ANALISIS CAMPURAN PARASETAMOL, SALISILAMIDA, DAN KAFEIN DALAM TABLET
SECARA KROMATOGRAFI CAIR KINERJA TINGGI (KCKT)
SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S. Farm.)
Program Studi Ilmu Farmasi
Oleh : Lanny Setyawati NIM : 038114043
FAKULTAS FARMASI
UNIVERSITAS SANATA DHARMA YOGYAKARTA
ANALISIS CAMPURAN PARASETAMOL, SALISILAMIDA, DAN KAFEIN DALAM TABLET
SECARA KROMATOGRAFI CAIR KINERJA TINGGI (KCKT)
SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S. Farm.)
Program Studi Ilmu Farmasi
Oleh : Lanny Setyawati NIM : 038114043
FAKULTAS FARMASI
UNIVERSITAS SANATA DHARMA YOGYAKARTA
2007
Skripsi berjudul
ANALISIS CAMPURAN PARASETAMOL, SALISILAMIDA, DAN KAFEIN DALAM TABLET
SECARA KROMATOGRAFI CAIR KINERJA TINGGI (KCKT)
oleh : Lanny Setyawati NIM : 038114043
telah disetujui oleh :
Pembimbing
Christine Patramurti, M. Si., Apt. tanggal
”Bagi Dialah, yang dapat
melakukan jauh lebih banyak
dar i pada yang kit a doakan at au
pikir kan, seper t i yang t er nyat a
dar i kuasa yang beker ja di
dalam kit a, bagi Dialah
kemuliaan di dalam jemaat dan
di dalam Kr ist us Yesus t ur un
t emur un sampai
selama-lamanya. A min. (Efesus 3 :
20-21)”
Kupersembahkan karyaku ini kepada :
Papa dan mama yang selalu ada bersamaku dan
menyayangiku,
Koko yang selalu mendukungku,
Sahabat-sahabatku yang berbagi suka duka bersama
Serta almamaterku yang selalu kukenang
PRAKATA
Puji syukur penulis panjatkan kepada Allah Bapa di Surga, hanya karena berkat dan kasih-Nyalah maka skripsi ini dapat diselesaikan oleh penulis. Skripsi ini disusun untuk memenuhi salah satu syarat memperoleh gelar Sarjana Farmasi (S. Farm.) di Fakultas Farmasi Universitas Sanata Dharma Yogyakarta.
Selama penyusunan skripsi ini, banyak pihak yang telah membantu penulis dalam menyelesaikannya, maka pada kesempatan ini penulis mengucapkan terima kasih kepada :
1. Ibu Rita Suhadi, M. Si., Apt. selaku Dekan Fakultas Farmasi Universitas Sanata Dharma Yogyakarta.
2. Ibu Christine Patramurti, M. Si., Apt. selaku pembimbing yang telah banyak meluangkan waktunya dalam memberikan arahan dan dorongan semangatnya selama penyusunan skripsi ini.
3. Drs. Sulasmono, Apt. dan Dra. M. M. Yetty Tjandrawati, M. Si. selaku dosen penguji yang telah memberikan kritik dan saran untuk skripsi ini.
4. Seluruh staf laboratorium kimia : Pak Mukmin, Pak Prapto, Mas Parlan, dan Mas Kunto yang telah membantu penulis selama penelitian di laboratorium. 5. Natalia, sahabat dari awal kuliah sampai saat ini, makasih atas dukungan,
semangat, kebersamaan, dan doanya.
6. Mellissa, Niken, Lee-a, dan Anita, makasih atas kebersamaan, doa dan waktu yang telah dijalani bersama selama ini.
7. Teman-teman kost ‘Dewi’ : Indah, Yohana, Nophie, Renny, Eunike, C’ Maria, C’ Meta, C’ Ricka, C’ Listy, Aning, Ratih, Selvi, Chika, serta teman kost yang lain, makasih untuk bantuan, kebersamaan, canda tawa, dan doanya.
8. Teman-teman persekutuan Magelang : Jo-john, C’ Dewi , DePe, Albert, C’ Ika, koh Paul, dll, makasih atas doa dan dukungannya.
9. Nanda, mbak Dinta dan C’ Yiyin, makasih atas diskusi dan bantuannya.
10. Teman-teman di kelas A angkatan 2003 khususnya kelompok praktikum B, makasih atas kebersamaannya selama ini.
11. Tiap orang yang pernah datang dan pergi dalam hidupku, yang memberi warna dalam hidupku dan menjadikanku sebagai pribadi seperti sekarang ini.
Penulis menyadari bahwa skripsi yang disusun ini masih banyak memiliki kekurangan. Oleh karena itu penulis mengharapkan kritik dan saran untuk perbaikan dan perkembangan selanjutnya. Akhir kata, semoga skripsi ini berguna bagi kemajuan ilmu pengetahuan.
Penulis
PERNYATAAN KEASLIAN KARYA
Saya menyatakan dengan sesungguhnya bahwa skripsi yang saya tulis ini tidak memuat karya atau bagian karya orang lain, kecuali yang telah disebutkan dalam kutipan dan daftar pustaka, sebagaimana layaknya karya ilmiah.
Yogyakarta, Februari 2007 Penulis
Lanny Setyawati
INTISARI
Saat ini banyak beredar obat analgesik antipiretik dengan bahan aktif yang digunakan adalah kombinasi dari beberapa zat aktif. Salah satu kombinasi yang ada di pasaran adalah kombinasi parasetamol, salisilamida, dan kafein. Penetapan ketiga zat aktif ini masih dilakukan dengan KLT densitometri yang mana ketiga zat aktif ini harus dipisahkan terlebih dahulu. Hal tersebut yang mendasari penelitian mengenai penetapan kadar ketiga zat aktif tersebut dalam tablet tanpa pemisahan masing-masing zat aktif tersebut. Tujuan dari penelitian ini adalah untuk mengetahui apakah metode KCKT dapat digunakan dalam penetapan kadar parasetamol, salisilamida, dan kafein dalam tablet dan juga mengetahui apakah kadar ketiga zat aktif tersebut sesuai dengan yang tertera pada etiket.
Penelitian ini merupakan penelitian non eksperimental deskriptif, menggunakan metode Kromatografi Cair Kinerja Tinggi (KCKT) fase terbalik dengan kolom C18, fase gerak metanol:aquabidest:amonia (70:29:1), kecepatan
alir 1,5 ml/menit, dan detektor UV pada panjang gelombang 270 nm.
Dari hasil penelitian ini didapatkan bahwa metode KCKT dapat digunakan untuk menetapkan kadar parasetamol, salisilamida, dan kafein dalam tablet. Kadar masing-masing senyawa dalam tablet sesuai dengan yang tertera pada etiket, yaitu untuk parasetamol, salisilamida, dan kafein berturut-turut adalah (194,3 ± 2,35) mg/tablet, (196,66 ± 3,24) mg/tablet, dan (51,15 ± 0,34) mg/tablet.
Kata kunci : parasetamol, salisilamida, kafein, KCKT fase terbalik
ABSTRACT
Nowadays, analgesic antipyretic medicine which uses the combination of some active substance as its substance revolves a lot. One of the combination that is exist in the market is the combination of paracetamol, salicylamide, and caffeine. The determination of the concentration of those three active substances is still be done with thin layer chromatography-densitometry in which these three active substances have to be separated first. It becomes the basic of this research. the aims of this research are to know whether HPLC method can be used in determining the concentration of paracetamol, salicylamide, and caffeine in tablet and to know whether the concentration of these three active substances suitable with the one which is stamped in the etiquette.
This research is descriptive non experimental research. This research uses reversed phase HPLC with C18 column, mobile phase of methanol : aquabidest : ammonia (70:29:1), flow rate 1,5 ml/min, and ultraviolet detector in 270 nm.
From the result of the research, it was found that HPLC method can be used to determine the concentration of paracetamol, salicylamide, and caffeine in tablet. The concentration of each compound in tablet should be suitable with the etiquette, that are (194,3 ± 2,35) mg/tablet for paracetamol, (196,66 ± 3,24) mg/tablet for salicylamide, and (51,15 ± 0,34) mg/tablet for caffeine.
Keywords : paracetamol, salicylamide, caffeine, reversed phase HPLC
DAFTAR ISI
HALAMAN JUDUL... ii
HALAMAN PERSETUJUAN PEMBIMBING ... iii
HALAMAN PENGESAHAN... iv
HALAMAN PERSEMBAHAN ... v
PRAKATA... vi
PERNYATAAN KEASLIAN KARYA ... viii
INTISARI... ix
ABSTRACT... x
DAFTAR ISI... xi
DAFTAR TABEL... xiv
DAFTAR GAMBAR ... xv
DAFTAR LAMPIRAN... xvii
BAB I. PENGANTAR ... 1
A. Latar Belakang ... 1
1. Permasalahan ... 2
2. Keaslian penelitian ... 3
3. Manfaat penelitian... 3
B. Tujuan Penelitian ... 4
BAB II. PENELAAHAN PUSTAKA... 5
A. Tablet ... 5
B. Parasetamol ... 6
C. Salisilamida ... 8
D. Kafein... 9
E. Spektrofotometri UV... 11
F. KCKT... 13
1. Definisi dan instrumentasi... 13
2. Pembagian jenis kromatografi ... 17
3. Kromatografi partisi ... 19
4. Pemisahan puncak dalam kromatografi ... 21
5. Analisis kualitatif dan kuantitatif ... 27
G. Keterangan Empiris... 28
BAB III. METODOLOGI PENELITIAN ... 29
A. Jenis dan Rancangan Penelitian ... 29
B. Definisi Operasional ... 29
C. Bahan ... 29
D. Alat... 30
E. Tata Cara Penelitian ... 31
1. Pembuatan larutan baku parasetamol... 31
2. Pembuatan larutan baku salisilamida ... 31
3. Pembuatan larutan baku kafein ... 32
4. Pembuatan fase gerak... 32
5. Optimasi metode KCKT ... 32
6. Pembuatan larutan sampel... 33 7. Penetapan kadar parasetamol, salisilamida, dan kafein dalam
sampel ... 33
F. Analisis Hasil ... 34
BAB IV. HASIL DAN PEMBAHASAN ... 35
A. Pemilihan dan Penyiapan Sampel ... 35
B. Penyiapan Fase Gerak ... 36
C. Pembuatan Larutan Baku ... 36
D. Optimasi Metode... 37
1. Penentuan panjang gelombang overlapping... 37
2. Pembuatan kurva baku parasetamol, salisilamida, dan kafein... 44
E. Analisis Kualitatif ... 50
F. Analisis Kuantitatif ... 51
BAB V. KESIMPULAN DAN SARAN... 53
A. Kesimpulan ... 53
B. Saran... 53
DAFTAR PUSTAKA ... 54
LAMPIRAN... 57
BIOGRAFI PENULIS ... 85
DAFTAR TABEL
Tabel I. Nilai indeks polaritas pelarut ... 16
Tabel II. Data kurva baku parasetamol ... 48
Tabel II. Data kurva baku salisilamida ... 49
Tabel III. Data kurva baku kafein ... 49
Tabel IV. Data waktu retensi (tR) masing-masing senyawa baku dan dalam sampel ... 51
Tabel V. Data kadar parasetamol, salisilamida, dan kafein dalam tablet .. 52
DAFTAR GAMBAR
Gambar 1. Struktur parasetamol ... 7
Gambar 2. Struktur salisilamida ... 8
Gambar 3. Struktur kafein ... 9
Gambar 4. Peralatan KCKT ... 14
Gambar 5. Mekanisme pemisahan kromatografi partisi ... 19
Gambar 6. Reaksi silanisasi ... 20
Gambar 7. Reaksi pembuatan kolom oktadesilsilan ... 20
Gambar 8. Pemisahan dua senyawa... 21
Gambar 9. Difusi eddy ... 24
Gambar 10. Transfer massa fase diam ... 25
Gambar 11. Transfer massa fase gerak ... 25
Gambar 12. Penentuan peak asymetry dan peak tailing factors... 26
Gambar 13. Distribusi analit dalam fase gerak dan fase diam... 26
Gambar 14. Gugus kromofor dan auksokrom parasetamol ... 38
Gambar 15. Gugus kromofor dan auksokrom salisilamida ... 39
Gambar 16. Gugus kromofor kafein ... 39
Gambar 17. Spektrum serapan parasetamol... 40
Gambar 18. Spektrum serapan salisilamida... 41
Gambar 19. Spektrum serapan kafein ... 42
Gambar 20. Gabungan spektrum serapan parasetamol, salisilamida, dan kafein... 43
Gambar 21. Kromatogram campuran baku parasetamol, salisilamida, dan
kafein ... 45
Gambar 22. Gugus non polar pada parasetamol, salisilamida, dan kafein ... 46
Gambar 23. Reaksi penggaraman parasetamol dengan adanya amonia ... 47
Gambar 24. Reaksi penggaraman salisilamida dengan adanya amonia... 47
Gambar 25. Kromatogram parasetamol, salisilamida, dan kafein dalam tablet ... 50
DAFTAR LAMPIRAN
Lampiran 1. Sertifikat analisis parasetamol... 58
Lampiran 2. Sertifikat analisis salisilamida... 59
Lampiran 3. Sertifikat analisis kafein... 60
Lampiran 4. Data penimbangan bahan ... 61
Lampiran 5. Kromatogram baku parasetamol ... 62
Lampiran 6. Kromatogram baku salisilamida ... 67
Lampiran 7. Kromatogram baku kafein... 72
Lampiran 8. Kromatogram parasetamol, salisilamida, dan kafein dalam sampel ... 77
Lampiran 9. Contoh perhitungan kadar larutan baku parasetamol... 78
Lampiran 10. Contoh perhitungan kadar larutan baku salisilamida ... 79
Lampiran 11. Contoh perhitungan kadar larutan baku kafein ... 80
Lampiran 12. Contoh perhitungan kadar parasetamol, salisilamida, dan kafein dalam sampel ... 81
Lampiran 13. Perhitungan CV parasetamol, salisilamida, dan kafein dalam sampel ... 84
BAB I PENGANTAR
A. Latar Belakang
Saat ini, sediaan obat analgesik yang banyak beredar merupakan bentuk sediaan obat dengan kombinasi beberapa zat aktif. Kombinasi ini ditujukan untuk memperoleh efek terapetik yang lebih baik. Salah satu kombinasi yang digunakan adalah parasetamol, salisilamida, dan kafein. Kombinasi ini berkhasiat sebagai analgesik antipiretik dalam obat seperti tablet Anaflu, tablet Anarin, tablet Cold, tablet Corexin, dan tablet Refagan. Pada penelitian ini digunakan produk obat dengan perbandingan parasetamol : salisilamida : kafein adalah 4 : 4 : 1.
Dalam sediaan obat yang mengandung parasetamol, salisilamida, dan kafein diperlukan suatu metode yang dapat diacu dan digunakan untuk penetapan kadar ketiga senyawa tersebut. Suatu metode analisis yang tepat menjadi sangat penting karena sebagai alat untuk mengontrol suatu sediaan obat memenuhi persyaratan atau tidak yang dalam hal ini adalah jumlah zat aktif yang sesuai dengan yang tertera pada etiket. Penetapan kadar ketiga zat aktif tersebut dalam tablet masih dilakukan dengan metode KLT densitometri (Anonim, 1998). Dengan metode ini masih perlu dilakukan pemisahan senyawa untuk dapat ditetapkan kadar bahan aktifnya. Hal inilah yang mendasari penulis untuk mencoba menggunakan metode lain dalam penetapan kadar ketiga zat aktif tersebut dalam tablet. Selain itu, belum adanya metode resmi yang dapat diacu
untuk penetapan kadar ketiga zat aktif tersebut dalam tablet secara simultan juga merupakan masalah yang harus dipikirkan.
Pada penetapan kadar parasetamol, salisilamida, dan kafein dalam tablet ini digunakan metode KCKT (Kromatografi Cair Kinerja Tinggi). Pemilihan metode KCKT karena metode KCKT merupakan metode analisis kualitatif dan kuantitatif yang dapat digunakan untuk analisis senyawa multikomponen dalam sampel yang berupa campuran (Johnson dan Stevenson, 1978).
Pada penelitian ini, penulis mengacu dari penelitian yang telah dilakukan oleh Sugianto (2007) mengenai optimasi pemisahan campuran parasetamol, salisilamida, dan kafein dengan metode KCKT. Pada penelitian tersebut telah didapatkan bahwa metode KCKT yang digunakan telah teruji memiliki validitas yang baik dalam penetapan kadar campuran parasetamol, salisilamida, dan kafein. Hal itu yang mendasari penulis untuk melakukan penelitian mengenai penetapan kadar parasetamol, salisilamida, dan kafein dalam produk obat yang dalam hal ini adalah tablet.
1. Permasalahan
Berdasarkan latar belakang yang ada, dapat dirumuskan permasalahan sebagai berikut :
a. apakah metode KCKT dapat digunakan untuk menetapkan kadar parasetamol, salisilamida, dan kafein dalam tablet ?
2. Keaslian penelitian
Metode Kromatografi Cair Kinerja Tinggi (KCKT) fase terbalik telah banyak dilakukan untuk menetapkan kadar dalam campuran obat. Penelitian mengenai campuran parasetamol, salisilamida, dan kafein telah dilakukan dengan metode spektrofotometri derivativ baik menggunakan aplikasi metode peak to peak (Friamata, 2006) maupun zero-crossing (Wulandari, 2006). Namun metode HPLC belum pernah dilakukan untuk penetapan kadar parasetamol, salisilamida, dan kafein dalam tablet. Penelitian ini mengacu pada penelitian dari Sugianto (2007) mengenai optimasi pemisahan campuran parasetamol, salisilamida, dan kafein dengan metode KCKT.
3. Manfaat penelitian
Penelitian ini dapat bermanfaat sebagai berikut :
a. manfaat praktis. Penelitian ini diharapkan dapat mengetahui apakah kadar parasetamol, salisilamida, dan kafein dalam tablet sesuai dengan yang tertera pada etiket.
B. Tujuan Penelitian
Berdasarkan latar belakang dan permasalahan yang ada, maka penelitian ini bertujuan untuk :
1. mengetahui apakah metode KCKT dapat digunakan untuk menetapkan kadar parasetamol, salisilamida, dan kafein dalam tablet.
BAB II
PENELAAHAN PUSTAKA
A. Tablet
Dalam Farmakope Indonesia (1995) menyebutkan definisi dari tablet
adalah suatu sediaan padat mengandung bahan obat dengan atau tanpa bahan
pengisi. Tablet adalah sediaan obat padat takaran tunggal yang dicetak dari serbuk
kering, kristal, atau granulat, yang umumnya dengan penambahan bahan
pembantu yang pembuatannya menggunakan mesin yang sesuai dengan tekanan
yang tinggi. Tablet merupakan bentuk sediaan yang banyak digunakan saat ini.
Keuntungan dari bentuk tablet antara lain adalah relatif murah dan relatif mudah
digunakan pada masyarakat (Voigt, 1984).
Tablet dapat berbeda-beda dalam ukuran, bentuk, berat, kekerasan,
ketebalan, daya hancurnya, dan dalam aspek lainnya tergantung pada cara
pemakaian tablet dan metode pembuatannya. Kebanyakan tablet digunakan pada
pemberian obat secara oral, dan kebanyakan dari tablet ini dibuat dengan
penambahan zat warna, zat pemberi rasa, dan lapisan-lapisan dalam berbagai
jenis. Tablet lain yang penggunaannya dengan cara sublingual, bukal, atau melalui
vaginal, tidak boleh mengandung bahan tambahan seperti pada tablet yang
digunakan secara oral (Ansel, 1985).
Kualitas tablet dapat dipantau dari evaluasi sifat fisik tablet (Aulton dan
Summer, 1994), meliputi :
1. Keseragaman kandungan. Salah satu syarat sediaan obat adalah harus
memiliki sifat kandungan yang konstan dalam tiap takarannya. Sediaan farmasi
berbentuk tablet harus memenuhi uji keragaman bobot untuk menggambarkan
keseragaman kandungan zat aktif yang terkandung di dalam tiap tabletnya (Aulton
dan Summer, 1994). Namun keragaman bobot tidak dapat menggambarkan
keseragaman dosis jika sediaan obat tersebut mengandung bahan aktif dengan
jumlah kurang dari 50% dari berat tablet atau sediaan obat yang mengandung
bahan aktif kurang dari 50 mg (Anonim, 2005).
2. Disintegrasi atau waktu hancur. Tablet dinyatakan hancur jika mereka terlarut
atau hancur menjadi partikel dalam suatu medium penguji yaitu air bersuhu
tertentu (misal 37°C) (Voigt, 1984).
3. Kekerasan. Dikehendaki tablet yang cukup keras agar tablet tidak pecah saat
pengemasan dan distribusi. Namun tidak terlalu keras agar tablet dapat hancur dan
menimbulkan efek.
4. Kerapuhan. Benturan-benturan pada proses pengemasan dan pengangkutan
tidak cukup kuat untuk memecahkan tablet, tetapi dapat menghilangkan beberapa
partikel obat dari permukaan tablet (Aulton dan Summer, 1994).
B. Parasetamol
Parasetamol yang memiliki nama lain asetaminofen atau
4’-hidroksiasetanilida memiliki bobot molekul sebesar 151,16 (Anonim, 1995).
HN
OH C O
CH3
Gambar 1. Struktur Parasetamol (Anonim, 1995)
Parasetamol mengandung tidak kurang dari 98,0% dan tidak lebih dari
101,0% C8H9NO2, dihitung terhadap zat anhidrat. Parasetamol merupakan serbuk
hablur berwarna putih, tidak berbau, dan berasa sedikit pahit (Anonim, 1995).
Parasetamol mempunyai titik lebur antara 169°C dan 172°C. Satu bagian
parasetamol larut dalam 70 bagian air, 20 bagian air panas, 7 bagian etanol, dan
50 bagian kloroform. Parasetamol tidak larut dalam eter (Clarke, 1969).
Parasetamol memiliki serapan maksimum pada daerah ultraviolet.
Parasetamol memiliki serapan maksimum pada panjang gelombang 250 nm
( = 900) dalam etanol dan pada panjang gelombang 255 nm ( = 710)
dalam larutan NaOH 0,1 N (Auterhoff, 1981). Dalam metanol, parasetamol
memiliki serapan maksimum pada panjang gelombang 249 nm ( = 900)
(Clarke, 1969). atau serapan jenis adalah serapan dari larutan 1% zat terlarut
dalam sel dengan ketebalan 1 cm (Anonim, 1995).
%
Parasetamol merupakan metabolit fenasetin dengan efek antipiretik yang
telah digunakan sejak tahun 1893. Efek antipiretik ditimbulkan oleh gugus
aminobenzen. Parasetamol juga digunakan sebagai analgesik. Namun
penggunaannya sebagai analgesik. Efek analgesik dari parasetamol yaitu
meredakan rasa nyeri ringan hingga sedang (Wilmana, 1995). Dosis oral untuk
nyeri dan demam 2-3 kali sehari 0,5-1 g, maksimum 4g/hari (Tjay dan Rahardja,
2002).
Tablet parasetamol mengandung parasetamol, C8H9NO2, tidak kurang
dari 90,0% dan tidak lebih dari 110,0% dari jumlah yang tertera pada etiket
(Anonim, 1995).
C. Salisilamida
Salisilamida atau 2-hidroksi benzamida memiliki bobot molekul 137,14
(Anonim, 1995). Rumus bangun salisilamida dapat dilihat pada gambar 2.
C
OH O
NH2
Gambar 2. Struktur Salisilamida (Anonim, 1995)
Salisilamida merupakan serbuk hablur berwarna putih, dan praktis tidak
berbau. Salisilamida mengandung tidak kurang dari 98,0% dan tidak lebih dari
102,0% C7H7NO2, dihitung terhadap zat anhidrat (Anonim, 1995).
Salisilamida memiliki titik lebur pada suhu antara 139°C dan 142°C.
Satu bagian salisilamida larut dalam 500 bagian air, lebih larut dalam air panas, 15
bagian etanol, 35 bagian eter, dan 100 bagian kloroform (Clarke, 1969).
Salisilamida dalam etanol memiliki serapan maksimum pada panjang
larutan NaOH 0,1N, salisilamida memiliki serapan maksimum pada panjang
gelombang 242 nm ( 1% =536) dan 328 nm ( = 435) (Clarke, 1969).
1cm
A A11cm%
Salisilamida adalah amida asam salisilat yang memperlihatkan efek
analgesik antipiretik mirip asetosal. Efek analgesik antipiretik salisilamida lebih
lemah dari salisilat, karena salisilamida dalam mukosa usus mengalami
metabolisme lintas pertama sehingga hanya sebagian kecil saja yang masuk dalam
sirkulasi sebagai zat aktif. Salisilamida dapat menghambat glukuronidasi obat
analgesik lain di hati seperti Na salisilat dan parasetamol, sehingga pemberian
bersamaan dapat meningkatkan efek terapi dan toksisitas obat (Wilmana, 1995).
Dosis yang digunakan adalah 3-4 kali sehari 0,5-1 g (Tjay dan Rahardja, 2002).
Tablet salisilamida mengandung salisilamida, C7H7NO2, tidak kurang
dari 90,0% dan tidak lebih dari 110,0% dari jumlah yang tertera pada etiket
(Anonim, 2005).
D. Kafein
Kafein atau 1,3,7-trimetil xantin berbentuk anhidrat dengan bobot
molekul 194,19 atau hidrat dengan mengandung 1 molekul air dengan bobot
molekul 212,21 (Anonim, 1995). Rumus bangun kafein dapat dilihat pada
gambar 3.
Kafein mengandung tidak kurang dari 98,5% dan tidak lebih dari 101,0%
C8H10N4O2, dihitung terhadap zat anhidrat. Kafein merupakan serbuk putih atau
bentuk jarum mengkilat putih yang biasanya menggumpal, tidak berbau, dan
berasa pahit (Anonim, 1995).
Kafein memiliki titik lebur antara 235°C dan 237°C. Satu bagian kafein
larut dalam 60 bagian air, 2 bagian air panas, 130 bagian etanol, dan 7 bagian
kloroform. Kafein larut dalam eter dan lebih larut dalam larutan asam (Clarke,
1969).
Kafein dalam etanol memiliki serapan maksimum pada panjang
gelombang 273 nm( = 519) dan dalam larutan NaOH 0,1N memiliki serapan
maksimum pada panjang gelombang 272 nm ( = 470) (Clarke, 1969).
%
Kafein merupakan golongan xantin yang menyebabkan relaksasi otot
polos, terutama otot polos bronkus, merangsang sistem saraf pusat (SSP), otot
jantung, dan meningkatkan diuresis (Wilmana, 1995). Parasetamol ataupun
asetosal dikombinasikan dengan kafein untuk memperkuat daya analgesiknya
(Anonim, 2000). Dosis yang digunakan pada rasa letih 1-3 kali sehari 100-200
mg, sebagai adjuvans bersama analgetika 50 mg 1 kali (Tjay dan Rahardja, 2002).
Tablet kafein mengandung kafein, C8H10N4O2, tidak kurang dari 90,0%
E. Spektrofotometri UV
Spektroskopi adalah salah satu teknik analisis fisiko-kimia yang
mengamati tentang interaksi atom atau molekul dengan radiasi elektromagnetik
(REM). Spektrofotometri ultraviolet adalah salah satu teknik analisis spektroskopi
yang memakai sumber radiasi elektromagnetik ultraviolet dekat (190-380 nm)
dengan memakai instrument spektrofotometer (Mulja dan Suharman, 1995).
Apabila suatu molekul dikenai radiasi elektromagnetik maka akan terjadi
eksitasi ke tingkat energi yang lebih tinggi yang dikenal sebagai orbital elektron
antibonding. Ada empat tipe transisi elektronik yang mungkin terjadi yaitu
σÆσ*, nÆσ*, nÆπ*, dan πÆπ*. Eksitasi elektron (σÆσ*) memberikan energi
yang terbesar dan terjadi pada daerah ultraviolet jauh yang diberikan oleh ikatan
tunggal, misalnya alkana. Eksitasi elektron (πÆπ*) diberikan oleh ikatan rangkap
dua dan tiga, juga terjadi pada daerah ultraviolet jauh. Eksitasi elektron (nÆσ*)
terjadi pada gugus karbonil (dimetil keton dan asetaldehid) yang terjadi pada
daerah ultraviolet jauh (Mulja dan Suharman, 1995).
Suatu molekul dapat menyerap radiasi elektromagnetik jika memiliki
kromofor, yaitu gugus penyerap dalam molekul. Molekul yang mengandung
kromofor disebut kromogen. Pada senyawa organik dikenal pula gugus
auksokrom, yaitu gugus yang tidak menyerap radiasi namun bila terikat bersama
kromofor dapat meningkatkan penyerapan oleh kromofor atau mengubah panjang
gelombang serapan maksimum (Christian, 2004).
Spektrofotometer ultraviolet melibatkan energi elektronik yang cukup
banyak untuk analisis kuantitatif dibandingkan kualitatif. Analisis kuantitatif
selalu melibatkan pembacaan absorban radiasi elektromagnetik oleh molekul, atau
radiasi elektromagnetik yang diteruskan, yang disebut absorban (A) tanpa satuan
dan transmitan dengan satuan persen (%T). Bouger, Lambert, dan Beer membuat
formula secara matematik hubungan antara transmitan atau absorban terhadap
intensitas radiasi atau konsentrasi zat yang dianalisis dan tebal larutan yang
mengabsorpsi sebagai :
b
It = intensitas radiasi yang diteruskan
ε = daya serap molar (Lt.mol-1 .cm-1)
c = konsentrasi (mol. Lt-1)
b = tebal larutan (cm)
A = serapan / absorbansi
F. KCKT 1. Definisi dan instrumentasi
Kromatografi adalah prosedur pemisahan senyawa campuran
berdasarkan perbedaan kecepatan migrasi, karena adanya perbedaan koefisien
distribusi masing-masing senyawa di antara dua fase yang saling bersinggungan
dan tidak saling campur, yang disebut sebagai fase gerak (mobile phase) yang
berupa zat cair atau zat gas, dan fase diam (stationary phase) yang berupa zat cair
atau zat padat (Noegrohati, 1994). Kromatografi pertama kali ditemukan oleh
TSWETT pada 1903. TSWETT telah menggunakan kromatografi untuk
pemisahan senyawa yang berwarna sehingga metode tersebut dinamai
kromatografi (kroma yang berarti berwarna). Namun pembatasan untuk senyawa
berwarna tidak berlangsung lama dan hampir kebanyakan pemisahan secara
kromatografi saat ini digunakan pada senyawa yang tidak berwarna
(Sastrohamidjojo, 2002).
Kromatografi Cair Kinerja Tinggi (KCKT) merupakan salah satu metode
kromatografi cair yang fase geraknya dialirkan secara cepat dengan bantuan
tekanan, dan hasilnya dideteksi dengan instrument (Willard, Merritt, Dean, dan
Settle, 1988). Pada mulanya teknik kromatografi ini disebut dengan High
Pressure Liquid Chromatography karena pada instrument ini terdapat sistem
pompa tekanan tinggi yang mampu mengalirkan fase gerak pada tekanan tinggi
sampai 300 atmosfer dan tekanan pada bagian atas kolom kurang dari 70 atmosfer
(Anonim, 1995). Pada akhir tahun 1970, perkembangan instrument ini dapat
baik sehingga sistem ini lebih dikenal dengan Kromatografi Cair Kinerja Tinggi
(Kromidas, 2000).
KCKT merupakan teknik analisis yang paling sering digunakan dalam
analisis farmasi untuk pemisahan, identifikasi, dan determinasi dalam campuran
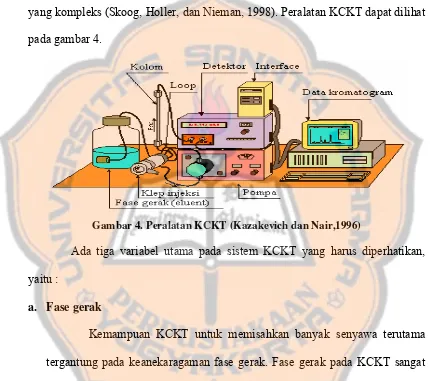
yang kompleks (Skoog, Holler, dan Nieman, 1998). Peralatan KCKT dapat dilihat
pada gambar 4.
Gambar 4. Peralatan KCKT (Kazakevich dan Nair,1996)
Ada tiga variabel utama pada sistem KCKT yang harus diperhatikan,
yaitu :
a. Fase gerak
Kemampuan KCKT untuk memisahkan banyak senyawa terutama
tergantung pada keanekaragaman fase gerak. Fase gerak pada KCKT sangat
berpengaruh pada tambatan dan pemisahan senyawa (Munson, 1984). Fase
gerak untuk analisis secara KCKT harus bersifat murni, tanpa cemaran, tidak
bereaksi dengan kemasan, dapat melarutkan cuplikan (solut), viskositas
rendah, memungkinkan memperoleh kembali cuplikan dengan mudah (jika
diperlukan), dan harganya wajar (Johnson dan Stevenson, 1978). Fase gerak
detektor, sehingga menghasilkan sinyal palsu, dan mempengaruhi kolom
(Gritter, Bobbit, dan Schwarting, 1985).
Kepolaran pelarut merupakan ukuran kekuatan pelarut untuk
mengelusi suatu senyawa. Kandungan utama fase gerak pada kromatografi
fase terbalik adalah air. Kecenderungan air untuk melarutkan sampel dapat
diubah dengan menambahkan garam untuk menimbulkan pengaruh
penggaraman, asam, basa, dapar untuk melarutkan atau mengendapkan asam
atau basa, pereaksi pengompleks untuk menimbulkan jenis pengaruh pelarutan
yang khas untuk gugus fungsi tertentu atau golongan senyawa tertentu, atau
pelarut organik yang dapat bercampur dengan air. Pemodifikasi organik yang
banyak digunakan adalah metanol, asetonitril, dan tetrahidrofuran (Gritter et
al., 1985; Munson, 1984).
Kepolaran pelarut dinyatakan dalam bentuk P’ (indeks polaritas).
Besarnya polaritas campuran pelarut dapat dihitung dengan persamaan
berikut:
dengan Ф adalah fraksi pelarut dalam campuran dan n adalah jenis pelarut
yang digunakan (Skoog et al., 1998).
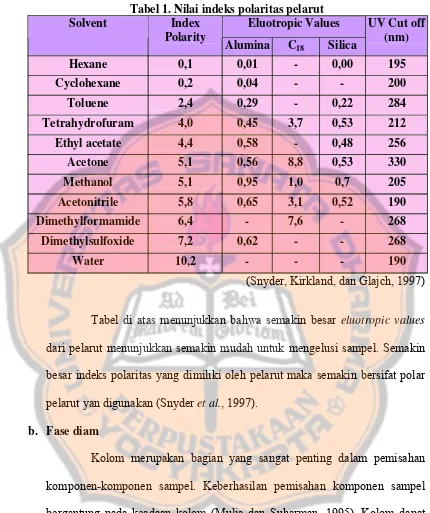
Berikut ini ditampilkan beberapa nilai indeks polaritas dari beberapa
Tabel 1. Nilai indeks polaritas pelarut
Cyclohexane 0,2 0,04 - - 200
Toluene 2,4 0,29 - 0,22 284
Tetrahydrofuram 4,0 0,45 3,7 0,53 212
Ethyl acetate 4,4 0,58 - 0,48 256
Acetone 5,1 0,56 8,8 0,53 330
Methanol 5,1 0,95 1,0 0,7 205
Acetonitrile 5,8 0,65 3,1 0,52 190
Dimethylformamide 6,4 - 7,6 - 268
Dimethylsulfoxide 7,2 0,62 - - 268
Water 10,2 - - - 190
(Snyder, Kirkland, dan Glajch, 1997)
Tabel di atas menunjukkan bahwa semakin besar eluotropic values
dari pelarut menunjukkan semakin mudah untuk mengelusi sampel. Semakin
besar indeks polaritas yang dimiliki oleh pelarut maka semakin bersifat polar
pelarut yan digunakan (Snyder et al., 1997).
b. Fase diam
Kolom merupakan bagian yang sangat penting dalam pemisahan
komponen-komponen sampel. Keberhasilan pemisahan komponen sampel
bergantung pada keadaan kolom (Mulja dan Suharman, 1995). Kolom dapat
dibagi menjadi dua kelompok berdasarkan diameternya, yaitu :
1) Kolom analitik, memiliki diameter pada bagian dalam 2-6 mm. Panjang
biasanya 50-100 cm dan untuk kemasan mikropartikel berpori biasanya
10-30 cm.
2) Kolom preparatif, dengan diameter 6 mm atau lebih dan panjang kolom
25-100 cm (Johnson dan Stevenson, 1978).
c. Detektor
Detektor yang baik hendaknya memiliki kepekaan tinggi, rentang
respon liniernya lebar, tidak dipengaruhi perubahan suhu dan aliran,
memberikan hasil dengan keterulangan yang baik, dan tidak banyak derau.
Secara umum, detektor dibagi menjadi 2 kategori, yaitu :
1) Bulk property detectors, merupakan detektor yang mengukur perubahan
sifat fisik fase gerak dan solut. Detektor tipe ini cenderung relatif tidak
sensitif dan menghendaki temperatur yang terkendali. Contoh detektor
jenis ini yaitu detektor indeks bias.
2) Solute property detectors, merupakan detektor yang hanya mengukur sifat
fisik solut. Detektor tipe ini 1000 kali lebih sensitif dan mampu mengukur
solut sampai satuan nanogram atau lebih kecil lagi. Contoh detektor jenis
ini yaitu detektor fluoresensi, detektor penyerapan (UV-Vis), dan detektor
elektrokimia (Munson, 1984; Willard et al., 1988).
2. Pembagian jenis kromatografi
a. Kromatografi cair-cair atau kromatografi partisi
Pada kromatografi partisi, fase diam dapat polar atau non polar. Bila
fase diam polar dan fase gerak nonpolar disebut kromatografi partisi fase
normal, sedangkan bila fase diam nonpolar dan fase gerak polar dinamakan
kromatografi partisi fase terbalik (Munson, 1984).
b. Kromatografi adsorpsi
Kromatografi ini menggunakan fase diam padat dan fase gerak cair
atau gas. Solut dapat diadsorpsi pada permukaan partikel padat (Harris, 1999).
c. Kromatografi pertukaran ion
Anion atau kation diikatkan secara kovalen pada fase diam padat,
biasanya resin. Ion-ion solut muatan berlawanan menyerang fase diam dengan
kekuatan elektrostatik. Fase geraknya cair (Harris, 1999).
d. Kromatografi eksklusi
Pada kromatografi ini tidak ada interaksi tarik menarik antara fase
diam dan solut. Fase gerak cair atau gas melalui gel berpori. Ukuran pori
cukup kecil untuk mengeluarkan molekul solut yang besar. Molekul solut
yang kecil akan masuk ke dalam pori gel, sedangkan molekul yang besar akan
mengalir tanpa memasuki pori gel (Harris, 1999).
e. Kromatografi afinitas
Digunakan untuk interaksi spesifik antara satu jenis molekul solut
dan sebuah molekul yang lain yang secara kovalen terikat pada fase diam.
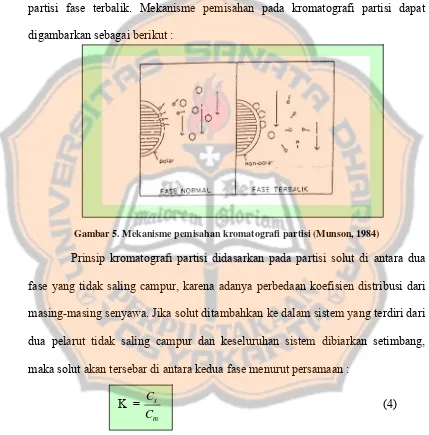
3. Kromatografi partisi
Pada salah satu dari dua fase kromatografi partisi, yaitu fase gerak dan
fase diam harus lebih polar dibanding yang lain. Bila fase diam lebih polar,
disebut kromatografi partisi fase normal. Bila sebaliknya dinamakan kromatografi
partisi fase terbalik. Mekanisme pemisahan pada kromatografi partisi dapat
digambarkan sebagai berikut :
Gambar 5. Mekanisme pemisahan kromatografi partisi (Munson, 1984)
Prinsip kromatografi partisi didasarkan pada partisi solut di antara dua
fase yang tidak saling campur, karena adanya perbedaan koefisien distribusi dari
masing-masing senyawa. Jika solut ditambahkan ke dalam sistem yang terdiri dari
dua pelarut tidak saling campur dan keseluruhan sistem dibiarkan setimbang,
maka solut akan tersebar di antara kedua fase menurut persamaan :
K = m s
C C
(4)
K adalah koefisien distribusi, Cs adalah konsentrasi solut dalam fase diam, dan
Cm adalah konsentrasi solut dalam fase gerak (Johnson dan Stevenson, 1978).
Kolom yang biasa digunakan pada kromatografi partisi fase terbalik
diamnya terikat secara kimia pada penyangga, sehingga tidak mudah terbawa oleh
fase gerak. Penyangga pada kemasan fase terikat biasanya terbuat dari silika yang
sudah diseragamkan, berpori, dan umumnya partikel mempunyai diameter 3,5
atau 10 μm (Skoog et al., 1998).
KCKT partisi fase terbalik biasanya mengandung bagian organik yang
terikat secara kimia dengan gugus silanol pada permukaan silika. Bagian organik
tersebut umumnya hidrokarbon rantai panjang, sehingga fase gerak umumnya
polar. Gugus silanol permukaan dapat direaksikan dengan berbagai cara untuk
menempelkan berbagai jenis gugus organik. Kemasan fase terikat dengan tipe
ikatan siloksan (Si-O-Si-O) dibuat dengan mereaksikan organoklorosilan dengan
gugus silanol pada permukaan silika gel yang terhidrolisis sebagai berikut :
Si OH + Cl Si(CH3)2R Si O Si(CH3)2R + HCl
Gambar 6. Reaksi silanisasi
Reaksi tersebut digunakan untuk membuat isian kolom oktadesilsilan (ODS) dari
gugus silanol dan oktadesilklorosilan sebagai berikut :
Si OH Cl Si CH3
(CH2)17CH3
CH3
Si O Si (CH2)17CH3 + HCl +
Gambar 7. Reaksi pembuatan kolom oktadesilsilan
Pada kromatografi partisis fase terbalik dengan kemasan fase terikat, R pada
siloksan biasanya berupa gugus C18 atau C8. Panjang pendeknya rantai karbon
4. Pemisahan puncak dalam kromatografi
Keberhasilan atau kegagalan analisis tergantung pada pemilihan kolom
dan kondisi kerja yang tepat. Ukuran kinerja dari kolom dapat dilihat dari
kemampuan kolom dalam memisahkan senyawa. Kolom yang efisien mencegah
pelebaran puncak atau menghasilkan puncak yang sangat sempit. (Johnson dan
Stevenson, 1978).
Faktor resolusi adalah ukuran pemisahan dari 2 puncak. Daya pisah, R,
dapat diukur dengan persamaan :
2
Harga tR2 dan tR1 adalah waktu retensi senyawa, diukur pada titik maksimum
puncak dan Δt adalah selisih antara tR2 dan tR1. Harga w2 dan w1 ialah lebar alas
puncak. Pemisahan dua senyawa dapat digambarkan sebagai berikut :
Gambar 8. Pemisahan dua senyawa (Johnson dan Stevenson, 1978)
Harga R > 1,5 disebut baseline resolution, yaitu pemisahan sempurna
dari dua puncak dengan ukuran yang sama. Dalam prakteknya, pemisahan dengan
harga R = 1,0 (kedua puncak berhimpit lebih kurang 2%) dianggap memadai
Pemisahan dari puncak-puncak dalam kromatografi erat hubungannya
dengan efisiensi kolom. Pada efisiensi kolom terdapat dua teori yang menjelaskan
mengenai pemisahan puncak pada kromatografi, yaitu :
a. Teori lempeng
Dalam teori lempeng dinyatakan bahwa kolom kromatografi
digambarkan sebagai suatu seri lapisan tipis horizontal yang disebut lempeng
teoritis. Setiap molekul analit akan mengalami keseimbangan antara fase diam
dan fase gerak. Pemisahan akan lebih baik jika terjadi keseimbangan
berkali-kali dalam jumlah yang tinggi. Hal ini terjadi jika jumlah lempeng teoritis juga
tinggi. Oleh karena itu jumlah teoritis dapat digunakan sebagai ukuran
efisiensi kolom (Noegrohati, 1994). Hubungan antara waktu retensi (tR), lebar
alas peak (W), dan jumlah lempeng teoritik (N) dapat dinyatakan dengan
persamaan (Johnson dan Stevenson, 1978; Munson, 1984):
2
Bilangan lempeng teoritik (N) berbanding lurus dengan panjang
kolom (L). Karena panjang kolom yang bermacam-macam, maka diperlukan
ukuran keefisienan kolom yang tidak tergantung pada panjang kolom. HETP
(Height Equivalent to a Theoritical Plate) atau H merupakan ukuran
keefesienan kolom yang lebih disukai karena memungkinkan perbandingan
antara kolom yang panjangnya berlainan, yang dapat diukur dengan
persamaan (Munson, 1984):
H = HETP =
N L
b. Teori laju
Teori lempeng hanya menggambarkan laju migrasi secara kuantitatif,
tetapi tidak dapat menggambarkan pengaruh variable-variabel lain yang
menyebabkan terjadinya pelebaran peak, oleh karena itu perlu diketahui teori
laju. Pada waktu migrasi, solut mengalami transfer antara fase diam dan fase
gerak berkali-kali. Karena solut hanya dapat bergerak jika berada dalam fase
gerak, migrasi di dalam kolom juga tidak teratur, dan mengakibatkan laju
rata-rata solut relatif terhadap fase gerak juga sangat bervariasi, sehingga terjadi
pelebaran peak solut (Noegrohati, 1994).
Menurut teori laju ini, efisiensi kolom dinyatakan dengan persamaan
Van Deemter yang dapat dinyatakan sebagai berikut (Willard et al., 1988):
μ
Dimana λ = tetapan ukuran ketidakteraturan kemasan
dp = diameter rata-rata partikel penyangga
D = kedifusian linarut dalam fase gerak
k’ = faktor kapasitas
μ = kecepatan alir
γ = faktor koreksi kelikuan saluran dalam kolom
Dari persamaan diatas dapat dilihat terdapat tiga varibel yang
1) Difusi Eddy , yang dinyatakan sebagai A (2λdp). Difusi Eddy
menggambarkan ketidakhomogenannya kecepatan alir dan panjang
lintasan di sekitar partikel yang terpack-ing (Gambar 8). Lintasan alir yang
tidak sama pasti ditemukan dalam setiap kolom terpack-ing. Suatu
molekul solut dapat melewati kolom dekat dinding kolom di mana
kerapatan kolom rendah dengan cepat mencapai akhir kolom, khususnya
pada kolom dengan diameter kecil. Sedangkan suatu molekul solut yang
melewati bagian tengah kolom, akan mencapai akhir kolom lebih lambat.
Hal ini menyebabkan perbedaan laju tiap molekul melalui kolom
berbeda-beda. Untuk meminimalkan difusi Eddy ini, maka diameter rata-rata
partikel dalam kolom harus sekecil mungkin dan seseragam mungkin.
Gambar 9. Difusi Eddy (Willard et al., 1988)
2) Difusi longitudinal. Nilai B (2γD/μ) menyatakan efek dari difusi
longitudinal, pergerakan acak dari molekul dalam fase gerak. Pengaruh
dari difusi longitudinal terhadap ketinggian lempeng menjadi signifikan
hanya pada kecepatan fase gerak yang rendah/lambat. Kecepatan difusi
dari solut yang tinggi pada fase gerak dapat menyebabkan molekul solut
terdispers secara aksial sementara dengan lambat bermigrasi melalui
3) Transfer massa. Transfer massa dinyatakan dengan nilai Cstasionery dan
Cmobile. Cstationary merupakan hasil dari ditahannya solut karena adanya fase
diam. Suatu molekul bergerak lambat dalam fase diam, sementara molekul
lainnya melaju melalui kolom bersama dengan fase gerak. Untuk
mengatasi hal ini diperlukan fase diam yang lebih encer (tidak terlalu
kental). Peristiwa ini dapat digambarkan sebagai berikut:
Gambar 10. Tranfer massa fase diam (Willard et al., 1988)
Cmobile menggambarkan adanya peristiwa dimana solut dalam fase diam
bertemu dengan fase gerak yang masih baru. Hal ini dapat digambarkan
sebagai berikut (Willard et al., 1988) :
Gambar 11. Transfer massa fase gerak (Willard et al., 1988)
Pada analisis secara KCKT, kondisi percobaan yang menghasilkan
puncak yang simetris selalu lebih disukai, karena puncak yang asimetris dapat
menghasilkan pengukuran bilangan lempeng teoritik dan faktor resolusi yang
tidak akurat, perhitungan yang tidak teliti, penurunan derajat resolusi dan
tidak reprodusibel. Parameter yang digunakan untuk menilai bentuk puncak
adalah peak asymmetry factor (As), yang diukur pada 10% tinggi puncak. Puncak
yang simetri memiliki nilai As sama dengan 1, sedangkan puncak dengan nilai As
pada rentang 0,95-1,1 masih dikatakan baik. Parameter lain yang dapat digunakan
yaitu peak tailing factor, yang diukur pada 5% tinggi puncak (Snyder et al.,
1997).
Gambar 12. Penentuan peak asymmetry dan peak tailing factors (Snyder et al., 1997)
Distribusi analit dalam fase gerak dan fase diam pada saat terjadi tailing dan
leading dapat dilihat sebagai berikut :
Gugus silanol yang tidak bereaksi karena adanya halangan sterik dapat
memberikan kepolaran yang tidak dikehendaki dan menyebabkan pengekoran
pada puncak kromatogram. Untuk mengurangi jumlah gugus silanol yang masih
bebas, reaksi dilanjutkan dengan penambahan trimetilklorosilan yang dapat
mencapai gugus silanol karena ukurannya yang lebih kecil dibanding
organoklorosilan lain. Penambahan trimetilklorosilan dapat menutupi banyak
gugus silanol yang masih bebas, namun tidak semua gugus tersebut dapat tertutupi
(Skoog et al., 1998).
Puncak kromatogram yang tidak simetri (tailing dan leading) sering
dijumpai bila konsentrasi solut dalam fase gerak terlalu besar. Senyawa-senyawa
polar juga berpotensi menimbulkan tailing apabila masih terdapat residu gugus
silanol pada fase diam. Penyebab tailing yang lain yaitu ketidaksesuian antara
solut dan kolom, pengemasan kolom yang tidak seragam, dan faktor yang terjadi
di luar kolom, seperti injektor (Noegrohati, 1994).
5. Analisis kualitatif dan kuantitatif
KCKT digunakan untuk analisis kualitatif dan kuantitatif dari suatu
sampel/cuplikan selain untuk memisahkan senyawa dalam sampel. Hasil dari
pemisahan adalah kromatogram. Dari kromatogram diperoleh informasi mengenai
waktu retensi suatu senyawa (Noegrohati, 1994).
Waktu retensi yang menunjukkan identitas suatu senyawa merupakan
selang waktu yang diperlukan senyawa mulai pada saat injeksi sampai keluar dari
memiliki waktu retensi yang spesifik pada kondisi tertentu seperti kolom, suhu,
laju, dan sebagainya sehingga dapat digunakan sebagai salah satu dasar uji
kualitatif (Noegrahati, 1994). Analisis kualitatif dilakukan dengan cara
membandingkan waktu retensi senyawa murni dengan waktu retensi senyawa
yang dimaksud dalam sampel (Gritter et al., 1985).
Analisis kuantitatif dilakukan berdasarkan perbandingan tinggi atau luas
puncak kromatogram senyawa sampel terhadap senyawa standar. Bila variasi
keadaan kolom tidak menyebabkan pelebaran puncak, maka analisis berdasarkan
tinggi puncak dapat memberikan ketelitian tinggi. Analisis berdasarkan luas
puncak tidak dipengaruhi oleh pelebaran puncak. Oleh karena itu cara ini lebih
disukai dalam perhitungan kuantitatif (Noegrohati, 1994).
G. Keterangan Empiris
Penelitian ini bersifat non eksperimental deskriptif yang bertujuan untuk
mengetahui apakah metode KCKT dapat digunakan untuk menetapkan kadar
parasetamol, salisilamida, dan kafein dalam tablet, serta untuk mengetahui apakah
kadar parasetamol, salisilamida, dan kafein dalam tablet sesuai dengan yang
BAB III
METODOLOGI PENELITIAN
A. Jenis dan Rancangan Penelitian
Penelitian ini mengikuti jenis dan rancangan penelitian non
eksperimental deskriptif karena tidak ada perlakuan terhadap subyek uji.
B. Definisi Operasional
1. Parasetamol, salisilamida, dan kafein yang ditetapkan kadarnya adalah
parasetamol, salisilamida, dan kafein yang terdapat dalam tablet merk x.
2. Tablet yang dianalisis adalah tablet merk x yang mencantumkan kandungan
parasetamol, salisilamida, dan kafein dengan perbandingan 4:4:1 dalam
labelnya, yang memiliki nomor produksi yang sama.
3. Sistem Kromatografi Cair Kinerja Tinggi (KCKT) fase terbalik yang
digunakan adalah seperangkat alat KCKT dangan fase diam kolom reversed
phase C18 dan fase gerak campuran metanol : aquabidest : amonia (70:29:1).
4. Kadar parasetamol, salisilamida, dan kafein dalam tablet ditetapkan dalam
satuan mg/tablet.
C. Bahan
Bahan yang digunakan pada penelitian ini meliputi parasetamol kualitas
working standard (Brataco), salisilamida kualitas working standard (Brataco),
kafein kualitas working standard (Brataco), metanol (p.a, E. Merck), amonia (p.a,
E. Merck), aquabidest (dari Laboratorium Kimia Organik Fakultas Farmasi
Universitas Sanata Dharma Yogyakarta), dan tablet merk x yang memiliki nomor
produksi yang sama.
D. Alat
Alat yang digunakan pada penelitian ini, yaitu :
1. spektrofotometer UV/Vis merk Perkin-Elmer Lambda 20
2. seperangkat KCKT yang meliputi :
a. pompa merk Shimadzu model LC-10 AD No. C20293309457 J2
b. detektor UV/Vis merk Shimadzu model SPD-10 AV No. C20343502697
KG
c. CBM-101 merk Shimadzu No. C50363502311 SA
d. kolom Waters BondapacTM C18 (dengan panjang 30 cm; P61271BO2 P/N
27324; diameter partikel 5-10 μm)
e. injektor jenis katup suntik model 77215i
f. seperangkat komputer merk ACER
3. syringe merk Hamilton Pat No. 2933087
4. degassingultrasonic merk Retsch tipe T460 No. V935922012 EY
5. vacum merk Gast model DOA-P104-BN
6. organic solvent membrane filter merk Whatman dengan ukuran pori 0,5 μm
dan diameter 47 mm.
7. inorganic solvent membrane filter merk Whatman dengan ukuran pori 0,45
8. membrane filter holder merk Whatman dengan kapasitas 300 ml
9. penyaring Millipore
10.micropipette merk Socorex ukuran 200-1000μl
11.neraca analitik merk Scaltec SBC 22 max 60/210 g; d = 0,01/0,1 mg; e = 1 mg
12.seperangkat alat gelas yang lazim digunakan untuk analisis.
E. Tata Cara Penelitian 1. Pembuatan larutan baku parasetamol
a.Pembuatan larutan stok parasetamol. Lebih kurang 40,0 mg
parasetamol ditimbang seksama dan dilarutkan dengan metanol hingga 100,0 ml.
b.Pembuatan seri konsentrasi larutan baku parasetamol. Larutan stok
parasetamol dari langkah di atas dipipet 1,0; 1,5; 2,0; 2,5; 3,0 ml dan diencerkan
dengan metanol hingga 10,0 ml sehingga didapatkan konsentrasi sebesar 4; 6; 8;
10; 12 mg%. Larutan tersebut disaring dengan Millipore dan degassing selama 15
menit.
2. Pembuatan larutan baku salisilamida
a.Pembuatan larutan stok salisilamida. Lebih kurang 40,0 mg
salisilamida ditimbang seksama dan dilarutkan dengan metanol hingga 100,0 ml.
b.Pembuatan seri konsentrasi larutan baku salisilamida. Larutan stok
salisilamida dari langkah di atas dipipet 1,0; 2,0; 2,5; 3,0; 4,0 ml dan diencerkan
10; 12; 16 mg%. Larutan tersebut disaring dengan Millipore dan degassing selama
15 menit.
3. Pembuatan larutan baku kafein
a.Pembuatan larutan stok kafein. Lebih kurang 10,0 mg kafein
ditimbang seksama dan dilarutkan dengan metanol hingga 100,0 ml.
b.Pembuatan seri konsentrasi larutan baku kafein. Larutan stok kafein
dari langkah di atas dipipet 1,0; 1,5; 2,0; 2,5; 3,0 ml dan diencerkan dengan
metanol hingga 10,0 ml sehingga didapatkan konsentrasi sebesar 1; 1,5; 2; 2,5; 3
mg%. Larutan tersebut disaring dengan Millipore dan degassing selama 15 menit.
4. Pembuatan fase gerak
Fase gerak dibuat dengan mencampur 70 bagian metanol, 29 bagian
aquabidest, dan 1 bagian amonia. Larutan ini kemudian disaring dan degassing
selama 15 menit.
5. Optimasi metode KCKT
a.Penentuan λOverlappingdengan spektrofotometri UV. Dari larutan stok
parasetamol, salisilamida, dan kafein dibuat larutan dengan konsetrasi 0,8 dan 1
mg%. Larutan dibaca serapannya pada rentang panjang gelombang 200-300 nm
sehingga dapat dilihat spektrum serapan dan serapan maksimum dari parasetamol,
salisilamida, dan kafein. Masing-masing spektrum yang didapat digunakan untuk
b.Pembuatan kurva baku parasetamol, salisilamida, dan kafein.
Masing-masing seri konsentrasi larutan baku parasetamol, salisilamida, dan kafein
disuntikkan dalam injector port menggunakan KCKT syringe dengan fase diam
kolom revered phase C18, fase gerak metanol : aquabidest : amonia (70:29:1), dan
flow rate 1,5 ml/menit. Kromatogram yang telah dihasilkan kemudian diamati.
Dengan metode regresi linier, memplotkan kadar (mg%) terhadap harga AUC dari
masing-masing seri larutan kadar sehingga didapat persamaan y = bx + a (y =
harga AUC, x = konsentrasi, b = slope, a = intersept).
6. Pembuatan larutan sampel
Sebanyak dua puluh tablet yang telah diketahui bobot rata-ratanya
digerus, ditimbang lebih kurang sejumlah sampel dengan seksama yang setara
dengan 40,0 mg parasetamol, 40,0 mg salisilamida, dan 10,0 mg kafein. Serbuk
sampel dilarutkan dalam metanol hingga 100,0 ml. Dari larutan sampel dipipet 2,0
ml dan diencerkan dengan metanol hingga didapatkan volume 10,0 ml. Menyaring
dengan Millipore dan degassing selama 15 menit.
7. Penetapan kadar parasetamol, salisilamida, dan kafein dalam sampel
Menyuntikkan larutan sampel dalam injector port dengan menggunakan
KCKT syringe dengan fase diam kolom revered phase C18, fase gerak metanol :
aquabidest : amonia (70:29:1), dan flow rate 1,5 ml/menit. Amati kromatogram
yang dihasilkan. Dengan memasukkan harga AUC sampel dalam masing-masing
kadar parasetamol, salisilamida, dan kafein dalam sampel (mg%). Kemudian data
disajikan dalam bentuk Χ±SDdengan satuan mg/tablet.
F. Analisis Hasil
Analisis yang dilakukan dalam penelitian ini adalah :
1. Analisis kualitatif
Analisis kualitatif dilakukan dengan membandingkan waktu retensi (tR)
yang didapatkan dalam sampel dengan waktu retensi (tR) senyawa baku.
2. Analisis kuantitatif
Analisis kuantitatif yang dilakukan adalah penetapan kadar dari
parasetamol, salisilamida, dan kafein berdasarkan analisis data AUC sampel dan
kurva baku dari masing-masing senyawa. Data kadar disajikan dalam bentuk
SD
±
Χ dengan satuan mg/tablet yang kemudian dibandingkan dengan yang
tertera pada etiket, apakah kadarnya sudah sesuai dengan yang tertera pada
BAB IV
HASIL DAN PEMBAHASAN
A. Pemilihan dan Penyiapan Sampel
Pada penelitian ini yang digunakan sebagai sampel adalah tablet dengan merk x yang beredar di Magelang. Pemilihan sampel ini didasarkan pada obat yang menuliskan komposisi zat aktifnya adalah parasetamol, salisilamida, dan kafein. Di dalam masyarakat banyak sediaan obat yang mengandung zat aktif tersebut, namun dengan perbandingan ketiga zat aktif yang berbeda-beda dan ada juga yang tidak hanya mengandung ketiga zat aktif itu saja. Pada penelitian ini, penulis membatasi sampel yang digunakan, yaitu sediaan obat berupa tablet yang mengandung hanya ketiga zat aktif tersebut dengan perbandingan parasetamol, salisilamida, dan kafein adalah 4:4:1. Sediaan yang memenuhi batasan tersebut hanya ada satu produk, sehingga penulis hanya menggunakan satu produk sebagai sampel. Menurut Sevilla (1993), pengambilan sampel minimal 10% dari populasi dan untuk populasi kecil dibutuhkan minimal 20 %. Dalam hal ini, pengambilan sampel pada penelitian ini sudah memenuhi aturan dalam pengambilan sampel tersebut.
Pemilihan sampel yang dilakukan adalah pemilihan sediaan dengan merk x yang memiliki nomor produksi yang sama. Dari kemasan diambil 20 tablet obat yang dihitung bobot rata-ratanya. Pada penelitian ini dilakukan replikasi sebanyak 6 kali.
B. Penyiapan Fase Gerak
Pemilihan fase gerak dan fase diam dalam penelitian ini mengacu pada penelitian dari Sugianto (2006) mengenai optimasi penetapan kadar campuran parasetamol, salisilamida, dan kafein dengan metode KCKT yang telah terbukti memiliki validitas yang baik.
Fase gerak yang digunakan pada penelitian ini adalah campuran antara metanol, aquabidest, dan amonia dengan perbandingan 70:29:1. Campuran fase gerak ini bersifat polar, sedangkan fase diam yang digunakan adalah kolom C18
yang bersifat non polar sehingga sistem kromatografi yang digunakan adalah kromatografi partisi fase terbalik. Dalam fase gerak ini, metanol memiliki jumlah yang paling besar karena didasarkan pada kelarutan parasetamol, salisilamida, dan kafein yang besar pada etanol. Pada penelitian tidak digunakan etanol namun metanol karena metanol dapat melarutkan ketiga komponen tersebut dan memiliki viskositas yang lebih rendah daripada etanol sehingga dapat mengurangi tekanan pada kolom dan meningkatkan efisiensi kolom untuk memisahkan komponen campuran.
C. Pembuatan Larutan Baku
Larutan baku dibuat dalam 5 seri konsentrasi untuk tiap komponen yang diuji. Konsentrasi untuk parasetamol adalah 4mg%, 6mg%, 8mg%, 10mg%, dan 12mg%. Konsentrasi untuk salisilamida adalah 4mg%, 8mg%, 10mg%, 12mg%, dan 16mg%, sedangkan untuk kafein adalah 1mg%, 1,5mg%, 2mg%, 2,5mg%, dan 3mg%. Pemilihan seri konsentrasi kurva baku ini dimaksudkan agar kadar yang terdapat dalam sampel dapat masuk dalam rentang seri konsentrasi larutan baku yang digunakan sehingga persamaan kurva baku yang diperoleh dapat digunakan untuk menetapkan kadar tiap komponen dalam sampel.
D. Optimasi Metode 1. Penentuan panjang gelombang overlapping
Penentuan panjang gelombang overlapping dimaksudkan untuk mengetahui panjang gelombang di mana parasetamol, salisilamida, dan kafein memiliki serapan yang optimal. Sebelum menentukan panjang gelombang
overlapping, panjang gelombang serapan maksimum dari masing-masing senyawa harus ditentukan lebih dahulu. Hal ini bertujuan untuk mendapatkan panjang gelombang masing-masing senyawa dalam metanol yang menunjukkan serapan maksimum.
gelombang antara 200-300 nm karena parasetamol, salisilamida, dan kafein memiliki panjang gelombang serapan maksimum pada rentang tersebut.
Senyawa yang ditetapkan kadarnya secara spektrofotometri ultraviolet harus memiliki gugus kromofor dalam strukturnya agar dapat menyerap sinar radiasi ultraviolet. Penyerapan sinar radiasi oleh suatu senyawa tergantung pada struktur elektronik dari senyawa tersebut. Pada gugus kromofor yang dimiliki oleh parasetamol, salisilamida, dan kafein terdapat ikatan rangkap yang mengandung elektron π yang bila dikenai sinar radiasi elektromagnetik akan mudah tereksitasi ke tingkat yang lebih tinggi yaitu obital π*. Selain gugus kromofor, terdapat juga gugus auksokrom yang langsung terikat pada gugus kromofor. Gugus auksokrom memiliki pasangan elektron bebas pada orbital n yang dapat berinteraksi dengan elektron π pada kromofor sehingga adanya auksokrom ini akan mengubah panjang gelombang serta intensitas serapan maksimum dari senyawa. Gambar gugus kromofor dan auksokrom masing-masing senyawa dapat dilihat pada gambar berikut.
OH
HN C
O
CH3
Gambar 14. Gugus kromofor dan auksokrom parasetamol
Keterangan : = kromofor
OH
C O
NH2
Gambar 15. Gugus kromofor dan auksokrom salisilamida
Keterangan : = kromofor
= auksokrom
N
N
N
N CH3
CH3
H3C
O
O
Gambar 16. Gugus kromofor kafein
Keterangan : = kromofor
Gambar 17. Spektrum serapan parasetamol ( λmaks = 250,8 nm)
Keterangan : A = konsentrasi 0,8mg%; B = konsentrasi 1mg%
Gambar 18. Spektrum serapan salisilamida ( λmaks = 241,5 nm )
Keterangan : A = konsentrasi 0,8mg%; B = konsentrasi 1mg%
Gambar 19. Spektrum serapan kafein ( λmaks = 272,2 nm )
Keterangan : A = konsentrasi 0,8mg%; B = konsentrasi 1mg%
Analisis tidak dilakukan pada panjang gelombang serapan maksimum salah satu senyawa karena hanya sensitif terhadap perubahan konsentrasi senyawa yang bersangkutan. Karena hal tersebut diperlukan panjang gelombang
overlapping dari ketiga senyawa yang diuji. Spektrum yang dihasilkan dari tiap senyawa pada konsentrasi yang sama (1mg%) ditumpang tindihkan sehingga didapatkan gambar sebagai berikut.
Gambar 20. Gabungan spektrum serapan parasetamol (a), salisilamida(b), dan kafein (c) dengan konsentrasi 1mg%
panjang gelombang ini serapan kafein cukup optimal, sedangkan untuk serapan parasetamol dan salisilamida kurang optimal namun tetap dapat terdeteksi karena konsentrasinya yang cukup besar dalam sampel.
2. Pembuatan kurva baku parasetamol, salisilamida, dan kafein
Tiap seri konsentrasi larutan baku parasetamol, salisilamida, dan kafein diinjeksikan pada KCKT dengan kondisi :
Instrument : Shimadzu LC-10 AD
Kolom : C18 merk Bondapack dengan panjang kolom 30 cm
No. P6127IBO2
Fase gerak : metanol : aquabidest : amonia (70:29:1) Flow rate : 1,5 ml/menit
AUFs/Attenuation : 0,01/ 7
Detektor : UV pada 270 nm.
Gambar 21. Kromatogram campuran baku parasetamol, salisilamida, dan kafein (4:4:1)
Pada kromatogram terlihat adanya perbedaan waktu retensi (tR) tiap
OH
Gambar 22. Gugus non polar pada parasetamol (A), salisilamida (B), dan kafein (C) yang berinteraksi dengan fase diam
Keterangan : = gugus non polar
Dari gambar tersebut dapat dilihat bahwa kafein memiliki lebih banyak gugus non polar daripada parasetamol dan salisilamida. Hal inilah yang menyebabkan kafein akan lebih tertahan pada fase diam daripada parasetamol dan salisilamida. Pada kromatogram yang dihasilkan didapatkan juga bahwa kafein memiliki waktu retensi yang paling lama daripada senyawa lainnya. Parasetamol dan salisilamida memiliki gugus non polar yang sama yaitu adanya benzen. Hal ini menyebakan interaksi kedua senyawa ini dengan fase diamnya mirip atau serupa sehingga pemilihan fase gerak sangat berpengaruh dalam pemisahan kedua senyawa ini.
daripada interaksinya pada fase diam. Kafein tidak tergaramkan karena sifatnya yang basa seperti amonia sehingga tidak terjadi reaksi penggaraman dengan adanya amonia. Reaksi penggaraman parasetamol dan salisilamida dengan adanya amonia dapat digambarkan sebagai berikut.
OH
Gambar 23. Reaksi penggaraman parasetamol dengan adanya amonia
OH
Gambar 24. Reaksi penggaraman salisilamida dengan adanya amonia
Penentuan persamaan kurva baku untuk masing-masing senyawa dilakukan 3 kali replikasi. Persamaan kurva baku menyatakan hubungan linier antara konsentrasi dan AUC yang dihasilkan. Sebagai parameter linieritasnya digunakan koefisien korelasi (r). Koefisien korelasi menunjukkan korelasi antara konsentrasi dan AUC. Dalam penelitian ini, dari 3 kali replikasi dipilih salah satu replikasi yang kemudian digunakan sebagai data kurva baku. Pemilihan data kurva baku ini didasarkan pada nilai r yang digunakan, yaitu nilai r yang lebih besar dari nilai r tabel untuk lima data dengan derajat bebas (db) = 3 yaitu sebesar 0,878 (pada taraf kepercayaan 95%). Selain itu pemilihan data kurva baku juga didasarkan pada nilai SE (standard error), yaitu nilai SE yang paling kecil karena semakin kecil SE maka kesalahan yang terjadi dalam penelitian juga semakin kecil. Persamaan untuk masing-masing baku dari parasetamol, salisilamida, dan kafein dapat dilihat pada tabel berikut.
Tabel II. Data kurva baku parasetamol
Rep. 1 Rep. 2 Rep. 3 *
Tabel III. Data kurva baku salisilamida
Keterangan : = merupakan data kurva baku yang digunakan
Tabel IV. Data kurva baku kafein
Rep. 1 * Rep. 2 Rep. 3 Keterangan : = merupakan data kurva baku yang digunakan
parasetamol. Persamaan kurva baku parasetamol yang diperoleh adalah Y = 154419,6 X + 30736. Dari data kurva baku salisilamida dapat dilihat bahwa pada replikasi kedua menunjukkan nilai r yang terbaik dan nilai SE terkecil sehingga persamaan tersebut yang digunakan untuk menghitung kadar salisilamida. Persamaan kurva baku salisilamida yang diperoleh adalah Y = 6589,43 X + 2005,15, sedangkan dari data kurva baku kafein diperoleh persamaan kurva baku Y= 180160,9 X + 7076,85 yang merupakan data kurva baku kafein replikasi pertama.
E.Analisis Kualitatif
Pada KCKT, analisis kualitatif dilakukan dengan membandingkan waktu retensi (tR) sampel dengan waktu retensi (tR) baku/pembanding (gb. 20). Pada
penelitian ini dilakukan analisis kualitatif dari sampel. Gambar kromatogram sampel dapat dilihat pada gambar berikut.
Dari gambar kromatogram sampel tersebut dapat dilihat waktu retensi tiap senyawa dan dibandingkan dengan waktu retensi baku yang dapat ditampilkan sebagai berikut.
Tabel V. Data waktu retensi (tR) masing-masing senyawa baku dan dalam sampel
Senyawa tR baku (menit) tR sampel (menit)
Parasetamol Salisilamida
Kafein
1,777 1,248 2,337
1,932 1,295 2,337
Dari tabel di atas dapat disimpulkan bahwa dalam sampel mengandung parasetamol, salisilamida, dan kafein. Hal ini karena baik pada baku maupun pada sampel, masing-masing senyawa memiliki waktu retensi yang relatif sama.
F.Analisis Kuantitatif
Tabel VI. Data kadar parasetamol, salisilamida, dan kafein dalam tablet
Parasetamol Salisilamida Kafein
Sampel
AUC (Y) Kadar
(mg/tablet) AUC (Y)
Kadar
(mg/tablet) AUC (Y)
Kadar
Berdasarkan tabel tersebut dapat dilihat kadar parasetamol, salisilamida, dan kafein dalam tablet secara berturut-turut adalah (194,3 2,35)mg/tablet, (196,66±3,24)mg/tablet, dan (51,1
±
BAB V
KESIMPULAN DAN SARAN A. Kesimpulan 1. Metode KCKT dengan kondisi :
Instrument : Shimadzu LC-10 AD
Kolom : C18 merk Bondapack dengan panjang kolom 30 cm
No. P6127IBO2
Fase gerak : metanol : aquabidest : amonia (70:29:1) Flow rate : 1,5 ml/menit
AUFs/Attenuation : 0,01/ 7
Detektor : UV pada 270 nm
dapat digunakan untuk penetapan kadar parasetamol, salisilamida, dan kafein dalam tablet.
2. Kadar parasetamol, salisilamida, dan kafein dalam tablet secara berturut-turut adalah (194,3 ± 2,35) mg/tablet, (196,66 ± 3,24) mg/tablet, dan (51,15 ± 0,34) mg/tablet. Kadar tersebut sesuai dengan jumlah yang tertera pada etiket obat.
B. Saran
Perlu dilakukan perbandingan hasil penelitian penetapan kadar parasetamol, salisilamida, dan kafein dalam tablet menggunakan metode Kromatografi Cair Kinerja Tinggi (KCKT) dan KLT densitometri.
DAFTAR PUSTAKA
Anonim, 1995, Farmakope Indonesia, Edisi IV, 4, 254, 649, 650, 753,999, 1009-1010 Departemen Kesehatan Republik Indonesia, Jakarta.
Anonim, 1998, Metode Analisa Obat 1997/1998, 41-46, Pusat Pemeriksaan Obat dan Makanan, Yogyakarta.
Anonim, 2000, Informatorium Obat Nasional Indonesia 2000, 184, Departemen Kesehatan Republik Indonesia Direktorat Jendral Pengawasan Obat dan Makanan, Jakarta.
Anonim, 2005, The United States Pharmacopeia, 28th ed.,20, 2459, 2711, United States Parmacopial Convention Inc., Canada.
Ansel, H. C., 1985, Introduction to Pharmaceutical Dosage Forms, diterjemahkan oleh Farida Ibrahim, 244-245, Penerbit Universitas Indonesia, Jakarta. Aulton, M., and Summer, M., 1994, Pharmaceutics : The Science of Dosage
Form Designs, 2nd ed., 305-306, Churchill Living Stone, London.
Auterhoff, H., Kovar, K.A., 1981, Identifizierung von Arzneistoffen, diterjemahkan oleh N. C. Sugiarso, 165, 166, Penerbit ITB, Bandung.
Christian, G. D., 2004, Analytical Chemistry, 6th ed., 465, Jhon Willey & Sons, Inc., USA.
Clarke, E.G.C., 1969, Isolation and Identification of Drugs, 234, 465, 538, The Pharmaceutical Press, London.
Friamata, R. D., 2006, Penetapan Kadar Kafein dalam Campuran Parasetamol, Salisilamida dan Kafein secara Spektrofotometri Derivatif dengan Aplikasi Metode Peak to Peak, Skripsi, Fakultas Farmasi Universitas Sanata Dharma, Yogyakarta.
Gritter, R.J., Bobbit, J.M., and Schwarting, A.E., 1985, Introduction to Chromatography, diterjemahkan oleh Kosasih Padmawinata, Edisi II, 205-219, Penerbit ITB, Bandung.
Harris, D. C., 1999, Quantitative Chemical Analysis, 2nd ed., 643, 648, 661, 664, W. H. Freeman and Company, New York.