TESIS
DETEKSI MOLEKULAR GEN SERTA UJI AKTIVITAS
ENZIM KATABOLIK DARI BAKTERI
Actinobacillus
sp. P3(7)
TERHADAP SUBSTRAT HIDROKARBON
MIRANTI PUSPITASARI
NIM. 081414253001
PROGRAM STUDI MAGISTER KIMIA
FAKULTAS SAINS DAN TEKNOLOGI
UCAPAN TERIMA KASIH
Segala puji syukur dipanjatkan ke hadirat Allah SWT yang telah
melimpahkan rahmat serta hidayah-Nya, sehingga penulisan tesis berjudul
“Deteksi Molekular Gen serta Uji Aktivitas Enzim Katabolik dari Bakteri
Actinobacillus sp. P3(7) terhadap Substrat Hidrokarbon” dapat terselesaikan
dengan baik. Penulisan tesis ini banyak mendapatkan bantuan moril maupun
materil dari berbagai pihak, sehingga ucapan terima kasih disampaikan dengan
tulus kepada:
1. Dr. Sri Sumarsih, M.Si. selaku Dosen Pembimbing I sekaligus Dosen Wali.
Terima kasih telah berkenan meluangkan waktu untuk selalu membimbing,
memberikan saran, memberikan motivasi, serta memberikan kesempatan
belajar dan berdiskusi.
2. Dr. Ni’matuzahroh selaku Dosen Pembimbing II. Terima kasih telah
berkenan meluangkan waktu untuk selalu membimbing, memberikan saran,
memberikan motivasi, serta memberikan kesempatan untuk belajar dan
memberikan pengalaman penelitian sehingga tesis ini dapat terselesaikan
dengan baik.
3. Tim penguji, Dr. Ir. Suyanto, M.Si., Prof. Dr. Afaf Baktir, MS., serta Dr.
Muji Harsini, M.Si. Terima kasih telah berkenan memberikan evaluasi dan
saran terkait penelitian dan penulisan tesis.
4. Ketua Program Studi Magister Kimia Fakultas Sains dan Teknologi
Universitas Airlangga, M. Zakki Fahmi, S.Si., M.Si., Ph.D. Terima kasih
telah memberikan masukan, saran, serta arahannya.
5. Tenaga kependidikan Departemen Kimia yang telah membantu kelancaran
penelitian.
6. Orang tua dan keluarga yang telah memberikan doa, kasih sayang, dan
dukungan yang tiada henti.
7. Tim penelitian bidang biokimia serta angkatan 2014/2015 atas bantuannya
ABSTRAK
Deteksi Molekular Gen serta Uji Aktivitas Enzim Katabolik dari Bakteri Actinobacillus sp. P3(7) terhadap Substrat Hidrokarbon
Penelitian ini bertujuan untuk mendeteksi keberadaan gen katabolik pada DNA genom bakteri hidrokarbonoklastik Actinobacillus sp. P3(7) serta uji aktivitas enzim katabolik terhadap substrat hidrokarbon. Fragmen gen diamplifikasi dari DNA genom menggunakan primer spesifik untuk gen alkM,
tod, dan ndo. Isolat ditumbuhkan pada media Sea Salt diperkaya yeast extract dan hidrokarbon, yaitu heksadekana, toluena, dan naftalen. Enzim katabolik diuji aktivitasnya terhadap substrat masing-masing. Hasil amplifikasi gen katabolik menunjukkan bahwa isolat memiliki fragmen gen alkM, tod, dan ndo dengan ukuran berturut-turut sebesar 900 bp, 600 bp, dan 650 bp. Isolat selama proses inkubasi menggunakan ketiga substrat hidrokarbon untuk proses pertumbuhan dan perkembangan hingga 10 hari inkubasi. Induksi ekspresi enzim alkana monooksigenase, toluena dioksigenase, dan naftalen dioksigenase terjadi selama proses degradasi namun pada waktu inkubasi yang berbeda. Aktivitas tertinggi alkana monooksigenase, toluena dioksigenase, dan naftalen dioksigenase berturut-turut sebesar 4,630 U/mL; 5,338 U/mL; dan 6,367 U/mL yang dicapai pada kondisi 10 hari, 8 hari, dan 10 hari inkubasi.
ABSTRACT
Molecular Detection of Gen and Activity Assay of Catabolic Enzyme from Actinobacillus sp. P3(7) toward Hydrocarbon Substrates
This research aims to detect the presence of catabolic genes in hydrocarbonoclastic bacteria Actinobacillus sp. P3 (7) and the catabolic enzyme activity assay against hydrocarbon substrate. Gene fragmen amplified from genomic DNA using specific primers for alkM, tod, and ndo genes. The isolate then grown on Sea Salt media enriched with yeast extract and hydrocarbons, namely hexadecane, toluene, and naphthalene. Catabolic enzymes tested for its activities against their respective substrates. Catabolic gene fragmen amplification results showed that the isolates have gene fragmen of alkM, tod, and ndo gene with consecutive size of 900 bp, 600 bp, and 650 bp. Isolates during the process of incubation using hydrocarbon substrate for growing and developing process until the 10 days of incubation. Induction of alkane monooxygenases, toluene dioxygenase, and naphthalene dioxygenase enzyme expression occur during the degradation process but at different incubation time. The highest activity of the alkane monooxygenases, toluene dioxygenase, and naphthalene dioxygenase are 4,630 U/mL; 5,338 U/mL; dan 6,367 U/mL, respectively, reached on the 10 days, 8 days, and the 10 days of incubation.
DAFTAR ISI
1.1. Latar Belakang Permasalahan 1
1.2. Rumusan Masalah 4
1.3. Tujuan Penelitian 4
1.4. Manfaat Penelitian 4
BAB II TINJAUAN PUSTAKA 6
2.1. Biodegradasi Hidrokarbon 6
2.2. Bakteri Hidrokarbonoklastik 7
2.3. Actinobacillus sp. 8
2.4. Enzim Monooksigenase dan Dioksigenase 9
2.4.1. Alkana Monooksigenase 10
2.4.2. Toluen Dioksigenase 12
2.4.3. Naftalen Dioksigenase 13
2.5. Gen Penyandi Enzim Monooksigenase dan Dioksigenase 15
2.5.1. Gen Penyandi Alkana Monooksigenase 16
2.5.2. Gen Penyandi Toluen Dioksigenase 16
2.5.3. Gen Penyandi Naftalen Dioksigenase 17
BAB III KERANGKA KONSEPTUAL DAN HIPOTESIS
PENELITIAN 18
3.1. Kerangka Konseptual 18
3.2. Hipotesis Penelitian 19
BAB IV METODE PENELITIAN 21
4.1. Lokasi dan Waktu Penelitian 21
4.2. Bahan dan Alat Penelitian 21
4.2.1. Isolat Bakteri 21
4.2.2. Bahan Penelitian 21
4.2.3. Alat penelitian 21
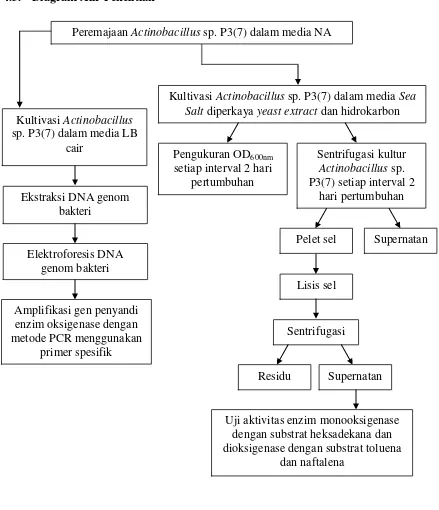
4.3. Diagram Alir Penelitian 22
4.4.1. Pembuatan Media 23
4.4.2. Peremajaan Mikroba 23
4.4.3. Kultivasi Mikroba dalam Media LB 23
4.4.4. Ekstraksi DNA Genom 24
4.4.5. Penentuan Kadar DNA Genom 25
4.4.6. Amplifikasi Gen Penyandi Enzim Monooksigenase dan
Dioksigenase 25
4.4.7. Elektroforesis DNA 25
4.4.8. Kultivasi Mikroba dalam Media Sea Salt diperkaya
Yeast Extract dan Substrat Hidrokarbon 27
4.4.9. Pengukuran OD600nm Mikroba 27
4.4.10. Uji Aktivitas Enzim Oksigenase 28
BAB V HASIL PENELITIAN DAN PEMBAHASAN 32
5.1. Ekstraksi DNA Genom Actinobacillus sp. P3(7) 32 5.2. Amplifikasi Gen Katabolik pada Actinobacillus sp. P3(7) 33 5.3. Respon Pertumbuhan Actinobacillus sp. P3(7) pada
Berbagai Substrat Hidrokarbon 35
5.4. Biomassa yang dihasilkan Kultur Actinobacillus sp. P3(7) 42 5.5. pH Akhir Kultur Actinobacillus sp. P3(7) 43 5.6. Kadar Protein Sel Actinobacillus sp. P3(7) 44 5.7. Aktivitas Enzim dari Actinobacillus sp. P3(7) 45
5.7.1. Alkana Monooksigenase 45
5.7.2. Toluena Dioksigenase dan Naftalena Dioksigenase 47 5.7.3. Pengaruh Penambahan Substrat terhadap Aktivitas
Enzim Katabolik dari Actinobacillus sp. P3(7) 49
BAB VI KESIMPULAN 52
6.1. Kesimpulan 52
6.2. Saran 52
DAFTAR PUSTAKA 53
DAFTAR TABEL
No. Judul Tabel Halaman
Tabel 4.1. Primer yang digunakan untuk amplifikasi fragmen gen……….. 26
Tabel 4.2. Set kondisi PCR………... 26
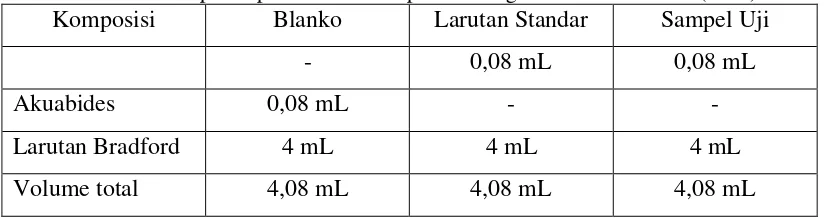
Tabel 4.3. Komposisi penentuan kadar protein dengan metode Bradford……… 29
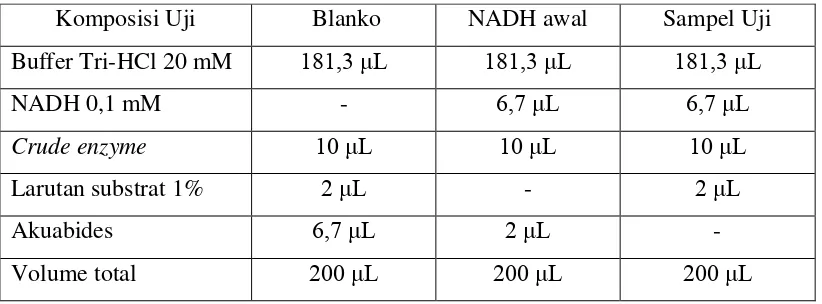
Tabel 4.4. Komposisi uji aktivitas enzim oksigenase………... 31
Tabel 5.1. Perbandingan antara hasil penelitian dengan hasil dari
DAFTAR GAMBAR
No Judul Gambar Halaman
Gambar 2.1. Fotomikrograf Actinobacillus sp. P3(7) ... 8
Gambar 2.2. Struktur monooksigenase pada sistem alkana monooksigenase 11 Gambar 2.3. Struktur oksigenase pada sistem toluena dioksigenase ... 12
Gambar 2.4. Struktur oksigenase pada sistem naftalena dioksigenase ... 14
Gambar 2.5. Ikatan koordinasi pada naftalena dioksigenase ... 14
Gambar 3.1. Kerangka konsep penelitian ... 20
Gambar 4.1. Diagram alir penelitian ... 22
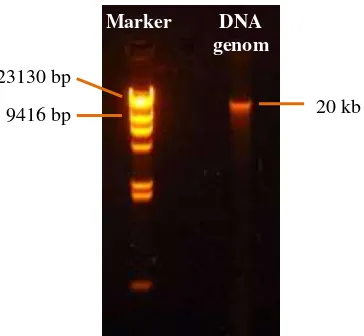
Gambar 5.1. Hasil elektroforesis DNA genom Actinobacillus sp. P3(7) ... 32
Gambar 5.2. Hasil amplifikasi dengan berbagai variasi suhu annealing ... 33
Gambar 5.3. Kurva pertumbuhan bakteri Actinobacillus sp. P3(7) ... 36
Gambar 5.4. Warna kultur pada berbagai variasi waktu inkubasi ... 37
Gambar 5.5. Struktur senyawa katekol, protokatekuat, dan asam salisilat .... 38
Gambar 5.6. Profil massa sel dari kultur dengan penambahan substrat hidrokarbon dan kultur tanpa penambahan substrat hidrokarbon 42 Gambar 5.7.Profil pH dari kultur dengan penambahan substrat hidrokarbon dan kultur tanpa penambahan substrat hidrokarbon ... 44
Gambar 5.8. Profil kadar protein sel dari kultur dengan penambahan substrat hidrokarbon dan kultur tanpa penambahan substrat hidrokarbon 45 Gambar 5.9. Kurva produksi enzim alkana monooksigenase selama proses kultivasi Actinobacillus sp. P3(7) dalam media mengandung substrat heksadekana ... 46
Gambar 5.10. Kurva produksi enzim toluena dioksigenase selama proses kultivasi Actinobacillus sp. P3(7) dalam media mengandung substrat toluena ... 47
Gambar 5.11. Kurva produksi enzim naftalena dioksigenase selama proses kultivasi Actinobacillus sp. P3(7) dalam media mengandung substrat naftalena ... 48
DAFTAR LAMPIRAN
No Judul Lampiran
Lampiran 1. Koloni Actinobacillus sp. P3(7) koleksi Laboratorium FST
Universitas Airlangga
Lampiran 2. Data penentuan OD600nm kultur Actinobacillus sp. P3(7)
Lampiran 3. Data penentuan biomassa kultur Actinobacillus sp. P3(7)
Lampiran 4. Data penentuan pH akhir kultur Actinobacillus sp. P3(7)
Lampiran 5. Data penentuan kadar protein sel Actinobacillus sp. P3(7)
DAFTAR SINGKATAN
bp : base pair
BLAST : Basic Local Alignment Search Tool
DNA : Deoxyribonucleic acid
NADH : Nicotinamide adenine dinucleotide
Trp : Triptofan
Ile : Isoleusin
Thr : Treonin
Leu : Leusin
His : Histidin
Asp : Asam aspartat
PCR : Polymerase Chain Reaction
AlkB : Alkana monooksigenase untuk rantai C medium (C5-C12)
AlkM : Alkana monooksigenase untuk rantai C panjang (C>12)
CYP : Sitokrom P450
NA : Nutrient Agar
SS : Sea Salt
EtBr : Etidium bromida
DMSO : Dimetil sulfoksida
OD : Optical density (densitas optik)
BSA : Bovine serum albumin
cAMP : Cyclic adenosine monophosphate
BAB I PENDAHULUAN
1.1. Latar Belakang Permasalahan
Lumpur minyak bumi (oil sludge) adalah campuran logam, minyak,
padatan, dan air yang membentuk emulsi air/minyak yang stabil dan terdeposisi
pada bagian dasar tangki penyimpanan minyak mentah maupun yang telah
melalui tahap pengolahan. Oil sludge terbentuk karena adanya proses oksidasi
minyak dan air oleh udara sehingga menghasilkan sedimentasi pada dasar tangki
dan menjadi hasil samping dalam proses pengolahan dan pemurnian minyak
bumi. Sedimen yang terbentuk menghambat aliran minyak dalam pipa dan bersifat
korosif terhadap permukaan tangki penyimpanan minyak sehingga dapat
mempercepat kerusakan komponen tangki. Oil sludge juga mengandung
hidrokarbon alifatik, hidrokarbon aromatik, serta senyawa organik yang
mengandung atom N, O, atau S yang sangat berbahaya bagi lingkungan dan
kesehatan sehingga perlu dilakukan pengolahan untuk menekan atau
menghilangkan kandungan berbahaya dari oil sludge (Hu et al., 2013).
Berbagai metode degradasi oil sludge secara fisika maupun kimia telah
banyak digunakan dan memiliki kelebihan yaitu efisien serta mampu menurunkan
kadar hidrokarbon pada oil sludge secara maksimal namun memiliki kelemahan
yaitu tidak ekonomis terutama jika diaplikasikan dalam skala industri. Salah satu
metode yang dapat menjadi pilihan untuk pengolahan oil sludge adalah
biodegradasi dengan menggunakan mikroorganisme yaitu bakteri, yeast, atau
fungi. Bakteri banyak dipilih dibandingkan yeast atau fungi karena memiliki
kecepatan pertumbuhan yang tinggi serta kemudahan untuk memperbanyak
jumlah selnya. Komponen oil sludge sebagian besar tersusun dari senyawa
hidrokarbon sehingga dapat digunakan oleh bakteri yang memiliki kemampuan
untuk mendegradasi hidrokarbon sebagai satu-satunya sumber karbon yang
tersedia untuk proses metabolisme, yang disebut sebagai bakteri
hidrokarbonoklastik (Hu et al., 2013). Spesies bakteri yang memiliki kemampuan
bakteri, baik bakteri Gram positif maupun Gram negatif, seperti Aeromonas
hydrophila, Acinetobacter faecalis tipe II, Actinobacillus sp. P(3)7, Pseudomonas
aeruginosa, Pseudomonas putida, Pseudomonas cepacea, Pseudomonas
fluorescens-25, dan Pseudomonas pseudomallei(Ni’matuzahroh et al., 2009).
Kemampuan bakteri dalam mendegradasi hidrokarbon dipengaruhi oleh
adanya enzim katabolik yang mampu memecah senyawa hidrokarbon menjadi
senyawa metabolit yang mampu masuk ke dalam siklus asam sitrat. Enzim
katabolik yang paling berperan penting dalam proses katabolisme hidrokarbon
yang masuk ke dalam sel bakteri adalah enzim yang mengkatalisis reaksi oksidasi
hidrokarbon tahap pertama. Tahap pertama katabolisme alkana dan aromatik oleh
bakteri masing-masing diinisiasi oleh enzim monooksigenase dan dioksigenase
(Jauhari et al., 2014).
Aktivitas enzim monooksigenase dan dioksigenase yang diisolasi dari
beberapa spesies bakteria dalam mendegradasi substrat hidrokarbon telah berhasil
dilaporkan. Aktivitas alkana monooksigenase dari Pseudomonas sp. BP10 dan
Stenotrophomonas nitritireducens E9 yang diisolasi dari petroleum sludge serta
konsorsiumnya dalam mendegradasi heksakosan (C26) masing-masing mencapai 527 ηmol/mg, 563 ηmol/mg, dan 607 ηmol/mg protein (Jauhari et al., 2014), sedangkan aktivitas alkana monooksigenase dari Pseudomonas aeruginosa PSA5,
Rhodococcus sp. NJ2 dan Ochrobactrum intermedium P2 yang diisolasi dari
petroleum sludge dalam mendegradasi heksadekana (C16) masing-masing
mencapai 89,83 μmol/g, 185 μmol/g, dan 186 μmol/g protein (Mishra dan Singh,
2012). Aktivitas naftalena dioksigenase dari Pseudomonas sp. NCIB9816 dan
Rhodococcus sp. NCIMB12038 dalam mendegradasi naftalen masing-masing
sebesar 37,9 U/mg protein dan 0,731 U/mg protein (Ensley dan Gibson, 1983;
Larkin et al., 1999). Aktivitas enzim alkana monooksigenase dan aromatik
dioksigenase dari spesies bakteri lain perlu diteliti untuk mengetahui
keanekaragaman aktivitas enzimatis bakteri hidrokarbonoklastik lainnya.
Berbagai spesies bakteri hidrokarbonoklastik berhasil diisolasi dari lokasi
yang tidak terkontaminasi maupun yang terkontaminasi oleh hidrokarbon, namun
belum banyak diteliti. Enzim yang berperan dalam tahap pertama degradasi
hidrokarbon, seperti alkana monooksigenase, toluena monooksigenase, naftalena
dioksigenase yang masing-masing disandi oleh gen alk, tod, serta ndo dapat
diamplifikasi dari DNA genom bakteri menggunakan primer spesifik sehingga
dapat diketahui sekuens gen yang menyandi enzim-enzim penting dalam
katabolisme hidrokarbon. Fragmen gen alkB yang menyandi alkana
monooksigenase untuk substrat C5-C12 berhasil diamplifikasi dari Rhodococcus
sp. dengan ukuran 701 bp menggunakan primer spesifik untuk bakteri
Rhodococcus sp. yang didesain berdasarkan daerah lestari gen alkB dari hasil
penjajaran gen alkB dari berbagai spesies Rhodococcus sp. (Tancsics et al., 2015)
serta fragmen gen alkB, ndo, C12O, dan C23O berhasil diamplifikasi dari DNA
genom Sphingomonas koreensis ASU06 yang diisolasi dari sampel tanah dari
perusahaan pengolahan minyak di Assiut, Mesir. Produk PCR yang didapatkan
berturut-turut yaitu 100; 487; 350; dan 900 bp (Hesham et al., 2014). Fragmen
gen alkM yang menyandi alkana monooksigenase untuk substrat C13-C30 juga
berhasil diamplifikasi dari Acinetobacter baumannii OS1 yang diisolasi dari
sampel oil sludge dari pengolahan minyak di Manila, Filipina, dengan ukuran 715
bp. Analisis BLAST menunjukkan adanya kemiripan sekuens gen alkM sebesar
99% dengan gen alkM A. baumannii AB307-0294 (Hedreyda dan Sarmago,
2014).
Salah satu bakteri yang memiliki kemampuan untuk mendegradasi
hidrokarbon adalah Actinobacillus sp. P3(7) hasil isolasi dari sampel tanah
pengilangan minyak Desa Wonocolo, Bojonegoro oleh Ni’matuzahroh et al.
(2009). Actinobacillus sp. P3(7) mampu hidup pada tanah yang tercemar minyak
bumi sehingga diperkirakan mampu menggunakan hidrokarbon sebagai sumber
karbon untuk proses metabolisme dengan bantuan enzim-enzim katabolik.
Actinobacillus sp. P3(7) memiliki aktivitas emulsifikasi substrat crude oil sebesar
100% dan mampu menurunkan tegangan permukaan hingga 51,2 mN/m melalui
proses pelepasan biosurfaktan. Pelepasan biosurfaktan akan menyebabkan
terjadinya emulsifikasi hidrokarbon dan penurunan tegangan antarmuka
hidrokarbon dalam fasa air sehingga hidrokarbon dapat masuk ke dalam sel dan
dimetabolisme oleh bakteri (Fatimah et al., 2009). Jenis-jenis gen katabolik dan
enzim katabolik yang berperan dalam proses degradasi hidrokarbon tahap pertama
untuk substrat heksadekana, toluena, dan naftalena serta nilai aktivitas
masing-masing enzim tersebut perlu dikaji lebih lanjut untuk mengetahui tingkat
degradasi enzimatis hidrokarbon oleh bakteri Actinobacillus sp. P3(7).
1.2. Rumusan Masalah
Rumusan masalah yang dikaji dalam penelitian ini berdasarkan uraian latar
belakang di atas yaitu sebagai berikut.
1. Apakah fragmen gen penyandi enzim monooksigenase dan dioksigenase
dapat diamplifikasi dari DNA genom Actinobacillus sp. P3(7)?
2. Berapa nilai aktivitas enzim monooksigenase dari Actinobacillus sp. P3(7)
dalam mengkatalisis reaksi oksidasi heksadekana sebagai substrat?
3. Berapa nilai aktivitas enzim dioksigenase dari Actinobacillus sp. P3(7) dalam
mengkatalisis reaksi oksidasi toluena dan naftalena sebagai substrat?
1.3. Tujuan Penelitian
Tujuan yang ingin dicapai dalam penelitian ini berdasarkan permasalahan
yang akan dikaji yaitu sebagai berikut.
1. Mengamplifikasi fragmen gen penyandi enzim monooksigenase dan
dioksigenase dari DNA genom Actinobacillus sp. P3(7).
2. Menentukan nilai aktivitas enzim monooksigenase dari Actinobacillus sp.
P3(7) dalam mengkatalisis reaksi oksidasi heksadekana sebagai substrat.
3. Menentukan nilai aktivitas enzim dioksigenase dari Actinobacillus sp. P3(7)
dalam mengkatalisis reaksi oksidasi toluena dan naftalena sebagai substrat.
1.4. Manfaat Penelitian
Manfaat yang diharapkan dari penelitian ini yaitu sebagai berikut.
1. Memberikan informasi mengenai pemanfaatan bakteri sebagai alternatif agen
2. Memberikan informasi mengenai gen-gen penyandi enzim pendegradasi
hidrokarbon sebagai identifikasi awal untuk mengenali selektifitas substrat
hidrokarbon untuk bakteri tertentu.
3. Memberikan informasi mengenai enzim-enzim yang berperan dalam proses
BAB II
TINJAUAN PUSTAKA
2.1. Biodegradasi Hidrokarbon
Biodegradasi merupakan proses konversi atau transformasi senyawa toksik
menjadi senyawa yang kurang toksik atau tidak toksik melalui aktivitas
metabolisme dari mikroorganisme, ragi, atau tumbuhan yang menggunakan
polutan sebagai sumber karbon dan energi. Produk degradasi hidrokarbon yang
memasuki siklus asam sitrat berfungsi sebagai substrat metabolisme energi dan
sebagai zat pembangun untuk proses biosintesis sel dan proses pertumbuhan
bakteri. Proses degradasi dapat dibagi menjadi dua model yaitu melalui
metabolisme aerob yang membutuhkan molekul oksigen serta metabolisme
anaerob yang tidak membutuhkan oksigen (Fritsche dan Hofrichter, 2008).
Laju biodegradasi komponen hidrokarbon oleh bakteri dipengaruhi oleh: i)
sifat biodegradabilitas senyawa, misalnya senyawa alifatik dengan C10-C18 lebih
mudah didegradasi oleh bakteri dibandingkan dengan rantai karbon yang lebih
pendek atau lebih panjang, sedangkan alifatik rantai panjang didegradasi lebih
lambat karena memiliki bioavailabilitas yang rendah karena senyawa tersebut sulit
larut dalam air sehingga tidak mudah digunakan oleh mikroba sebagai sumber
karbon, dan alifatik rantai pendek mudah larut dalam air namun sangat toksik
untuk sel mikroba; serta ii) kemudahan senyawa untuk diakses oleh mikroba dan
oleh aktivitas biologis sehingga proses degradasi menjadi berjalan lebih cepat
(Mishra dan Singh, 2012).
Biodegradasi komponen hidrokarbon oleh bakteri dimediasi oleh beberapa
jenis enzim degradatif. Jenis enzim degradatif yang terlibat dalam degradasi
hidrokarbon dapat dibagi menjadi dua berdasarkan mekanisme kerja enzim, yaitu
enzim periferal dan enzim fisi. Enzim periferal bekerja untuk mengenali dan
mengkonversi hidrokarbon menjadi molekul yang lebih mudah masuk ke dalam
sel sehingga lebih mudah didegradasi, contohnya yaitu enzim lipase. Enzim fisi
bekerja mendegradasi molekul tersebut melalui jalur metabolisme sel, contohnya
Enzim yang dihasilkan oleh bakteri hidrokarbonoklastik dapat mendegradasi
hidrokarbon melalui satu atau lebih jalur metabolik karena enzim-enzim tersebut
tidak mampu mendegradasi semua jenis senyawa hidrokarbon (Mishra et al.,
2014; Macaulay, 2014). Enzim fisi yang bekerja pada proses degradasi alkana
bergantung dari panjang rantai karbon alkana tersebut, sehingga bakteri yang
mampu mendegradasi alkana umumnya memiliki beberapa gen yang menyandi
berbagai variasi enzim alkana monooksigenase (Van Beilen et al., 2003).
2.2. Bakteri Hidrokarbonoklastik
Aktivitas hidup bakteri memerlukan senyawa karbon sebagai salah satu
sumber nutrisi dan energi untuk melangsungkan proses metabolisme dan
perkembangbiakan. Beberapa bakteri memiliki kemampuan yang khas yaitu
menggunakan senyawa hidrokarbon sebagai satu-satunya sumber karbon dan
energi. Bakteri jenis ini tersebar luas di alam dan dikenal sebagai bakteri
hidrokarbonoklastik. Bakteri ini dapat memetabolisme hidrokarbon dengan
dikatalisis enzim-enzim katabolik pendegradasi hidrokarbon yang dihasilkan
secara intraseluler (Fritsche dan Hofrichter, 2008).
Beberapa genus bakteri hidrokarbonoklastik yang mampu menggunakan
hidrokarbon sebagai sumber karbon antara lain Achromobacter, Acinetobacter,
Aeromonas, Alcaligenes, Arthrobacter, Bacillus, Benecdea, Brevibacterium,
Candida, Corynebacterium, Flavobacterium, Methylobacterium, Methylococcus,
Methylocystis, Methylomonas, Micromonospora, Micrococcus, Mycobacterium,
Nocardia, Pseudomonas, Rhodotula, Spirillium, Sporobolomyces, dan Vibrio.
Beberapa bakteri mampu menggunakan hidrokarbon sebagai sumber karbon
akibat adanya proses adaptasi setelah terjadi kontaminasi minyak di laut, yaitu
genus Oleispira, Marinobacter, Thalassolitus, Alcanivorax, dan Cycloclasticus.
Hal ini menunjukkan bahwa kemampuan degradasi hidrokarbon dapat muncul
secara alami akibat adanya proses evolusi (Chen, 2013). Spesies bakteri yang
berhasil diisolasi dan memiliki kemampuan dalam mendegradasi hidrokarbon
dalam minyak mentah antara lain Acinetobacter faecalis tipe II, Actinobacillus sp.
Pseudomonas cepacea, Pseudomonas fluorescens-25, dan Pseudomonas
pseudomallei(Ni’matuzahroh et al., 2009).
2.3. Actinobacillus sp.
Actinobacillus sp. merupakan bakteri Gram negatif, imotil, tidak
menghasilkan spora, serta berbentuk oval hingga batang. DNA genom bakteri
Actinobacillus sp. mengandung 40% mol guanin dan 47% mol sitosin. Taksonomi
dari spesies Actinobacillus sp. dapat dilihat di bawah ini (Mutters et al., 1986).
Kingdom : Bacteria
Gambar 2.1. Fotomikrograf Actinobacillus sp. P(3)7
Spesies Actinobacillus sp. diketahui memiliki kemampuan untuk
mendegradasi senyawa hidrokarbon. Actinobacillus sp. mampu mendegradasi
fenol 100 mg/L hingga 100% pada kondisi optimum yaitu pada pH 7, suhu
inkubasi 35-37ºC, serta kecepatan agitasi 150 rpm. Asam suksinat serta glisin
sebagai sumber karbon dan sumber nitrogen merupakan jenis kosubstrat yang
paling efisien dalam menunjang proses degradasi hidrokarbon (Khleifat , 2007).
Actinobacillus sp. P3(7) hasil isolasi dari sampel tanah pengilangan minyak
Desa Wonocolo, Bojonegoro oleh Ni’matuzahroh et al. (2009) mampu hidup pada
tanah yang tercemar minyak bumi sehingga diperkirakan mampu menggunakan
hidrokarbon sebagai sumber karbon untuk proses metabolisme. Actinobacillus sp.
P3(7) memiliki aktivitas emulsifikasi substrat crude oil sebesar 100% dan mampu
menurunkan tegangan permukaan hingga 51,2 mN/m melalui proses pelepasan
biosurfaktan. Pelepasan biosurfaktan akan menyebabkan terjadinya emulsifikasi
hidrokarbon dan penurunan tegangan antarmuka minyak-air sehingga hidrokarbon
hidrokarbon dapat masuk ke dalam sel dan dimetabolisme oleh bakteri (Fatimah et
al., 2009).
Actinobacillus sp. juga memiliki kemampuan untuk mensekresikan enzim
lipase dan dapat dikombinasikan dengan Acinetobacter sp. P2(1), Bacillus subtilis
3KP, serta Pseudomonas putida yang memiliki kemampuan untuk menghasilkan
biosurfaktan, untuk proses pengolahan crude oil dengan metode sand pack
column. Masing-masing jenis kombinasi biosurfaktan dan lipase dari keempat
bakteri tersebut secara efektif mampu menurunkan kadar crude oil sebesar
16,73%; 12%; dan 11,9%. Keefektifan kombinasi biosurfaktan dan lipase dalam
menurunkan kadar crude oil sebanding dengan surfaktan sintetik yang digunakan
sebagai kontrol positif yaitu Tween-20 yang mampu menurunkan kadar crude oil
hingga 13,40%, sehingga kombinasi biosurfaktan dan lipase dari keempat bakteri
tersebut diharapkan dapat digunakan dalam proses oil recovery(Ni’matuzahroh et
al., 2015).
2.4. Enzim Monooksigenase dan Dioksigenase
Tahap pertama dalam mekanisme degradasi alkana oleh bakteri dalam
kondisi aerob adalah oksidasi alkana oleh kelas enzim monooksigenase yaitu
enzim yang mengkatalisis inkorporasi satu atom oksigen ke dalam substrat,
sedangkan tahap pertama dalam mekanisme degradasi hidrokarbon aromatik
selain benzena oleh bakteri dalam kondisi aerob adalah oksidasi aromatik oleh
kelas enzim dioksigenase yaitu enzim yang mengkatalisis inkorporasi dua atom
oksigen ke dalam substrat (Madigan et al., 2012).
Sistem enzim yang terlibat dalam oksigenasi hidrokarbon alkana pada
prokariot telah banyak ditemukan dan diklasifikasikan berdasarkan panjang rantai
karbon substrat serta karakteristik degradasi karena sistem enzim yang berbeda
dibutuhkan untuk mengoksidasi alkana dengan panjang rantai yang berbeda untuk
menginisiasi proses biodegradasi, antara lain metana monooksigenase (MMO
yang dikode oleh klaster gen mmo), sitokrom P450 (CYP yang dikode oleh klaster
yang dikode oleh klaster gen alkB), dan alkana monooksigenase untuk rantai C
panjang yaitu C>12 (AlkM yang dikode oleh klaster gen alkM) (Singh et al., 2012).
2.4.1. Alkana monooksigenase
Enzim alkana monooksigenase merupakan enzim yang paling banyak
ditemukan pada bakteri pendegradasi alkana dan dikode oleh klaster gen alk.
Enzim ini mengkatalisis reaksi tahap pertama dalam degradasi alkana, yaitu
oksidasi hidrokabon alkana linier (rantai medium C5-C12 dan rantai panjang C12-30)
melalui inkorporasi 1 atom O dari O2 dan penggunaan NADH. Reaksi katalisis
oksidasi alkana menjadi alkohol yaitu sebagai berikut.
CH3-(CH2)n-CH3 + O2 + NADH + H+→ CH3-(CH2)n-CH2OH + NAD+ + H2O +
H+
(Ji et al., 2013).
Enzim famili alkana monooksigenase memiliki tiga komponen, yaitu
monooksigenase, rubredoksin, rubredoksin reduktase, serta dua atom Fe.
Komponen monooksigenase adalah protein integral membran, sedangkan
komponen rubredoksin dan rubredoksin reduktase merupakan protein sitoplasma
dan larut pada sitoplasma. Elektron ditangkap oleh NADH kemudian rubredoksin
reduktase mentransfer elektron dari NADH ke rubredoksin. Rubredoksin
kemudian mereduksi monooksigenase yang menyebabkan katalisis adisi 1 atom O
dari O2 ke alkana membentuk alkohol (Singh et al., 2012).
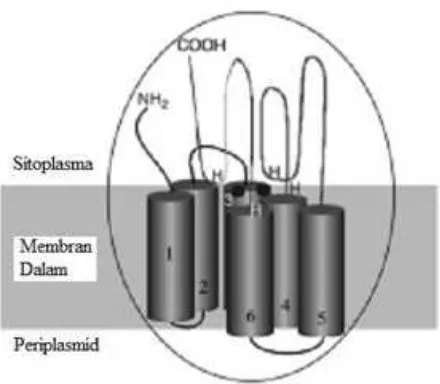
Komponen monooksigenase (gambar 2.2) memiliki 6 situs heliks
transmembran dan sisi aktif yang menghadap ke arah sitoplasma. Sisi aktif enzim
tersebut meliputi motif yang mengandung 4 residu histidin (simbol H) yang
membentuk kelat dengan dua atom Fe (simbol ●), dan bekerja dalam aktivasi
alkana dengan adanya O2 melalui pembentukan intermediet radikal. Satu dari
atom O pada O2 ditransfer ke gugus metil terminal dari alkana menghasilkan
alkohol, sedangkan atom O lainnya direduksi menjadi H2O melalui proses transfer
Gambar 2.2. Struktur monooksigenase pada sistem alkana monooksigenase. Simbol H: residu histidin, simbol ●: atom Fe, dan simbol batang: situs heliks transmembran (Van Beilen et al., 2003)
Enzim alkana monooksigenase memiliki spesifisitas substrat yang rendah
namun tetap bekerja secara regiospesifik (hanya bekerja pada posisi satu jenis
ikatan tertentu) dan stereospesifik (hanya menghasilkan salah satu jenis
stereoisomer). Alkana monooksigenase yang diisolasi dari Pseudomonas
oleovorans Gpo1 mampu mengkatalisis reaksi oksidasi pada alkana linier, alkana
bercabang, serta sikloalkana. Alkana linier seperti dekana, undekana, heksana,
serta heptana dioksidsasi menjadi alkohol primer. Alkana bercabang seperti
metilbutana, metilpentana, serta metilheksana dioksidasi pada atom karbon
sekunder. Oksidasi alkana tidak terjadi jika terdapat atom karbon tersier. Alkana
siklik tersubstitusi seperti metilsikloheksana dioksidasi pada posisi trans-4 dari
posisi substituen. Alkana selain jenis alkana linier dioksidasi dalam laju yang
sangat lambat. Enzim bekerja terhadap jenis substrat yang luas namun terbatas
pada substrat dengan struktur yang sederhana karena sisi aktif enzim yang
berukuran sempit sehingga substrat berukuran besar seperti dekalin serta indolin
dioksidasi dalam laju yang sangat lambat. Spesifisitas enzim ditentukan oleh
residu asam amino Trp-55 yang berada pada sisi aktif enzim (Van Beilen et al.,
2.4.2. Toluena dioksigenase
Toluena dioksigenase merupakan enzim yang mengkatalisis reaksi tahap
pertama dalam degradasi toluena, yaitu oksidasi toluena menjadi (+)-cis-(1S,2R
)-dihidroksi-3-metilsikloheksa-3,5-diena (cis-toluena dihidrodiol) melalui
penggunaan O2 dan NADH. Reaksi yang terjadi sebagai berikut.
Toluena + NADH + H+ + O2 → (+)-cis-(1S,2R
)-dihidroksi-3-metilsikloheksa-3,5-diena + NAD+
(Jiang et al., 1999).
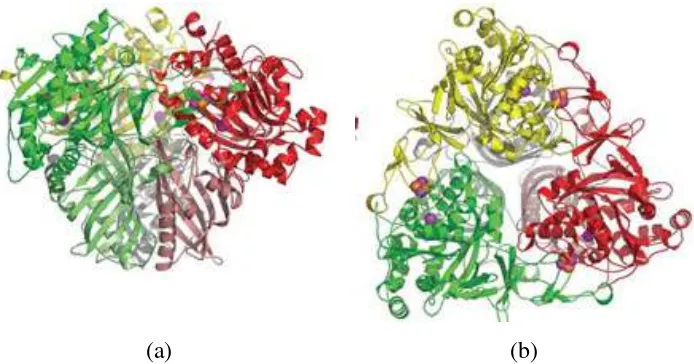
Toluena dioksigenase merupakan sistem enzim multikomponen yang terdiri
dari ferodoksin reduktase, ferodoksin, dan oksigenase (gambar 2.3). Elektron
ditangkap oleh NADH lalu ditransfer ke ferodoksin reduktase, kemudian
ditransfer ke ferodoksin. Ferodoksin kemudian mereduksi oksigenase yang
menyebabkan katalisis adisi 2 atom O dari O2 ke senyawa toluena secara
stereospesifik membentuk (+)-cis-(1S,2R)-dihidroksi-3-metilsikloheksa-3,5-diena.
Komponen oksigenase pada enzim toluena dioksigenase merupakan
heteroheksamer yang terdiri dari subunit katalitik (sub-unit α) dan subunit
struktural (sub-unit β). Sub-unit katalitik mengandung Rieske center [2Fe-2S] dan
Fe mononuklir pada sisi aktif (Friemann et al., 2009).
(a) (b)
Toluena dioksigenase bekerja pada rentang jenis substrat yang luas yaitu
berbagai jenis alkilbenzena sederhana seperti toluena, etilbenzena, maupun
dimetilbenzena, namun tetap memberikan sifat spesifisitas yaitu merubah substrat
tersebut menjadi produk dihidrodiol secara stereospesifik dan regiospesifik. Sifat
ini menjadikan toluena dioksigenase bernilai tinggi dalam proses sintesis senyawa
yang dikontrol berdasarkan sifat enantiomerik. Residu asam amino yang berperan
dalam menentukan spesifisitas substrat yaitu Ile-301, Thr-305, Ile-307, dan
Leu-309. Residu ini tidak berada pada sisi pengikatan substrat namun pada bagian
saluran yang dilalui substrat untuk menuju sisi aktif (Bagneris et al., 2005).
2.4.3. Naftalen dioksigenase
Naftalena dioksigenase merupakan enzim yang mengkatalisis reaksi tahap
pertama dalam degradasi naftalen, yaitu oksidasi naftalen menjadi (+)-cis-(1R,2S
)-dihidroksi-1,2-dihidronaftalen (naftalen cis-dihidrodiol) melalui penggunaan O2
dan NADH. Reaksi yang terjadi yaitu sebagai berikut.
Naftalena + NADH + H+ + O2→ (+)- cis-(1R,2S)-dihidroksi-1,2-dihidronaftalena
+ NAD+
(Lee, 2005).
Naftalena dioksigenase merupakan sistem enzim multikomponen yang
terdiri dari ferodoksin reduktase, ferodoksin, dan oksigenase. Elektron ditangkap
oleh NADH lalu ditransfer ke ferodoksin reduktase, kemudian ditransfer ke
ferodoksin. Ferodoksin kemudian mereduksi oksigenase yang menyebabkan
katalisis adisi 2 atom O dari O2 ke senyawa naftalena secara stereospesifik
membentuk (+)-cis-(1R,2S)-dihidroksi-1,2-dihidronaftalen (Lee, 2005).
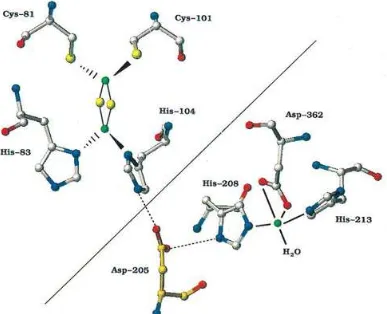
Struktur oksigenase pada sistem enzim naftalena dioksigenase juga terdiri
dari heksamer α3β3 (gambar 2.4). Tiap subunit pada komponen oksigenase mengandung Rieske center [2Fe-2S] dan sisi aktif berupa Fe yang berkoordinasi
dengan molekul air, 2 residu histidin, dan 1 residu aspartat bidentat membentuk
triad 2-His-1-Asp yang terlibat dalam aktivasi O2 dan proses katalisis. Salah satu
Fe pada Rieske center berkoordinasi dengan 2 residu sistein sedangkan Fe lainnya
Gambar 2.4. Struktur oksigenase pada sistem enzim naftalena dioksigenase. Sub-unit α berwarna ungu, hijau, dan biru. Sub-unit β berwarna ungu muda, hijau muda, dan biru muda. Atom Fe berwarna merah dan atom S berwarna kuning (Kauppi et al, 1998)
Gambar 2.5. Ikatan koordinasi pada naftalena dioksigenase. Ikatan terbentuk antara triad 2-His-1-Asp dan molekul air dengan atom Fe pada sisi aktif dan terbentuk antara 2 residu sistein dan 2 residu histidin dengan 2 atom Fe pada Rieske
Enzim naftalena dioksigenase memiliki spesifisitas substrat yang rendah
namun tetap bekerja secara regiospesifik (hanya bekerja pada posisi satu jenis
ikatan tertentu) dan stereospesifik (hanya menghasilkan salah satu jenis
stereoisomer) dalam menghasilkan produk dihidrodiol. Naftalena dioksigenase
mampu mengkatalisis reaksi oksidasi naftalena dan bifenil dalam laju yang sama,
namun mengkatalisis reaksi oksidasi fenantrena dalam laju yang lebih lambat.
Poliaromatik cincin 4 seperti krisen dan benz[a]antrasena juga dapat mengalami
reaksi oksidasi dengan dikatalisis oleh enzim nafttalena dioksigenase
menghasilkan struktur bis-cis-dihidrodiol (Parales et al., 2000; Jouanneau et al.,
2006).
2.5. Gen Penyandi Enzim Monooksigenase dan Dioksigenase
Bakteri mampu mendegradasi hidrokarbon karena bakteri memiliki gen
penyandi enzim-enzim katabolik yang berperan dalam jalur katabolisme
hidrokarbon, baik karena proses adaptasi akibat terjadinya kontaminasi maupun
muncul secara alamiah sebagai sifat bakteri. Gen katabolik ini dapat digunakan
sebagai dasar untuk mengidentifikasi dan mengevaluasi potensi isolat dalam
mendegradasi hidrokarbon dengan proses amplifikasi menggunakan metode PCR
(Chen, 2013; Mathew dan Hobani, 2015). Gen-gen katabolik penyandi enzim
hidrokarbon alifatik maupun aromatik yang telah berhasil diamplifikasi dari DNA
bakteri antara lain klaster gen alk, CYP, tod, ndo, phn, xyl, serta PAH-RHD yang
berturut-turut menyandi enzim alkana monooksigenase, sitokrom P450, toluena
dioksigenase, naftalen dioksigenase, fenantren dioksigenase, katekol
dioksigenase, serta PAH-ring hydroxylating dioxygenase (Marquez-Rocha et al.,
2005; Phillips et al., 2008; Hesham et al., 2014). Enzim pendegradasi hidrokarbon
merupakan enzim multikomponen sehingga gen struktural yang menyandi
komponen enzim tersebut tersusun dalam sebuah klaster gen (operon), baik dalam
bentuk operon polisistronik pada kromosom atau pada plasmid (Sahoo, 2010).
2.5.1. Gen penyandi alkana monooksigenase
Alkana monooksigenase untuk rantai C medium yaitu C5-C12 (AlkB) yang
dikode oleh klaster gen alkB dan alkana monooksigenase untuk rantai C panjang
yaitu C>12 (AlkM) yang dikode oleh klaster gen alkM merupakan dua jenis enzim
alkana monooksigenase yang telah berhasil dikarakterisasi. Klaster gen alkB
pertama kali diamplifikasi dari strain Pseudomonas oleovorans Gpo1 dan terdiri
dari 3 komponen enzim yaitu alkana monooksigenase (alkB), rubredoksin (alkG),
dan rubredoksin reduktase (alkT). Homolog alkB telah banyak tersebar di alam
pada sekitar 45 spesies bakteri (Chen, 2013). Klaster gen alkM pertama kali
diamplifikasi dari strain Acinetobacter sp. ADP1 dan terdiri dari gen yang
menyandi 3 komponen enzim yaitu alkana monooksigenase (alkM), rubredoksin
(rubA), dan rubredoksin reduktase (rubB). Gen alkM telah berhasil dideteksi pada
berbagai strain Acinetobacter, seperti Acinetobacter sp. M-1, A. calcoaceticus
NCIMB 8250, A. calcoaceticus EB104, Acinetobacter sp. 2769A, dan A.
calcoaceticus 69-V. Analisis filogenetik sekuens asam amino dari AlkM dan
AlkB menunjukkan tingkat diversitas sekuens yang sangat tinggi sehingga gen
alkM dapat dengan mudah dibedakan dari gen alkB. Gen alk juga memiliki
diversitas cukup tinggi pada genus bakteri yang berbeda terutama antara bakteri
Gram positif dan Gram negatif. Hal ini mengindikasikan jika probe DNA atau
primer oligonukleotida spesifik dapat didesain untuk mendeteksi dan memonitor
genotipe alkana monooksigenase secara spesifik menggunakan metode molekuler
(Phrommanich et al., 2009; Whyte et al., 2002).
2.5.2. Gen penyandi toluen dioksigenase
Gen penyandi sistem enzim toluena dioksigenase yaitu klaster gen tod
pertama kali diamplifikasi dari strain Pseudomonas putida F1. Gen tod terdiri dari
dua klaster gen (operon) yang mengandung gen struktural yang menyandi enzim
untuk proses degradasi toluen. Gen todA-C menyandi enzim toluen diksigenase
yang mengkatalisis tahap pertama reaksi degradasi toluen kemudia gen todD-E
yang menyandi enzim-enzim yang bekerja dalam tahap degradasi selanjutnya
2.5.3. Gen penyandi naftalen dioksigenase
Gen penyandi sistem enzim naftalen dioksigenase yaitu klaster gen nah
pertama kali diamplifikasi dari plasmid NAH7 dari strain Pseudomonas putida
G7. Gen nah terdiri dari dua klaster gen (operon) yang mengandung gen struktural
yang menyandi enzim untuk proses degradasi naftalen, yaitu operon nah1 (
nahA-F) dan operon nah2 (nahG-M). Operon nah1 terdiri dari gen nahA-F yang
mengkode enzim-enzim yang terlibat dalam konversi naftalen menjadi salisilat
dan operon nah2 terdiri dari gen nahG-M yang mengkode enzim-enzim yang
terlibat dalam metabolisme salisilat melalui jalur pemutusan meta menjadi piruvat
dan asetaldehid, sehingga gen penyandi enzim naftalen dioksigenase terletak pada
BAB III
KERANGKA KONSEPTUAL DAN HIPOTESIS PENELITIAN
3.1. Kerangka Konseptual
Lumpur minyak bumi (oil sludge) adalah campuran hidrokarbon, padatan,
logam dan air yang membentuk emulsi w/o yang stabil dan terdeposisi pada
bagian bawah/dasar tangki penyimpanan minyak. Oil sludge menjadi hasil
samping atau limbah dalam proses pengolahan dan pemurnian minyak bumi. Oil
sludge mengandung hidrokarbon alifatik, monoaromatik, dan poliaromatik yang
sangat berbahaya bagi lingkungan dan kesehatan sehingga perlu dilakukan
pengolahan untuk menekan atau menghilangkan kandungan berbahaya dari oil
sludge tersebut.
Degradasi oil sludge secara fisika dan kimia telah banyak digunakan dan
memiliki kelebihan yaitu efisien serta mampu menurunkan kadar polutan pada oil
sludge secara maksimal namun memiliki kelemahan yaitu tidak ekonomis
terutama jika diaplikasikan dalam skala industri. Salah satu metode yang menjadi
pilihan untuk pengolahan oil sludge adalah biodegradasi menggunakan
mikroorganisme yaitu bakteri, yeast, atau fungi. Bakteri banyak dipilih
dibandingkan yeast atau fungi karena memiliki kecepatan pertumbuhan yang
tinggi serta kemudahan untuk memperbanyak jumlah selnya. Hidrokarbon yang
didegradasi digunakan sebagai sumber karbon utama oleh bakteri dalam proses
metabolisme. Produk degradasi hidrokarbon yang memasuki siklus asam sitrat
berfungsi sebagai substrat metabolisme energi dan sebagai zat pembangun untuk
proses biosintesis sel serta proses pertumbuhan bakteri.
Salah satu bakteri yang memiliki kemampuan untuk mendegradasi oil
sludge adalah Actinobacillus sp. P3(7). Mekanisme degradasi oil sludge oleh
Actinobacillus sp. P3(7) adalah melalui sekresi biosurfaktan yang mempermudah
masuknya komponen hidrokarbon dari oil sludge ke dalam sel bakteri. Komponen
oil sludge yang telah masuk ke dalam sel kemudian dimetabolisme oleh
enzim-enzim katabolik sebagai sumber karbon, yaitu enzim-enzim monooksigenase dan
metabolisme oil sludge disandi oleh gen-gen penyandi enzim monooksigenase
dan dioksigenase. Fragmen gen ini dapat diangkat dengan proses amplifikasi
menggunakan metode PCR dengan primer spesifik sehingga dapat diketahui
estimasi ukuran fragmen gen tersebut.
3.2. Hipotesis Penelitian
1. Fragmen gen penyandi enzim monooksigenase dan dioksigenase dapat
diamplifikasi dari DNA genom Actinobacillus sp. P3(7).
2. Actinobacillus sp. P3(7) memiliki nilai aktivitas enzim monoksigenase
terhadap substrat hidrokarbon alifatik berupa heksadekana.
3. Actinobacillus sp. P3(7) memiliki nilai aktivitas enzim dioksigenase terhadap
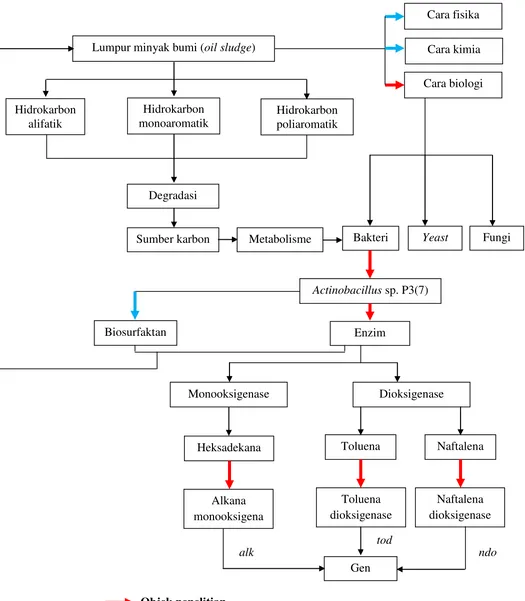
Lumpur minyak bumi (oil sludge)
Hidrokarbon alifatik
Hidrokarbon monoaromatik
Hidrokarbon poliaromatik
Cara kimia
Cara biologi
Degradasi
Sumber karbon Metabolisme Bakteri Yeast Fungi
Actinobacillus sp. P3(7)
Enzim Biosurfaktan
Monooksigenase Dioksigenase
Toluena Naftalena
Toluena dioksigenase
Naftalena dioksigenase Heksadekana
Alkana monooksigena
se
Gen
alk
tod
ndo
Gambar 3.1. Kerangka konsep penelitian Objek penelitian
BAB IV
METODE PENELITIAN
4.1. Lokasi dan Waktu Penelitian
Penelitian dilakukan di Laboratorium Biokimia Departemen Kimia Fakultas
Sains dan Teknologi Universitas Airlangga mulai bulan Januari 2016 hingga Juli
2016.
4.2. Bahan dan Alat Penelitian 4.2.1. Isolat bakteri
Bakteri yang digunakan adalah Actinobacillus sp. P3(7) hasil isolasi dari
pengeboran minyak Desa Wonocolo, Bojonegoro, Jawa Timur yang dilakukan
oleh Ni’matuzahroh et al. (2009) dan menjadi koleksi Laboratorium Mikrobiologi
Departemen Biologi FST Universitas Airlangga.
4.2.2. Bahan penelitian
Bahan yang digunakan dalam penelitian ini meliputi Nutrient Agar (NA),
Luria Bertani (LB), Sea Salt (SS), yeast extract, tripton, heksadekana, toluena,
naftalena, DMSO, GenEluteTM Bacterial Genomic DNA Kit, Tris-HCl, NADH,
primer (alk-F, alk-R, tod-F, tod-R, ndo-F, ndo-R), Q5® High-Fidelity 2X master
mix, PCR water, agarose, DNA marker Lambda DNA/HindIII, DNA marker 100
bp ladder, loading dye, Coomassie Brilliant Blue G 250, asam fosfor, etanol,
akuades, akuabides, dan etidium bromida (EtBr).
4.2.3. Alat penelitian
Peralatan yang digunakan dalam penelitian ini yaitu autoklaf, sentrifuse,
mikrosentrifuse, neraca analitik, inkubator, lemari pendingin, tabung Eppendorf,
mikropipet, sonikator, laminar air flow cabinet, pH meter, shaker incubator,
waterbath, hotplate, microplate reader, thermocycler (BioRad),
UV-transluminator, spektrofotometer UV-Vis (UV1800 Shimadzu), set alat
4.3. Diagram Alir Penelitian
Peremajaan Actinobacillus sp. P3(7) dalam media NA
Kultivasi Actinobacillus
sp. P3(7) dalam media LB cair
Kultivasi Actinobacillus sp. P3(7) dalam media Sea Salt diperkaya yeast extract dan hidrokarbon
Ekstraksi DNA genom bakteri
Elektroforesis DNA genom bakteri
Pengukuran OD600nm
setiap interval 2 hari pertumbuhan
Sentrifugasi kultur
Actinobacillus sp. P3(7) setiap interval 2
hari pertumbuhan
Amplifikasi gen penyandi enzim oksigenase dengan metode PCR menggunakan
primer spesifik
Pelet sel Supernatan
Lisis sel
Sentrifugasi
Residu Supernatan
Uji aktivitas enzim monooksigenase dengan substrat heksadekana dan dioksigenase dengan substrat toluena
dan naftalena
4.4. Cara Kerja
4.4.1. Pembuatan media
Pembuatan media untuk peremajaan isolat bakteri
Media Nutrient Agar (NA) sebanyak 0,56 gram dilarutkan dalam 20 mL
akuades, lalu dipanaskan menggunakan hotplate dan dihomogenkan
menggunakan magnetic stirrer hingga mendidih. Media dituang ke dalam 4
tabung reaksi masing-masing sebanyak 5 mL lalu disumbat dengan kapas yang
telah dibungkus dengan kain kassa steril dan ditutup dengan aluminium foil,
kemudian disterilisasi menggunakan autoklaf pada suhu 121ºC selama 20 menit.
Media didiamkan pada posisi miring dan suhu kamar sampai membeku.
Pembuatan media untuk produksi enzim
Media Sea Salt sebanyak 3,6 gram dan yeast extract sebanyak 2 gram
dilarutkan dalam 1 L akuades. Media yang telah jadi disterilisasi menggunakan
autoclave pada suhu 121ºC selama 20 menit.
Pembuatan media untuk preparasi DNA template dalam proses PCR
Media Luria Bertani sebanyak 100 mL dibuat dengan komposisi: 0,5 g
ekstrak yeast; 1 g tripton; dan 1 g NaCl. Semua bahan dilarutkan dalam 100 mL
akuades hingga homogen. Media yang telah jadi disterilisasi menggunakan
autoclave pada suhu 121ºC selama 20 menit.
4.4.2. Peremajaan mikroba
Sebanyak satu ose biakan murni Actinobacillus sp. P3(7) diinokulasikan
dengan metode gores (streak) pada media Nutrient Agar (NA) miring lalu
diinkubasi selama 24 jam pada suhu 37ºC. Peremajaan mikroba dilakukan secara
aseptik untuk menghindari kontaminasi (Ni’matuzahroh et al., 2009).
4.4.3. Kultivasi mikroba dalam media LB
Sebanyak 2 ose biakan bakteri Actinobacillus sp. P3(7) dari media NA
diinkubasi pada shaker incubator dengan kecepatan 120 rpm selama 24 jam pada
suhu 37ºC. Mikroba hasil kultivasi digunakan untuk ekstraksi DNA genom
mikroba.
4.4.4. Ekstraksi DNA genom
Proses ekstraksi DNA genom mengikuti tahapan dari GenEluteTM Bacterial
Genomic DNA Kit untuk bakteri Gram negatif. Penyiapan larutan Protease K 20
mg/mL dilakukan dengan cara menambahkan 20 mg Protease K ke dalam 1 mL
akuabides. Kultur bakteri Actinobacillus sp. P3(7) murni dibiakkan semalam pada
media LB lalu diambil sebanyak 1,5 mL dan disentrifuse dengan kecepatan
12.000-16.000 × g selama 2 menit. Supernatan dibuang sehingga didapatkan pelet
bakteri yang akan digunakan untuk ekstraksi DNA genom.
Pelet diresuspensikan ke dalam 180 μL Lysis Solution T, kemudian ditambahkan 20 μL larutan RNAse A lalu diinkubasi pada suhu ruang selama 2
menit. Sebanyak 20 μL Larutan Proteinase K ditambahkan ke dalam sampel lalu
diinkubasi pada suhu 55ºC selama 30 menit. Sebanyak 200 μL larutan Lysis Solution C (B8803) kemudian ditambahkan ke dalam sampel lalu diinkubasi pada
suhu 55ºC selama 10 menit dan siap untuk dilisis.
Preparasi kolom yang akan digunakan dalam proses ekstraksi dilakukan
dengan cara sebanyak 500 μL larutan Column Preparation ditambahkan ke dalam
masing-masing GenElute Miniprep Binding Column yang telah diset dengan
collection tube 2 mL kemudian disentrifuse dengan kecepatan 12.000 × g selama
1 menit. Eluat yang tertampung pada collection tube kemudian dibuang.
Sebanyak 200 μL etanol (95-100%) ditambahkan ke dalam lisat. Sel lisat kemudian dituang ke dalam binding column lalu disentrifuse dengan kecepatan ≥
6.500 × g selama 1 menit. Eluat yang tertampung pada collection tube kemudian
dibuang. Sebanyak 500 μL Wash Solution 1 (W0263) ditambahkan ke dalam
kolom lalu disentrifuse dengan kecepatan ≥ 6.500 × g selama 1 menit. Eluat yang tertampung pada collection tube kemudian dibuang. Sebanyak 500 μL Wash
Solution ditambahkan ke dalam kolom kemudian disentrifuse dengan kecepatan
dipastikan bebas dari etanol sebelum proses elusi DNA sehingga kolom
disentrifuse kembali dengan kecepatan 12.000-16.000 × g selama 1 menit jika
masih terdapat residu etanol. Eluat yang tertampung pada collection tube
kemudian dibuang. Collection tube diganti dengan yang berukuran 1,5 mL.
Sebanyak 100 μL Elution Solution (B6803) diteteskan tepat pada bagian tengah kolom kemudian diinkubasi pada suhu ruang selama 5 menit. Kolom lalu
disentrifuse pada kecepatan ≥ 6.500 × g selama 1 menit untuk mengelusi DNA. Sebanyak 100 μL Elution Solution (B6803) diteteskan kembali tepat pada bagian
tengah kolom kemudian diinkubasi pada suhu ruang selama 5 menit. Kolom lalu
disentrifuse pada kecepatan ≥ 6.500 × g selama 1 menit untuk mengelusi DNA. Eluat pada collection tube mengandung DNA genom murni. Penyimpanan jangka
panjang disimpan pada suhu -20ºC dan jangka pendek disimpan pada suhu 2-8ºC.
4.4.5. Penentuan kadar DNA genom
Konsentrasi dan kemurnian DNA genom ditentukan dengan metode
nano-drop, yaitu absorbansi sampel diukur menggunakan spektrofotometer pada
panjang gelombang 260 dan 280 nm. Kemurnian DNA dapat dikalkulasi
berdasarkan persamaan sebagai berikut.
𝐾𝑒𝑚𝑢𝑟𝑛𝑖𝑎𝑛𝐷𝑁𝐴=𝐴𝑏𝑠𝑜𝑟𝑏𝑎𝑛𝑠𝑖260 𝑛𝑚 𝐴𝑏𝑠𝑜𝑟𝑏𝑎𝑛𝑠𝑖280 𝑛𝑚
Nilai kemurnian pada rentang 1,75-1,9 menandakan kualitas DNA yang
bagus dengan tingkat kemurnian tinggi, sedangkan ≤ 1,75 menandakan adanya kontaminasi protein, dan ≥ 1,9 menandakan adanya kontaminasi RNA.
Konsentrasi DNA dapat dikalkulasi menggunakan persamaan:
𝐾𝑢𝑎𝑛𝑡𝑖𝑡𝑎𝑠𝐷𝑁𝐴 (𝑛𝑔/𝜇𝐿) =𝐴𝑏𝑠𝑜𝑟𝑏𝑎𝑛𝑠𝑖260 𝑛𝑚 × 50 ×𝑓𝑎𝑘𝑡𝑜𝑟𝑝𝑒𝑛𝑔𝑒𝑛𝑐𝑒𝑟𝑎𝑛 1000
4.4.6. Amplifikasi gen penyandi enzim monooksigenase dan dioksigenase
Proses amplifikasi dilakukan berdasarkan prosedur standar PCR
menggunakan thermal cycler. Volume reaksi yang digunakan sebesar 25 μL yang
DNA genom sebagai template, 12,5 μL Q5® High-Fidelity 2X master mix (yang
terdiri dari campuran dNTP, DNA polimerase, dan Mg2+) dan 9 μL PCR water.
Campuran dihomogenkan kemudian dimasukkan ke mesin PCR. Reaksi PCR
dilakukan dengan kondisi: 98ºC selama 30 detik, (98ºC selama 10 detik, 55ºC
selama 20 detik, 72ºC selama 45 detik) sebanyak 35 siklus, dan pada 72ºC selama
2 menit, lalu hold pada suhu 4ºC.
Tabel 4.1. Primer yang digunakan untuk amplifikasi fragmen gen (Marquez-Rocha et al., 2005)
Primer Sekuens Primer (5’→3’) Ukuran Fragmen (bp)
alkM-F
Pre-denaturasi 98°C 30 detik
Denaturasi
akuabides hingga volume akhir mencapai 1 L. Larutan kemudian diencerkan
dengan menambahkan 2 mL buffer TAE 50× ke dalam 98 mL akuabides sehingga
volume total larutan adalah 100 mL dan didapatkan larutan buffer TAE 1×.
Metode elektroforesis
Gel agarose 1% dibuat dengan cara sebanyak 0,35 g serbuk agarose
dimasukkan ke dalam Erlenmeyer 250 mL kemudian ditambahkan 35 mL buffer
TAE 1× (Tris-asetat 40 mM, asam asetat 20 mM, dan Na2EDTA 1 mM pH 8,6)
lalu dipanaskan di atas hot plate hingga larut. Larutan kemudian didinginkan
hingga suhu ± 45°C lalu dituang pada cetakan agar dan didiamkan hingga gel
memadat. Sebanyak 1 μL loading dye dicampur dengan 5 μL DNA sampel lalu
dimasukkan ke dalam sumuran pada gel. Sebanyak 5 μL DNA marker
dimasukkan pada sumuran terpisah. Elektroforesis dilakukan dalam buffer TAE
1x pada tegangan 70 V dan dihentikan ketika bromophenol blue dalam loading
dye telah bermigrasi sepanjang 2/3 dari panjang gel. Gel kemudian direndam
dalam larutan EtBr 250 μg/mL selama 15 menit dan dibilas dalam akuades selama 2 menit kemudian diamati pendarannya menggunakan UV-transluminator dan
difoto menggunakan kamera.
4.4.8. Kultivasi mikroba dalam media Sea Salt diperkaya yeast extract dan substrat hidrokarbon
Sebanyak 5% (v/v) suspensi sel dengan OD600nm = 0,5 dari media NA
dimasukkan ke dalam masing-masing Erlenmeyer 250 mL yang berisi 47,5 mL
media Sea Salt diperkaya yeast extract dan 1% heksadekana. Campuran
diinkubasi pada shaker incubator dengan kecepatan 100 rpm selama 14 hari pada
suhu 37ºC. Prosedur yang sama dilakukan dengan penambahan substrat berupa
100 ppm toluena dan 200 ppm naftalena. Erlenmeyer tanpa penambahan
hidrokarbon juga diinkubasi dalam kondisi yang sama sebagai kontrol
4.4.9. Pengukuran OD600nm mikroba
Pertumbuhan bakteri selama proses kultivasi berlangsung diukur dengan
cara mengambil sebanyak 4 mL suspensi sel dan OD suspensi diukur
menggunakan spektrofotometer UV-Vis pada λ 600 nm. Hal ini dilakukan setiap
interval dua hari.
4.4.10.Uji aktivitas enzim oksigenase
Pembuatan larutan stok buffer 20 mM Tris-HCl
Sebanyak 3,79 g Tris dilarutkan dalam 800 mL akuabides dan pH larutan
diset hingga pH 7 melalui penambahan HCl pekat, kemudian ditambahkan
akuabides hingga volume larutan mencapai 1000 mL sehingga didapatkan larutan
buffer 31,25 mM Tris-HCl sebagai larutan stok. Buffer ini kemudian diencerkan
untuk didapatkan buffer Tris-HCl 20 mM.
Panen bakteri
Sel dipanen dengan cara disentrifuse pada kecepatan 5000 rpm. Supernatan
dipisahkan dan pelet dicuci dua kali dengan 1 mL buffer Tris-HCl 20 mM pH 7,4.
Pelet kemudian diresuspensikan dalam 500 μL buffer Tris-HCl 20 mM pH 7,4.
Pelet lalu disonikasi menggunakan ultrasonic disintegrator dengan diameter
probe 3 mm, daya 80%, dan dalam interval 30 s on serta 15 s off selama 4 menit.
Hasil sonikasi lalu disentrifugasi selama 10 menit pada kecepatan 8000 rpm pada
suhu 4ºC. Supernatan yang diperoleh kemudian digunakan untuk uji aktivitas
enzim dan ditentukan kadar protein sel dengan metode Bradford (1976).
Penentuan kadar protein
Kadar protein ditentukan dengan metode Bradford (1978). Sebanyak 0,1 g
Coomassie Brilliant Blue G 250 dilarutkan dalam 50 mL etanol 95% (v/v),
kemudian ditambahkan 100 mL asam fosfor 85%, dan ditambahkan akuabides
hingga volume larutan mencapai 250 mL, lalu dihomogenkan dan disaring.
Larutan stok bovine serum albumin (BSA) 500 μg/mL dibuat dengan cara
melarutkan 0,005 g BSA dalam 10 mL akuabides. Larutan standar BSA dibuat
dengan cara sebanyak 0,2 mL; 0,4 mL; 0,8 mL; 1,2 mL; 1,6 mL; dan 2 mL larutan
stok BSA 500 μg/mL masing-masing dimasukkan ke dalam labu ukur 10 mL dan
diencerkan dengan akuabides hingga tanda batas volume sehingga didapatkan
larutan standar BSA dengan konsentrasi 10 μg/mL; 20 μg/mL; 40 μg/mL; 60
μg/mL; 80 μg/mL; dan 100 μg/mL. Masing-masing variasi konsentrasi larutan standar dipipet sebanyak 0,08 mL kemudian ditambahkan larutan Bradford
sebanyak 4 mL, lalu divorteks dan diinkubasi pada suhu ruang selama 2 menit.
Larutan standar BSA dengan variasi konsentrasi 10 μg/mL; 20 μg/mL; 40
μg/mL; 60 μg/mL; 80 μg/mL; dan 100 μg/mL dibaca absorbansinya menggunakan
spektrofotometer UV-Vis pada panjang gelombang 595 nm. Perlakuan diulang
sebanyak 2 kali untuk masing-masing variasi konsentrasi. Hasil absorbansi
kemudian dibuat kurva kalibrasi hubungan antara konsentrasi vs absorbansi
dengan persamaan garis linier y = mx + C. Koefisien y pada persamaan garis
menyatakan nilai absorbansi, sedangkan koefisien x menyatakan besarnya
konsentrasi larutan.
Sampel crude enzyme sebanyak 0,08 mL ditambahkan larutan Bradford
sebanyak 4 mL, lalu divorteks dan diinkubasi pada suhu ruang selama 2 menit.
Sampel dibaca absorbansinya menggunakan spektrofotometer UV-Vis pada
panjang gelombang 595 nm. Blanko menggunakan 0,08 mL akuabides yang
ditambahkan larutan Bradford sebanyak 4 mL.
Tabel 4.3. Komposisi penentuan kadar protein dengan metode Bradford (1978)
Komposisi Blanko Larutan Standar Sampel Uji
- 0,08 mL 0,08 mL
Akuabides 0,08 mL - -
Larutan Bradford 4 mL 4 mL 4 mL
Uji aktivitas enzim monooksigenase
Supernatan yang didapatkan digunakan untuk uji aktivitas enzim alkana
monooksigenase. Campuran reaksi mengandung buffer Tris-HCl 20 mM; NADH
0,1 mM; larutan heksadekana (1% heksadekana dalam 80% DMSO), serta ekstrak
enzim kasar. Reaksi dimulai dengan menambahkan 2 μL larutan heksadekana ke
dalam campuran reaksi. Campuran kemudian dihomogenkan menggunakan
vorteks selama 3 detik dan diinkubasi selama 6 menit. Absorbansi campuran
diukur menggunakan microplate reader pada panjang gelombang 340 nm. Nilai
absorbansi yang diperoleh selanjutnya digunakan untuk menentukan besarnya
aktivitas enzim (Jauhari et al., 2014; Mishra et al., 2014; Singh et al., 2013;
Mishra dan Singh, 2012).
Uji aktivitas enzim dioksigenase
Supernatan yang didapatkan digunakan untuk uji aktivitas enzim toluen
dioksigenase. Campuran reaksi mengandung buffer Tris-HCl 20 mM; NADH 0,1
mM; larutan toluena (1% toluena dalam 80% DMSO), serta ekstrak enzim kasar.
Reaksi dimulai dengan menambahkan 2 μL larutan toluena ke dalam campuran reaksi. Campuran kemudian dihomogenkan menggunakan vorteks selama 3 detik
dan diinkubasi selama 6 menit. Absorbansi campuran diukur menggunakan
microplate reader pada panjang gelombang 340 nm. Nilai absorbansi yang
diperoleh selanjutnya digunakan untuk menentukan besarnya aktivitas enzim. Hal
yang sama juga dilakukan untuk uji aktivitas enzim naftalena dioksigenase
menggunakan substrat naftalena (Jauhari et al., 2014; Mishra et al., 2014; Singh et
Tabel 4.4. Komposisi uji aktivitas enzim oksigenase (Jauhari et al., 2014; Mishra et al., 2014; Singh et al., 2013; Mishra dan Singh, 2012)
Komposisi Uji Blanko NADH awal Sampel Uji
Buffer Tri-HCl 20 mM 181,3 μL 181,3 μL 181,3 μL
NADH 0,1 mM - 6,7 μL 6,7 μL
Crude enzyme 10 μL 10 μL 10 μL
Larutan substrat 1% 2 μL - 2 μL
Akuabides 6,7 μL 2 μL -
Volume total 200 μL 200 μL 200 μL
Analisis data uji aktivitas enzim
Satu unit aktivitas enzim dinyatakan sebagai banyaknya enzim yang
membutuhkan 1 μmol NADH untuk mengoksidasi substrat per menit per mL
enzim. Besarnya aktivitas enzim ditentukan dari nilai absorbansi yang didapat
setelah pengukuran campuran reaksi enzimatis menggunakan microplate reader
dengan persamaan sebagai berikut:
𝐴𝑘𝑡𝑖𝑣𝑖𝑡𝑎𝑠𝑒𝑛𝑧𝑖𝑚 (𝑈/𝑚𝐿) = ∆𝐴340×𝑉𝑒 𝑚𝐿
𝑎340 𝑚𝐿𝜇𝑚𝑜𝑙−1𝑐𝑚−1 ×𝑉𝑠 𝑚𝐿 ×𝑡 𝑚𝑒𝑛𝑖𝑡 ×𝑙 𝑐𝑚
dengan:
a340 : absortivitas molarNADH, sebesar 6,22 mL μmol-1 cm-1
Ve : volume enzim, sebesar 1 mL
Vs : volume sampel enzim, sebesar 0,01 mL
t : waktu inkubasi, sebesar 5 menit
l : pathlength, sebesar 0,05 cm
BAB V
HASIL PENELITIAN DAN PEMBAHASAN
5.1.Ekstraksi DNA Genom Actinobacillus sp. P3(7)
Isolat ditumbuhkan pada media Luria Bertani yang termasuk dalam media
kompleks (undefined medium), yang berarti jenis dan kuantitas spesifik dari
senyawa penyusunnya tidak diketahui secara pasti. Dua komponen media LB
yaitu tripton dan yeast extract merupakan campuran kompleks dari
senyawa-senyawa yang tidak diketahui secara spesifik. Tripton merupakan sumber asam
amino dan peptida, sedangkan yeast extract (sediaan kering dari hasil digesi sel
yeast) merupakan sumber nitrogen, gula, nutrien organik, dan anorganik. Media
kompleks seperti LB tidak membutuhkan suplemen tambahan dan mendukung
pertumbuhan berbagai jenis spesies bakteri, salah satunya yaitu Actinobacillus sp.
P3(7) (Brown, 2010).
Proses ekstraksi DNA genom mengikuti tahapan dari GenEluteTM Bacterial
Genomic DNA Kit untuk bakteri Gram negatif dan didapatkan DNA genom
Actinobacillus sp. P3(7) dengan kemurnian sebesar 1,82 dan konsentrasi sebesar
130,88 ng/μL. Hasil visualisasi menggunakan elektroforesis menunjukkan adanya
pita tunggal DNA dengan ukuran sekitar 20 kb, yang menandakan kualitas DNA
yang bagus dengan tingkat kemurnian tinggi. DNA genom yang didapat kemudian
digunakan dalam proses amplifikasi gen katabolik.
Gambar 5.1. Hasil elektroforesis DNA genom Actinobacillus sp. P3(7); kiri: Marker Lambda DNA/HindIII
23130 bp
9416 bp 20 kb
5.2.Amplifikasi Gen Katabolik pada Actinobacillus sp. P3(7)
Amplifikasi gen katabolik dilakukan menggunakan primer spesifik
kemudian divisualisasikan menggunakan elektroforesis dengan pewarnaan EtBr.
Thermal gradient PCR dengan variasi Ta sebesar 50,0ºC; 53,2ºC; 55,6ºC; 58,0ºC;
60,0ºC; 62,4ºC; 65,2ºC; dan 68,1ºC dilakukan untuk mengetahui suhu annealing
(Ta) optimum dari setiap pasang primer yang digunakan sehingga proses
hibridisasi primer dapat berlangsung spesifik untuk mengamplifikasi sekuens gen
target.
alkM tod
ndo
Gambar 5.2. Hasil amplifikasi dengan berbagai variasi Ta, yaitu 50,0ºC; 53,2ºC; 55,6ºC;
58,0ºC; 60,0ºC; 62,4ºC; 65,2ºC; dan 68,1ºC; kanan: Marker DNA ladder 100 bp
100 bp 500 bp 1000 bp 100 bp 500 bp 1000 bp