Continuous growth of autocatalytic sets
Shigenobu Mitsuzawa
a,b,*, Sei-ichiro Watanabe
aaDepartment of Earth and Planetary Sciences,Graduate School of Science,Nagoya Uni6ersity,Furo-cho,Chigusa,Nagoya, Aichi464-8602, Japan
bTheoretical/Computational Physics Group,National Laboratory for High Energy Physics(KEK),1-1Oho,Tsukuba, Ibaraki305-0801,Japan
Received 30 August 2000; received in revised form 4 December 2000; accepted 8 December 2000
Abstract
We investigated a topology of the reaction network for polymerization of amino acids as the emergence and the continuous growth of autocatalytic sets. Reaction networks including transpeptidation reaction can yield newer and longer peptides with time having higher concentrations. Conditions necessary for the occurrence of transpeptidation can be met in actual amino acid/peptide systems. © 2001 Elsevier Science Ireland Ltd. All rights reserved.
Keywords:Origin of life; Polymerization of amino acids; Transpeptidation
www.elsevier.com/locate/biosystems
1. Introduction
One of the fundamental questions regarding the origin of life is about which came first, either proteins or nucleic acids. The view that nucleic acids, probably RNA, were the primordial molecules of life (Eigen et al., 1981) has been favored by many researchers, but has not been substantiated as yet (Miller, 1992). On the other hand, some researchers have suggested the view that metabolisms of peptides could have origi-nated prior to or independently of the replication of nucleic acids (Dyson, 1985; Kauffman, 1993). This view is based upon the vantage point that
peptides could be synthesized more easily than nucleic acids in abiotic conditions. Ito et al. (1990) suggested that polypeptides carrying molecular weights of 1000 – 4000 Da had secondary struc-tures and catalytic functions for many metabolic reactions. Seffens (1995) investigated the possibil-ity that peptide systems might have exhibited metabolic and replicative activities. Recently, re-action networks of self- and cross-replicating pep-tides consisting of about 30 residues have been constructed in the laboratory by Lee et al. (1996) and other researchers(Lee et al., 1997; Yao et al., 1998). These observations strongly support the possibility of peptide systems of abiotic origin.
In order to have functional peptide systems, a set of long peptide chains must be prepared abiot-ically. In this study, we investigate how longer peptide chains could be synthesized from amino * Corresponding author. Tel.:+81-298-796102; fax:+
81-298-645755.
E-mail address: [email protected] (S. Mit-suzawa).
acids that may be long enough to carry primitive metabolic capabilities.
Kauffman (1986) proposed an autocatalytic set of polymers. Simulation studies of autocatalytic sets (Bagley and Farmer, 1991), however, revealed that it is not possible to synthesize longer poly-mers with high concentrations unless the equi-librium constant of condensation is large enough. In the present study, we performed numerical experiments by varying the topology of reaction networks. Our major finding is that even if the equilibrium constant of condensation is small, certain reaction networks can synthesize many kinds of longer polymers with higher concentrations.
2. Numerical model
2.1. Basic equations
We start with the basic equations of polymer-ization reaction, based on Bagley and Farmer (1991), though with some modifications and sim-plifications as shown in Appendix A.
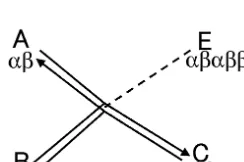
Each polymerization reaction is reversible (Fig. 1). Two polymers condense into a single longer polymer giving off a water molecule, and at the same time, the longer polymer cleaves itself into two shorter polymers. In this study, we denote the two reactants for condensation as ‘A’ and ‘B’, and the product as ‘C’. Note thatA+B"B+A in the respect of the sequence synthesized. The reaction is catalyzed by another polymer ‘E’. The rate equations forA,B,CandEof the catalyzed
where k is the equilibrium constant of the reac-tion, xN (Nis A, B, Cor E) is the concentration of the respective polymers, and n is the catalytic efficiency. For simplicity, we assume that all cata-lyzed reactions have the same catalytic efficiency n. (1+nxE) is the enhancement rate with a cata-lyst. When the net flow, B
A FC
E, is positive, the
reaction is a net condensation reaction. When it is negative, the reaction is a net cleavage reaction. When the net flow vanishes, the reaction is in equilibrium. In the absence of catalytic reaction, the rate equations for A, Band C are
For simplicity, we assume that all reactions, whether catalyzed or not, have the same equi-librium constant k.
With the use of the net flows of reaction, the kinetic equation of each polymer species i in the reaction system is expressed as
dxi
The first three terms on the right-hand side are the sums over the net flows of reactions in which polymer i denotes A, B, and C. The fourth term represents the flows of the polymers leaving the system into the outside. The term is proportional to the concentrations of polymers, in which we callkdthe dissipation rate constant. In this study, we set the same dissipation rate constant for all polymers. However, if we suppose that polymer-ization takes place within enclosures (Oparin, 1957), the longer polymers should have the smaller values of dissipation rate constant because it is difficult for longer polymers entrapped by enclosures to escape into the outside. Thus the Fig. 1. Graph of a pair of condensation and hydrolysis
assumption of the same dissipation rate constant for all polymers makes our model least vulnerable to polymer elongation of artifact origin. If i is a monomeric species, it is supplied from the outside. The fifth term represent the supply of the monomeric species, where ris the supply rate.
In the following numerical experiments, we cal-culate the concentration distributions of reactants at steady state. At steady state, the total concen-tration of monomers in the system,
m0=% i
Lixi
whereLiis the length of each polymer speciesi, is derived from Eq. (6) as
m0= Sr kd
(7)
whereSis the number of kinds of monomers, i.e.
iLi (i runs over monomeric species). We shall normalize the concentration by the quantity m0. For expressing the actual concentration, we use a hatted symbol of the parameter such as mˆ0. We express the magnitude of the flow between the system and the outside with use of the dissipation rate constant.
Now we shall estimate the actual value of the equilibrium constant in a real peptide system. For example, we consider aminoacyl-adenylate, e.g. Gly – AMP, for the monomer (Paecht-Horowitz et al., 1970). Cleavage of Gly – AMP into Gly and AMP gives off free energy of about 7 kcal/mol (Voet and Voet, 1995) and condensation of two Gly into Gly – Gly needs 3.6 kcal/mol (Borsook, 1953). Then the equilibrium constant of the fol-lowing reaction, Gly – AMP+Gly – AMPGly –
Gly – AMP+AMP, is
k=mˆ0exp(−G 0
/RT)=0.25, assuming mˆ0= 0.001 M (Borsook, 1953), G0
=(−7+3.6) kcal/
mol, and T=310 K.
2.2. Algorithms for setting up catalyzed reactions
In order to simulate behaviors of the reaction networks, we elaborate a list of the catalyzed reactions to be expected.
Procedure of the simulation is as follows: The initial condition is such that only monomers are present. Then,
Fig. 2. Graph of a transpeptidation reaction. 1. Pick up those polymers whose concentrations
newly exceed a given threshold T(=1/V) cor-responding to the presence of a single molecule in the system volume V.
2. Enumerate all possible catalyzed reactions in which selected polymers can participate. Then determine whether or not each reaction is physically realizable as referring to the list of the catalyzed reactions.
3. Solve Eq. (6) at steady state conditions. If some polymers newly exceed the threshold, go back to Step 1, or else stop the whole operation.
3. Numerical simulations
3.1. Topology of reaction network
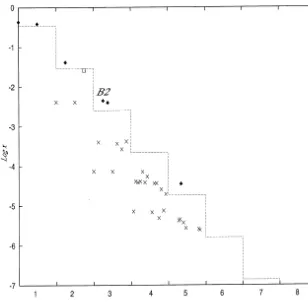
Fig. 3. Distribution of the concentrations of polymers in the reaction network including a transpeptidation reaction. Filled diamonds are the elites. Empty squares are the products of transpeptidation. Crosses are the polymers which are synthesized by uncatalyzed reactions. The polymers are sorted with their lengths in the horizontal axis.
3.2. Transpeptidation
Here we show the result of numerical simula-tion (Fig. 3) for the reacsimula-tion network shown in Fig. 2 with parameters S=2, k=0.25, n=
105,
T=10−5,
kd=10. We assume that all of the catalysts are substituted by one identical monomeric species, since our result will turn out to be indifferent to the variety of catalytic species. The dotted line in Fig. 3 denotes the concentra-tion profile of a polymer with L amino acids in equilibrium, i.e. kd=0, which is expressed as (Bagley and Farmer, 1991)
xL=
rS
Lk , r=1−
1+4k−1
2k (8)
We find that the concentrations of the elites which are denoted as filled diamonds show an
exponen-tial decrease of their concentrations with length and that the longer species due to the transpepti-dation, denoted as B2, has the higher concentration.
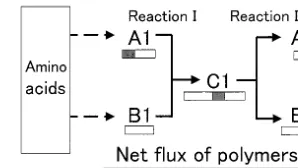
Next we consider the case where the product of the transpeptidation reaction is an elite (Fig. 4).
Fig. 5. Distribution of the concentrations of polymers in the reaction network of Fig. 4. This catalyzed reaction network is the one shown
in Fig. 2 supplemented by an additional catalyzed reaction, which exhibits a net condensation reac-tion yielding B2 as an elite synthesized from a monomer and a dimer elite. In this case, the concentration of B2 decreases down to the level of the elites (Fig. 5).
3.3. Continuous growth
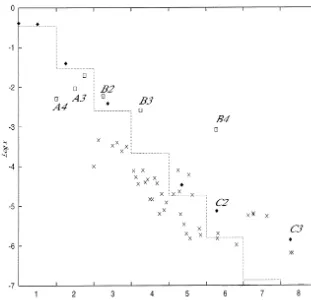
Next we consider the case that some transpepti-dation reactions are connected. That is to say, a product of a transpeptidation reaction becomes a substrate of another transpeptidation reaction. Fig. 6 demonstrates the reaction network. B2 is the longer product of the first transpeptidation reaction which is depicted in Fig. 2. An additional transpeptidation taking two of the B2 molecules as substrates yields A3 and B3. In the same
manner, the third one taking two of the B3 molecules as substrates yields A4 and B4. Theo-retically, the chain of transpeptidation reactions can extend itself to an infinite length. As shown in Fig. 7, the longer products of transpeptidations have higher concentrations than those of the elites.
The reaction network can continuously grow by repeating the cycle of incorporating a newer transpeptidation. Although the reaction networks
Fig. 7. Distribution of the concentrations of polymers in the reaction network of Fig. 6. we studied are limited, the possibility of having
transpeptidation reactions do not have to be lim-ited to ours.
4. Discussion
4.1. Necessary conditions for transpeptidation
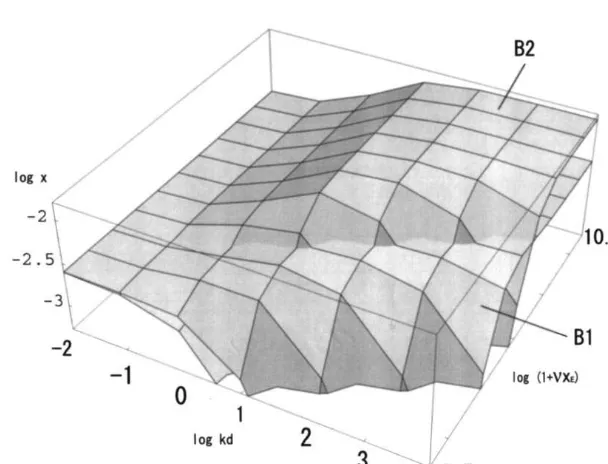
In Fig. 8, the concentrations of the elite, B1 are displayed and also the longer product of transpeptidation, B2 of Fig. 2 with parameterskd and (1+nxE). Here we investigate the condition of the system for satisfying that the concentration of B2 is higher than that of B1, which manifests the occurrence of transpeptidation to be effective. From Fig. 8, we read the condition
(1+nxE)\4 103.5 (9)
is necessary for transpeptidation to be effective. We now examine whether shorter oligopeptides can bear the high catalytic efficiencies satisfying Eq. (9), by adopting a Michaelis – Menten scheme. The Michaelis – Menten equation expresses the rate of a catalyzed hydrolysis reaction of a sub-strate as (Voet and Voet, 1995)
6cat=k
.catxˆETxˆS K.M+xˆS
(10)
where xˆS denotes the concentration of the sub-strate, and xˆET denotes the total concentration of the enzyme, that is, the sum of the concentrations of the free enzyme and of the enzyme – substrate complex. The values of rate constants for car-boxypeptidase A are k.cat=578 s−1 and k.
constant of hydrolysis of peptide without catalyst is k.non=3.0×10
−9 s−1 at 25°C (Radzicka and
Wolfenden, 1995). Shimizu (1996) reported that a dipeptide Ser – His, which is relevant to the cata-lytic site of chimotrypsin, has a peptidase activity. According to his experimental data, the strength of bonding interpreted as 1/K. M between Ser – His and a substrate is equal to that of chimotrypsin. The reaction rate interpreted ask.catof Ser – His is smaller by a factor of 0.001 than that of chimotrypsin. These facts and the above values of parameters give k.cat=0.578 s−
1
and K.M= 8.8×10−5
M for Ser – His. We assume that some other oligopeptides have weak peptidase activities as with Ser – His. The enhancement rate with a catalyst in our model, (1+nxE), is ex-pressed as
(1+nxE)=
6cat
k.nonxˆS
(11)
Inserting Eq. (10) into Eq. (11) with k.cat= 0.578 s−1, K.
M=8.8×10−5 M, k.non=3.0× 10−9 s−1, mˆ
0=0.001 M, and the assumption, xˆSK. M, gives
n=k.catmˆ0
K. Mk.non
(12)
=2.2×109
(13)
Based upon the results demonstrated in Fig. 3, it turns out that Eq. (9) is met if the catalysts are either any polymers with the length of under 5 or a part of polymers with the length of 5 for the case ofn=2.2×109. Thus we conclude that short peptides can be the catalysts whose catalytic effi-ciencies are high enough for transpeptidation to be effective.
In fact, the crucial parameter is the equilibrium constant which determines the gradient of the concentration against the length of a polymer (Eq. (8)). If the equilibrium constant is too small, the enhancement rate with a catalyst cannot be high enough for transpeptidation to become effec-tive because the concentration of a catalyst is too small. Recently, the high temperature and high pressure conditions like those expected near sub-marine hydrothermal systems have been consid-ered as a suitable site for polymerization of amino acids (Shock, 1992; Qian et al., 1993; Imai et al., 1999). Thus autocatalytic sets including
tidation might have emerged in such a condition that realizes a high value for the equilibrium constant with non-activated free amino acids.
4.2. Could the transpeptidation reaction system emerge?
Silver and James (1981a,b) reported that pepsin catalyzes the transpeptidation of oligopeptides. We expect that some short peptides which might have relevance to contemporary peptidase activi-ties could exhibit similar catalytic function to pepsin.
Our model assumes a constant flux of amino acids and peptides. It might be implausible to expect a constant flux of appropriate materials for a sustained period of time in a natural setting. One possible strategy for the reaction system to avoid attainment of equilibrium conditions could be the formation of an enclosure dispelling water molecules which persisted during some periods in equilibrium conditions (Bagley and Farmer, 1991). Another strategy is to put reaction net-works within submarine hydrothermal systems. The hydrothermal systems can be regarded as continuous-flow reactors guaranteeing the persis-tence of mass flux (Corliss, 1986).
In our simulations, we studied only two differ-ent kinds of monomers. If we consider more than two different kinds, much wider varieties of poly-mers and reactions would be expected, with a more enhanced likelihood of having transpeptidations.
Acknowledgements
We are grateful to Katsuro Ogawa and Shigeo Yoshida for valuable discussions. We also appre-ciate Tetsuyuki Yukawa for reading the manuscript and for useful comments.
Appendix A
In this work, we use a numerical model, based on Bagley and Farmer (1991) adding some mod-ifications and simplmod-ifications, which we summarize here.
A.1. Rate equation
Bagley and Farmer expressed the rate equations of a catalyzed reaction as
dxC
dt =(1+nxE)(kfxAxB−krHxC) (14)
where kf is the rate constant of the condensation reaction, kr is the rate constant of cleavage reac-tion, and H is the concentration of water. The equilibrium constant is
k= kf
krH
As with Bagley and Farmer, we assume for sim-plicity that all reactions have the same values ofkf and kr, respectively, and we normalize the time parameter by krH. Then Eq. (14) is rewritten as
dxC
dt =(1+nxE)(kxAxB−xC) (15)
A.2. Complexes in the catalyzed reactions
Bagley and Farmer’s model considers the com-plexes into which the catalyst and the reactants are bound together. For simplicity, our model did not consider this effect. We have confirmed that this assumption is appropriate in actual oligopep-tide syntheses.
References
Bagley, R.J., Farmer, J.D., 1991. Spontaneous emergence of a metabolism. In: Langton, C.G., Taylor, C., Farmer, J.D., Rasmussen, S. (Eds.), Artificial Life II. Addison-Wesley, Reading, MA, pp. 93 – 140.
Borsook, H., 1953. Peptide bond formation. Adv. Protein Chem. 8, 127 – 174.
Corliss, J.B., 1986. On the creation of living cells in submarine hot spring flow reactions: attractors and bifurcations in the natural hierarchy of dissipative systems. Origins Life Evol. Biosphere 16, 381 – 382.
Dyson, F.J., 1985. Origins of Life. Cambridge University Press, Cambridge.
Eigen, M., Gardiner, W., Schuster, P., Winckler-Oswatitch, R., 1981. The origin of genetic information. Sci. Am. 244, 88 – 118.
Ito, M., Handa, N., Yanagawa, H., 1990. Synthesis of polypeptides by microwave heating II. Function of polypeptides synthesized during repeated hydration – dehy-dration cycles. J. Mol. Evol. 31, 187 – 194.
Kauffman, S.A., 1986. Autocatalytic sets of proteins. J. Theor. Biol. 119, 1 – 24.
Kauffman, S.A., 1993. The Origin of Order. Oxford University Press, New York.
Lee, D.H., Granja, J.R., Martinez, J.A., Severin, K., Ghadiri, M.R., 1996. A self-replicating peptide. Nature 382, 525 – 528.
Lee, D.H., Severin, K., Yokobayashi, Y., Ghadiri, M.R., 1997. Emergence of symbiosis in peptide self-replication through a hypercyclic network. Nature 390, 591 – 594. Miller, S.L., 1992. The prebiotic synthesis of organic
com-pounds as a step toward the origin of life. In: Schopf, J.W. (Ed.), Major Events in the History of Life. Jones and Bartlett, Boston, pp. 1 – 28.
Oparin, A.I., 1957. The Origin of Life on the Earth. Academic Press, New York.
Paecht-Horowitz, M., Berger, J., Katchalsky, A., 1970. Prebi-otic synthesis of polypeptides by heterogeneous polycon-densation of amino-acid adenylates. Nature 228, 636 – 639. Qian, Y., Engel, M.H., Macko, S.A., Carpenter, S., Deming, J.W., 1993. Kinetics of peptide hydrolysis and amino acid
decomposition at high temperature. Geochim. Cosmochim. Acta 57, 3281 – 3293.
Radzicka, A., Wolfenden, R., 1995. A proficient enzyme. Science 267, 90 – 93.
Seffens, W., 1995. Model predictions of an origin of life hypothesis based on sea spray, 3rd Euro. A-Life, 452 – 456. Shimizu, M., 1996. The Essence and Origin of Life. Kyoritsu,
Tokyo.
Shock, E.V., 1992. Stability of peptides in high-temperature aqueous solutions. Geochim. Cosmochim. Acta 56, 3481 – 3491.
Silver, M.S., James, S.L.T., 1981a. Enzyme-catalyzed conden-sation reactions which initiate rapid peptic cleavage of substrates 1. How the structure of an activating peptide determines its efficiency. Biochemistry 20, 3177 – 3182. Silver, M.S., James, S.L.T., 1981b. Enzyme-catalyzed
conden-sation reactions which initiate rapid peptic cleavage of substrates 2. Proof of mechanism for three examples. Bio-chemistry 20, 3183 – 3189.
Voet, D., Voet, J.G., 1995. Biochemistry, 2nd edn. Wiley, New York.
Yao, S., Ghosh, I., Zutshi, R., Chmielewski, J., 1998. Selective amplification by auto- and cross-catalysis in a replicating peptide system. Nature 396, 447 – 450.