Frequency of the R3500Q mutation of the apolipoprotein B-100
gene in a sample screened clinically for familial
hypercholesterolemia in Hungary
A
´ kos Kalina
a,*, Albert Csa´sza´r
b, Andrew E. Czeizel
c, La´szlo´ Romics
d,
Ferenc Szabo´ki
a, Csaba Szalai
e, Istva´n Reiber
f, Attila Ne´meth
b, Susan Stephenson
g,
Roger R. Williams
gaCardiology Department,MA´ V Hospital,Ta´ncsics M.20,1014Budapest,Hungary bFirst Department of Medicine,Haynal Imre Medical Uni6ersity,Budapest,Hungary
cNational Institute of Public Health-WHO Collaborating Center for the Community Control of Hereditary Diseases,Budapest,Hungary dThird Department of Medicine,Semmelweis Medical Uni6ersity,Budapest,Hungary
eLaboratory of Heim Pa´l Children Hospital,Budapest,Hungary fFirst Department of Medicine,Szent Gyo˜rgy Hospital,Sze´kesfehe´r6a´r,Hungary
gUni6ersity of Utah Cardio6ascular Genetics,Salt Lake City,UT,USA
Abstract
Familial hypercholesterolemia (FH) and familial defective apolipoprotein B-100 (FDB) cause early onset of coronary heart diseases (CHD). According to the recommendations of the international MEDPED program, we tried to find FH cases. We analyzed 73 FH probands and their 304 first-degree relatives. A total of 39 probands were found from the 21 000 subjects screened (1:538) from family doctors’ registers recording all citizens, while the remaining 34 were derived from screened patients from lipid clinics. In our FH probands, four cases of FDB (R3500Q mutation) were diagnosed with allele-specific PCR, and the mutation was also detectable in five cases out of seven living family members. In the remaining 69 FH families, 156 people were diagnosed clinically with FH, and 31.8% of the males (against 13% of the not clinically diagnosed FH males,PB0.01), and 32.4% of the females (against 13.5% of the not clinically diagnosed FH females,PB0.01) suffered from early onset CHD. The plasma total cholesterol level of the FDB patients, especially in the younger patients, was very close to normal values. Therefore, the FDB patients seem to be under-represented in this type of survey. Because FDB is one of the independent causes of early onset CHD, the R3500Q mutation should be considered in families with a high frequency of cardiovascular diseases. © 2001 Elsevier Science Ireland Ltd. All rights reserved.
Keywords:Familial hypercholesterolemia; Familial defective apoB-100; Coronary heart diseases
www.elsevier.com/locate/atherosclerosis
1. Introduction
Familial hypercholesterolemia (FH) and familial de-fective apolipoprotein B-100 (FDB) are monogenic, autosomal, dominantly inherited diseases belonging to the type II/a primary hyperlipidemia group based on Fredrickson’s classification [1]. Both can cause early onset of cardio- and cerebrovascular diseases and early death [2]. In the European populations the percentage
of heterozygous FH patients is estimated at 0.2%, which means 20 000 FH patients in our country [3]. Similar values are known for the frequency of het-erozygous FDB [4]. In both diseases, occurrence of homozygous cases is very rare (1:1 000 000).
FH, due to the defective function of receptors, is caused by over 400 mutations. In the case of FDB, the receptor binding domain of apolipoprotein B-100, the main apoprotein of the LDL-molecule, is damaged, which may have genetic causes. The decrease of binding to the LDL-receptor also leads to accumulation of LDL-cholesterol in the plasma. The plasma cholesterol level in the heterozygous form of FH patients is twice * Corresponding author. Tel.: +36-1-3301030; fax: +
36-1-2695579.
E-mail address:[email protected] (A´ . Kalina).
as high as normal, while in FDB patients a smaller degree of elevation can be detected [5 – 7].
The objective of our study was to define, by an internationally accepted clinical method, the occurrence of FH in the Hungarian population, and to investigate by a genetic method, the frequency of FDB diagnosed as clinically FH.
2. Methods
We have been involved in the international
MEDPED program (Make Early Diagnoses-Prevent Early Deaths) to detect and treat FH families as coordi-nated by the center in Salt Lake City, USA [8]. The FH patients have been screened from the family doctors’ register of two districts in Budapest and our lipid clinics between 1996 and 1998. Every Hungarian citizen (both healthy and ill) has to be recorded in the family doc-tors’ registers.
We followed the recommendations of the MEDPED program (Table 1) as to the clinical diagnosis of FH. In accordance with MEDPED recommendations, deceased people were included in the group with FH diagnosis if CHD occurred in the males before the age of 55 and in the females at the age of 60. Based on case history, a myocardial infarction, positive coronary angiography, PTCA, ACBG, and sudden death, were considered as CHD. In the living patients, if there was no myocardial infarction, PTCA or ACBG, but there was chest pain in the case history, then treadmill or stress echocardiogra-phy was performed. In the case of any positive finding, we carried out coronary angiography, and CHD was estimated. In the present study, the data of 377 persons were examined separately, and 292 DNA analyses were provided.
The MEDPED clinical diagnosis of heterozygous FH has three categories based on the family history, case history, physical examination, and laboratory results. These are possible, probable, and certain FH. As the mean values of lipid findings typical of FH patients according to the age groups are not known in Hungary, we did not apply these categories. The LDL receptor gene investigation is the control for this.
We only focused our investigation on probands and first-degree relatives (parents, siblings and children). Blood samples were taken after a 12-h fast for lipid and DNA examination. The serum total cholesterol and triglyceride values were determined by enzymatic col-orimetric assay, and HDL cholesterol was measured enzymatically in the supernatant after precipitation of other lipoproteins with dextran sulfate magnesium. The LDL cholesterol was calculated using the Friedewald formula. DNA was isolated from leukocytes by the sodium chloride precipitation method [9] and the apoB R3500Q mutation was examined by allele-specific PCR
technique (UOL: 5% GAC CAC AAG CTT AGC TTG
G 3%, LOL: 5%GGG TGG CTT TGC TTG TAT G 3%,
ASO: 5% TGC AGC TTC ACT GAA GAC T 3%) [10].
Data are averages9S.D. Plasma lipid levels and ages were compared with the Mann – Whitney test. Qualita-tive variables were analyzed with the chi-square test. PB0.05 was considered statistically significant. The statistical analyses were done using the GraphPadPrism program (address: www.graphpad.com).
3. Results
The data of the probands and their relatives clinically diagnosed as having FH are summarized in Tables 2 and 3. We found 39 FH cases in the family doctors’ registers (1:538) and 34 FH cases in the lipid clinics’ registers. The 377 (alive: 292, deceased: 85) members of the 73 FH families were analyzed. Of the 73 FH probands, four (5.5%) were diagnosed as having FDB Table 1
MEDPED recommendations for clinical diagnosis of FH in the US (1996 version)
I. Criteria of the clinical diagnosis of FH concerning the patient (all the criteria have to be met!)
Cholesterol or LDL Age (years)
30–39 Over 40 cholesterol rate (mg/dl)
OR
\240 \220 \200
Very high LDL rate \260
(mg/dl)
B175 B95
b) Average triglyceride B150 B200 rate (mg/dl)
c) Secondary hypercholesterolemia due to diabetes mellitus, kidney disease (nephrosis syndrome, uremia), pregnancy, hypothyreosis, liver disease (primary biliar cirrhosis, obstruction jaundice), Cushing syndrome (steroid treatment), systemic lupus erythematoses, etc. can definitely be excluded
d) The total cholesterol rate of at least one child, grandchild, nephew, niece, etc. under 18 is\270 mg/dl OR at least one has tendon xanthoma
e) The evaluation of the family tree proves the dominant heritage of FH: one half of the siblings or descendants is affected by FH or their total cholesterol level is bi-modal (the high and normal values are clearly differentiated)
II. Criteria for the close relatives of the patient definitely diagnosed as affected with FH
Cholesterol or LDL Age (years)
Over 40 Under 18 18–29 30–39
\220 \240 \280
a) High total cholesterol \300
rate (mg/dl)
Table 2
Data of 88 males with clinically diagnosed FH and 100 males with clinically diagnosed non-FH of 69 families affected with FH
Living
Age (years) 40.5 60.3 15.4
3.3 27.0
25.0 2.3
BMI (kg/m2)
Total cholesterol 369 46 231 54
(mg/dl)
15
HDL (mg/dl) 46 48 16
35
Triglyceride (mg/dl) 138 185 46 28 140
Death CHD (year) 53 70 13
difference was found in triglyceride or HDL cholesterol values (Tables 2 and 3).
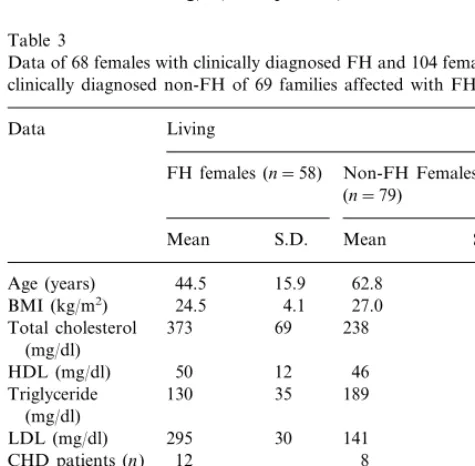
Of the hypercholesterolemic members in the FH families, 28 of the 88 males (31.8%), and 22 of the 68 females (32.4%), had or have CHD (Tables 2 and 3). The occurrence of CHD is lower in normocholes-terolemic cases in FH families. Only 13 of the 100 males (13%) and 14 of the 104 females (13.5%) had or have CHD (Tables 2 and 3). In both sexes, the occur-rence of CHD frequency is significantly higher (PB 0.01) in the hypercholesterolemic group. The onset of CHD occurred earlier in the FH group in males at the age of 4599 years versus the non-FH group at 6397 years (PB0.001), and in females at 5297 years versus at 6497 years (PB0.001).
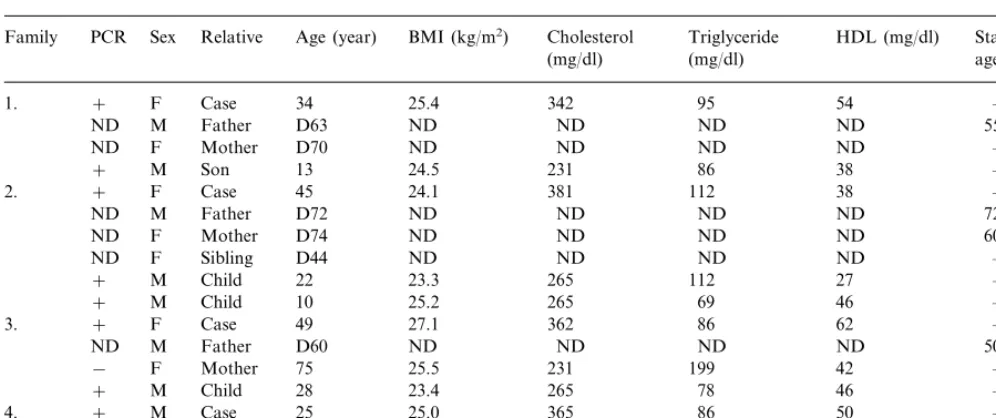
Of the 73 families with FH, four families with FDB were diagnosed by PCR examination (Table 4). We were able to conduct DNA tests on 11 of the 17 members of the four families (six of them died), and in nine cases, we found the defective apoB. Because of the small number of cases, we only highlighted the tenden-cies. The significant difference in total cholesterol and LDL cholesterol levels between the affected and non-af-fected family members detected in FH patients is not observed in FDB patients. The triglyceride and HDL cholesterol values among the family members did not differ either. The FDB patients are young, and clinical symptoms of CHD could not be demonstrated. Refer-ring to case histories, we assumed that three of the deceased people in the FDB families suffered from CHD.
4. Discussion
In 1986, Vega and Grundy [11] called attention to the fact that in five of their 15 FH cases the defect in the LDL-receptor was absent. The high plasma level of the LDL was explained by a binding deficiency arising from structural deformity of the LDL. Innerarity et al. [12] determined that this binding inability of the LDL can be based on a genetic defect of apoB-100, and they named this disease familial defective apolipoprotein B-100 (FDB). According to testing carried out by Soria et al. [13] the main genetic cause of FDB is a change of the codon CGG triplet responsible for the 3500th amino acid into CAG in APOB gene, causing arginine-glutamine change (R3500Q). Recently, in addition to the R3500Q mutation, other forms were described (R3531C and R3500W) with low rates of occurrence [14]. A total of 58% of the R3500Q cases have been reported as belonging to a similar haplotype that refers to the origin of the mutation from a common ancestor [15]. The prevalence of FDB caused by the R3500Q mutation in California was found to be 0.08% in the 5160 persons examined [16]. Rauh et al. [17] demon-by PCR examination. In the remaining 69 FH families,
the number of the first degree relatives was 360 (alive: 281, deceased: 79). In the FH families, there was a significant difference in total cholesterol and LDL cholesterol concentration between the affected (males total cholesterol: 369946, LDL cholesterol: 295928 mg/dl and females total cholesterol: 373969, LDL cholesterol: 295930 mg/dl) and unaffected (males total cholesterol: 231954, LDL cholesterol: 140926 mg/dl and females total cholesterol: 238950, LDL choles-terol: 141929 mg/dl) subjects (all PB0.0001). No
Table 3
Data of 68 females with clinically diagnosed FH and 104 females with clinically diagnosed non-FH of 69 families affected with FH
Living Data
FH females (n=58) Non-FH Females (n=79)
Mean S.D. Mean S.D.
44.5 15.9 62.8 17.7
Age (years)
24.5 4.1
BMI (kg/m2) 27.0 3.4
Total cholesterol 373 69 238 50
Table 4
Data of the four FDB affected familiesa
Family PCR Sex Relative Age (year) BMI (kg/m2) Cholesterol Triglyceride HDL (mg/dl) Start of CHD
age (year) (mg/dl)
(mg/dl)
F
1. + Case 34 25.4 342 95 54 –
M Father D63 ND
ND ND ND ND 55
ND F Mother D70 ND ND ND ND –
M Son 13 24.5 231 86 38 –
+
F Case 45 24.1
+ 381
2. 112 38 –
M Father D72 ND ND
ND ND ND 72
F Mother D74 ND
ND ND ND ND 60
ND F Sibling D44 ND ND ND ND –
M Child 22 23.3
+ 265 112 27 –
+ M Child 10 25.2 265 69 46 –
F Case 49 27.1 362 86
3. + 62 –
M Father D60 ND
ND ND ND ND 50
F Mother 75 25.5 231
− 199 42 –
M Child 28 23.4
+ 265 78 46 –
M
4. + Case 25 25.0 365 86 50 –
M Father 52 26.2
− 327 147 65 –
F Mother
+ 48 24.5 304 52 38 –
aD, deceased; ND, no data.
strated that in North America and Europe the R3500Q mutation could be found in one out of 500 – 700 indi-viduals of European origin. The exception is the Finnish population where such a mutation was not detected despite extensive testing [18]. Similarly, this mutation could not be found in Israel or Japan [19]. In our present study of 21 000 people, two R3500Q muta-tions (0.01%) have been found in the FH families selected clinically by the MEDPED method.
To distinguish between FH and FDB is very difficult based on clinical appearance as the phenotype of the two diseases is very similar (arcus cornealis, xanthe-lesma, tendon xanthoma) [20]. In the Netherlands FDB was diagnosed in 18 cases (1.85%) out of 970 clinically diagnosed FH patients. A British group found 17 (3%) FDB cases out of 562 FH patients with allele-specific PCR [14,21]. In the present study, we found FDB in four (5.5%) families (nine cases) out of 73 FH families (292 subjects). Earlier, we observed three R3500Q mu-tations in 119 patients with type II/a primary hyperlipi-demia (2.5%) without familial screening and two more cases were discovered among the FDB family members [22]. In Germany, among 243 patients with type II/a primary hyperlipidemia, 57 (23.5%) individuals with FH, and eight (3.3%) persons with FDB were found. A further ten FDB cases were diagnosed in the family members [23]. The generally observed low number of FDB cases can also be explained by the fact that the total cholesterol level of the patients — especially when young — are nearly normal.
The moderately elevated cholesterol level can partly be explained by the fact that the LDL precursors (VLDL, IDL), which still contain apolipoprotein E, are eligible for elimination across the LDL receptor that
can bind apoE in addition to apoB [21,24,25]. This route of elimination is not available in FH patients suffering from LDL receptor deficiency. Furthermore, some functional polymorphisms (PVU-II, Sfa NI, Ava II, RFLP), typical for LDL receptor gene, may be associated with this different (such as lower) cholesterol level in FDB patients [15,26]. In a meta-analysis com-paring 73 German, 87 Dutch, and 45 Danish FDB patients, the cholesterol level was highest in German patients. This was explained by polymorphisms of the LDL receptor gene and by different dietary and exer-cise habits [24]. The LDL concentration which in-creased to a smaller extent, does not mean a lower atherosclerotic risk as this LDL particulum can be oxidized more easily [27].
The frequency of FDB in patients with myocardial infarction is twice as high compared to persons without it [4]. CHD already occurs before the age of 50 in 40% of the males and 20% of the females with FDB repre-senting similarities with FH. In males over the age of 40, CHD occurs more frequently in FH than in FDB cases. In contrast, in females over 50, CHD is more frequent in FDB cases [28].
In the therapy of FH and FDB, the most important drugs are the HMG-CoA reductase inhibitors [29]. The efficiency of statins was the same in Dutch FH and FDB patients [21]. However, in Germany the simvas-tatin treatment of FDB patients showed a weaker effect [17].
clinical phenotype of FDB is very poor at early age, genetic screening of defective apoB is highly recom-mended for families which experience early onset CHD.
References
[1] Fredrickson DS. Plasma lipoproteins: micellar models and mu-tants. Trans Assoc Am Phys 1969;82:68 – 86.
[2] Thompson GR. Handbook of hyperlipidemia. Merck, 1990. [3] Czeizel AE, Kalina A´ , Williams RR. Euphenic prevention of
coronary artery disease. Am J Cardiol 1997;79:140 – 4.
[4] Brousseau T, Arvelier D, Cambou PJ, Evans AE, Luc G, Fruchart JC, et al. Familial defective apolipoprotein B-100 and myocardial infarction. The ECTIM study. Atherosclerosis 1995;116:269 – 71.
[5] Ludwig HE, McCarthy JB. Haplotype analysis of the human apolipoprotein B mutation associated with familial defective apolipoprotein B-100. Am J Hum Genet 1990;47:712 – 20. [6] McCarthy BJ, Soria L, Ludwig EM. An arginine 3500 glutamine
mutation in familial defective apoB-100 subjects with LDL de-fective in binding to the apoB,E (LDL) receptor. Circulation 1988;78(suppl II):11 – 66.
[7] Hansen SP, Meinertz H, Jensen KH, Fruergaard P, Launbjerg J, Klausen CI, et al. Characteristics of 46 heterozygous carriers and 57 unaffected relatives in five Danish families with familial defective apolipoprotein B-100. Arterioscler Thromb 1994;14:207 – 13.
[8] World Health Organization. World Health Organization Human Genetics Programme Division of Noncommunicable Diseases: familial hypercholesterolaemia. Geneva: WHO, 1998 WHO/
HGN/FH/CONS/98.7,6-1.
[9] Miller SA, Dykes DD, Polesky HF. A simple salting out proce-dure for extracting DNA from human nucleotide cells. Nucleic Acids Res 1988;16:1215.
[10] Schuster H, Rauh G, Mu¨ller S, Keller C, Wolfram G, Zo¨llner N. Allele-specific and asymmetric polymerase chain reaction am-plification in combination: a one step polymerase chain reaction protocol for rapid diagnosis of familial defective apolipoprotein B-100. Anal Biochem 1992;203:1 – 4.
[11] Vega GL, Grundy SM. In vivo evidence for reduced binding of low density lipoproteins to receptors as a cause of primary moderate hypercholesterolemia. J Clin Invest 1986;78:1410 – 4. [12] Innerarity TL, Weisgraber KH, Arnold KS, Mahley RW,
Krauss RM, Vega GL, et al. Familial defective apolipoprotein B-100: low density lipoprotein with abnormal receptor binding. Proc Natl Acad Sci USA 1987;84:6919 – 23.
[13] Soria LF, Ludwig EH, Clarke HR, Vega GL, Grundy SM, McCarthy BJ. Association between a specific apolipoprotein B mutation and familial defective apolipoprotein B-100. Proc Natl Acad Sci USA 1989;86:587 – 91.
[14] Talmud JP, Tamplin JO, Heath K, Gaffney D, Day MNI, Humphrise ES. Rapid testing for three mutations causing famil-ial defective apolipoprotein B100 in 562 patients with familfamil-ial hypercholesterolaemia. Atherosclerosis 1996;125:135 – 7. [15] Gallagher JJ, Myant BN. Variable expression of the mutation in
familial defective apolipoprotein B-100. Arterioscler Thromb
1993;13:973 – 6.
[16] Bersot PT, Russell JS, Thatcher RS, Pomernacko KN, Mahley WR, Weisgraber HK, et al. A unique haplotype of the protein B-100 allele associated with familial defective apolipo-protein B-100 in a Chinese man discovered during a study of the prevalence of the disorder. J Lipid Res 1993;34:1149 – 54. [17] Rauh G, Keller C, Schuster H, Wolfram G, Zo¨llner N. Familial
defective apolipoprotein B-100: a common cause of primary hypercholesterolemia. Clin Invest 1992;70:77 – 84.
[18] Hamalainen T, Palotie A, Aalto-Setala K, Komtula K, Tikka-men MJ. Absence of familial defective apolipoprotein B-100 in Finnish patients with elevated serum cholesterol. Atherosclerosis 1990;82:177 – 83.
[19] Friedlander Y, Dann EJ, Leitersdorf E. Absence of familial defective apolipoprotein B-100 in Israeli patients with domi-nantly inherited hypercholesterolemia and in offspring with parental history of myocardial infarction. Hum Genet 1993;91:299 – 300 (letter).
[20] Miserez RA, Keller U. Differences in the phenotypic characteris-tics of subjects with familial defective apolipoprotein B-100 and familial hypercholesterolemia. Arterioscler Thromb Vasc 1995;15:1719 – 28.
[21] Defesche CJ, Pricker LK, Hayden RM, van der Ende EB, Kastelein PJJ. Familial defective apolipoprotein B-100 is clini-cally indistinguishable from familial hypercholesterolemia. Arch Intern Med 1993;153:2349 – 56.
[22] Csa´sza´r A. Is there a Finno-Ugrian suicide gene? Lancet 1996;347:403 (letter).
[23] Schuster H, Rauh G, Kormann B, Hepp T, Humphris S, Keller C, et al. Familial defective apolipoprotein B-100. Comparison with familial hypercholesterolemia in 18 cases detected in Mu-nich. Arteriosclerosis 1991;10:577 – 81.
[24] Hansen SP, Defesche CJ, Kastelin PJJ, Gerdes UL, Fraza L, Gerdes C, et al. Phenotypic variation in patients heterozygous for familial defective apolipoprotein B (FDB) in three European countries. Arterioscler Thromb Vasc 1997;17:741 – 6.
[25] Marz W, Baumstartk MW, Schnarnagl H, Ruzicka V, Buxbaum S, Herwig J, et al. Accumulation of ‘small dense’ low density lipoprotein (LDL) in a homozygous patient with familial defec-tive apolipoprotein B-100 results from heterogeneous interaction of LDL sub-fractions with the LDL receptor. J Clin Invest 1993;92:2922 – 33.
[26] Schaefer RJ, Scharnagl H, Baumstark WM, Schweer H, Zech AL, Seyberth H, et al. Homozygous familial defective apolipo-protein B-100. Enhanced removal of apolipoapolipo-protein E containing VLDLs and decreased production of LDLs. Arterioscler Thromb Vasc 1997;17:348 – 52.
[27] Stalenhoef FHA, Defesche CJ, Kelinveld AH, Demacker MNP, Kastelein PJJ. Decreased resistance against in vitro oxidation of LDL from patients with familial defective apolipoprotein B-100. Arterioscler Thromb 1994;14:489 – 93.
[28] Tybjaerg-Hansen A, Humphries ES. Familial defective apolipo-protein B-100: a single mutation that causes hypercholes-terolemia and premature coronary artery disease. Atherosclerosis 1992;96:91 – 107.
[29] Illingworth DR, Vakar F, Mahley RW, Weisgraber KH. Hypoc-holesterolaemia effects of lovastatin in familial defective apolipo-protein B-100. Lancet 1992;339:598 – 600.