses unfolded juga menyebabkan beberapa residu menjadi lebih
fleksibel dari sebelumnya.
Saran
Peningkatan jari-jari girasi yang drastis dari protein 1GB1 setelah mengalami unfolded menyebabkan simulasi tidak dilanjutkan lebih lama karena
18
keterbatasan alat. Oleh karena itu diperlukan simulasi lanjutan pada temperatur 500K untuk beberapa nano sekon berikutnya. Selain itu simulasi pada temperatur 325K, 350K, 375K, 400K dan 450K dapat dilanjutkan untuk waktu yang lebih lama sehingga didapatkan informasi yang lebih dari dinamika protein.
DAFTAR PUSTAKA
1. Murray, R. K., Graner, D. K., and Rodwell, V. W. Harper’s Ilustrated Biochemistry 28th edition. 2009.
2. Ghifari, Abi Sofyan. Deteksi Struktur Protein Hingga Tingkat Atomik
(terhubung berkala) http://www.chem-is-try.org/artikel_kimia/deteksi-struktur-protein-hingga-tingkat-atomik/ (diakses pada tanggal 22 Nopember 2012).
3. Rico, Manuel, Protein structure, dynamics and function by NMR, Instituto de Química Física “Rocasolano” (CSIC), Spain.
4. Li, Ju. Handbook of Materials Modeling. Department of Materials Science and Engineering, Ohio State University, USA. 2005.
5. Gronenborn, A. M., Clore, G. M. Structural Studies of Immunoglobulin-Binding
Domains of Streptococcal Protein G, Immunomethods, Academic Press Inc.,
Maryland. 1993.
6.Pande, V. S., Rokhsar, D. S. Molecular dynamics simulations of unfolding and refolding of beta-hairpin fragment of protein G. Proc. Natl. Acad. Sci.
USA. 1999. Vol. 96, August 1999, pp. 9062-9067.
7. Lobanov, M. Yu., Bogatyreva, N. S., Galzitskaya, O. V. Radius Gyration as an Indicator of Protein Structure Compactness. Molecular Biology. 2008.
Vol. 8 No 4, March 2008 pp. 623-628
8. Aksimentiev, Alek, et. al. Using VMD. www.ks.uiuc.edu. 2012.
9. Schuffler, Peter. Analyse a molecular dynamics (MD) Trajectory
10. Levitt, M., Hirshberg, M., Sharon, R., Daggett, V. Potential energy function and parameters for simulations of the molecular dynamics of proteins and
nucleid acids in solution, Computer physics communication. 1995. Vol. 91,
November 1994, pp. 215-231
11. Randy, Ahmad. [Tesis]. Desain Peningkatan Termostabilitas Lipase B Candida
antarctica dengan Rekayasa Penambahan Ikatan Disulfida Pada Enzim.
Program Studi Ilmu Kimia. Fakultas MIPA. Universitas Indonesia. 2011. 12. Michele, D. Molecular Biophysics: Structures in Motion, Oxford University
Press, London. 2004.
13. Ruhle, Victor. Berendsen and Nose-Hoover thermostats. 2007.
14. Phillips, J. C., Braun, S., Wang, W., Gumbart, J., Tajkhorshid, E., Villa, E., Chipot, C., Skeel, R. D., Kale, L., Schulten, K., Scalable Molecular Dynamics with NAMD. Journal of Computational Chemistry, Vol. 26 No 16, May 2005, pp. 1781-1802.
15. Humprey, W., Dalke, A., Schulten, K., “VMD - Visual Molecular Dynamics” J.
19
Lampiran 1 Perubahan struktur sekunder protein 1GB1 selama simulasi pada (a) 325 K, (b) 350 K, (c) 375 K, (d) 400 K dan (e) 500 K
Keterangan: beta-sheet coil turn
alpha-helix
(a) (b)
(c) (d)
20
Lampiran 2 Karakteristik residu yang terdapat pada 1GB1
No Jenis Residu Kode
Residu
Nama Residu
1 Polar positif L Lysine
2 Polar negatif D Aspartic Acid
E Glutamic Acid
3 Polar netral T Threonine
N Asparagine
Q Glutamine
4 Non polar G Glycine
5 Hidrophobik A Alanine
I Isoleucine
L Leucine
M Methionine
F Phenylalanine
W Tryptophan
Y Tyrosine
V Valine
6 Aromatik F Phenylalanine
Y Tyrosine
Lampiran 3 Pasangan residu penyusun jembatan garam
Temperatur Pasangan Temperatur Pasangan
325K E15-K4 400K E15-K4
E27-K31 E27-K31
D40-K31 D40-K10
D47-K50 D40-K31
E56-K10 D47-K50
E56-K10
350K E15-K4 450K E15-K4
E27-K28 E15-K13
E27-K31 E27-K28
D40-K31 E27-K31
E56-K10 D40-K10
E56-K13 D40-K31
D47-K50 E56-K10
375K E15-K4 500K E15-K4
E27-K31 E15-K13
D40-K10 E27-K28
D40-K31 E27-K31
D47-K50 D47-K50
Lampiran 4 Konformasi akhir setiap simulasi (a) 325 K, (b) 350 K, (c) 375 K, (d) 400 K dan (e) 500 K
(a) (b)
(c) (d)