www.elsevier.com / locate / bres
Research report
Differential antagonism of endomorphin-1 and endomorphin-2 spinal
antinociception by naloxonazine and 3-methoxynaltrexone
a ,
*
a a bShinobu Sakurada
, Takafumi Hayashi , Masayuki Yuhki , Tsutomu Fujimura ,
b a c d
Kimie Murayama , Akihiko Yonezawa , Chikai Sakurada , Mitsuhiro Takeshita ,
e e c
James E. Zadina , Abba J. Kastin , Tsukasa Sakurada
a
Department of Physiology and Anatomy, Tohoku Pharmaceutical University, 4-4-1 Komatsushima, Sendai 981-8558, Japan b
Division of Biochemical Analysis, Central Laboratory of Medical Sciences, Juntendo University School of Medicine, 2-1-1 Hongo, Tokyo 113-8421,
Japan
c
Department of Biochemistry, Daiichi College of Pharmaceutical Sciences, 22-1 Tamagawa-cho, Minami-ku, Fukuoka 815-8511, Japan d
Department of Pharmaceutics, Tohoku Pharmaceutical University, 4-4-1 Komatsushima, Sendai 981-8558, Japan e
Veterans Affairs Medical Center and Tulane University School of Medicine, New Orleans, LA, USA Accepted 25 July 2000
Abstract
To determine the role of spinal mu-opioid receptor subtypes in antinociception induced by intrathecal (i.t.) injection of endomorphin-1 and -2, we assessed the effects ofb-funaltrexamine (a selective mu-opioid receptor antagonist) naloxonazine (a selective antagonist at the mu -opioid receptor) and a novel receptor antagonist (3-methoxynaltrexone) using the paw-withdrawal test. Antinociception of i.t.1
2 4 5
endomorphins and [D-Ala , MePhe , Gly(ol) ]enkephalin (DAMGO) was completely reversed by pretreatment withb-funaltrexamine (40
mg / kg s.c.). Pretreatment with a variety of doses of i.t. or s.c. naloxonazine 24 h before testing antagonized the antinociception of endomorphin-1, -2 and DAMGO. Judging from the ID50 values of naloxonazine, the antinociceptive effect of endomorphin-2 was more sensitive to naloxonazine than that of endomorphin-1 or DAMGO. The selective morphine-6b-glucuronide antagonist, 3-methoxynaltrex-one, which blocked endomorphin-2-induced antinociception at each dose (0.25 mg / kg s.c. or 2.5 ng i.t.) that was inactive against DAMGO, did not affect endomorphin-1-induced antinociception but shifted the dose–response curve of endomorphin-2 3-fold to the right. These findings may be interpreted as indicative of the existence of a novel mu-opioid receptor subtype in spinal sites, where antinociception of morphine-6b-glucuronide and endomorphin-2 are antagonized by 3-methoxynaltrexone. The present results suggest that endomorphin-1 and endomorphin-2 may produce antinociception through different subtypes of mu-opioid receptor. 2000 Elsevier Science B.V. All rights reserved.
Theme: Neurotransmitters, modulators, transporters, and receptors
Topic: Opioid receptors
Keywords: Naloxonazine;b-Funaltrexamine; 3-Methoxynaltrexone; Endomorphin-1; Endomorphin-2
1. Introduction reactivity for endomorphin-1 and endomorphin-2 were
widely and densely distributed throughout human and rat Recently two novel peptides have been isolated from brain whereas endomorphin-2-immunoreactivity is present bovine brain [41] that have high affinity and selectivity for in the superficial laminae of the spinal cord dorsal horn the mu-opioid receptor, and have been termed where primary afferent nociceptors terminate. These find-endomorphin-1 (Tyr-Pro-Trp-Phe-NH ) and endomorphin-2 ings indicate that endomorphin-2 plays a physiological role 2 (Tyr-Pro-Phe-Phe-NH ). Furthermore, both peptides2 in regulating nociceptive information in the spinal cord have been isolated from human brain [6] and immuno- [16–18]. Both endomorphin-1 and endomorphin-2 sig-nificantly increase nociceptive thresholds after both spinal and supraspinal administration [4,31,35,41] which are
*Corresponding author. Fax:181-22-275-2013.
E-mail address: [email protected] (S. Sakurada). blocked by mu-receptor-specific antagonists, naloxone and
b-funaltrexamine [41]. Neither supraspinal endomorphin-1 thesized in our laboratory. b-Funaltrexamine and nalox-or -2 elicited antinociception in CXBK mice which are onazine were purchased from Research Biochemical
Inter-2 4
also insensitive to morphine [4]. national (Natick, MA), and [D-Ala , MePhe ,
5
There is biochemical and pharmacological evidence Gly(ol) ]enkephalin (DAMGO) was from Sigma (St.
supporting the existence of mu-opioid receptor subtypes Louis, MO). Endomorphins and DAMGO were dissolved [5,20,40] which are localized in spinal and supraspinal in sterile artificial cerebrospinal fluid (CSF) containing 7.4 structures involved in the modulation of nociception [19]. g NaCl, 0.19 g KCl, 0.19 g MgCl , 0.14 g CaCl / 1000 ml2 2
At least two mu-opioid receptor subtypes have been and 3-methoxynaltrexone (2.5 ng / 2 ml) was dissolved in proposed: mu and mu opioid receptor subtypes [23,24].1 2 0.1% Tween in CSF. b-Funaltrexamine (40 mg / kg, s.c.)
b-Funaltrexamine irreversibly antagonizes both mu1 and and naloxonazine (6.91, 10.4, 15.6, 23.3, 35, 39.4, 43.7, mu2 opioid receptors and inhibits both supraspinal and 52.5, 65.7 and 78.7 mg / kg, s.c.) were dissolved in saline spinal antinociception, whereas naloxonazine selectively and injected s.c. in a volume of 0.1 ml / 10 g body weight antagonizes the mu -opioid receptor. It has been suggested1 24 h before testing. Under these conditions, b -funaltrex-that these receptor subtypes have different physiological amine (40 mg / kg, s.c.) antagonizes both mu - and mu -1 2
roles, with mu -opioid receptors mediating supraspinal1 mediated antinociception, and naloxonazine’s actions of antinociception, whereas mu -opioid receptors mediate2 s.c. 35 mg / kg are relatively selective for mu -receptors1
spinal antinociception measured by the tail-flick test. [13]. 3-Methoxynaltrexone (0.125, 0.1875, 0.25, 0.375, Recent report suggests that heroin and morphine-6b- 0.5, 0.75 and 1.0 mg / kg, s.c.) was dissolved in 0.1% glucuronide in the family of mu opioids both act through a Tween-80 solution in saline and administered in a volume novel third mu-opioid receptor subtype, distinct from those of 0.1 ml / 10 g body weight. 3-Methoxynaltrexone an-mediating morphine’s actions. 3-methoxynaltrexone selec- tagonizes heroin-induced antinociception at doses (2.5 ng
3
tively competes for [ H]morphine-6b-glucuronide binding i.t. or 0.25 mg / kg s.c.) that are inactive against morphine-antagonizes the antinociceptive actions of heroin and and DAMGO-induced antinociception [2,34].
morphine-6b-glucuronide without interfering with mu
(morphine and DAMGO), delta- or kappa-opioid receptor 2.4. Assessment of nociceptive threshold agonist-induced antinociception [2].
The objective of the present study was to determine The antinociceptive activity of opioid peptides against whether antinociceptive activities of i.t. administered the response to a thermal stimulus was assessed by the endomorphin-1 and endomorphin-2 are mediated through mouse paw-withdrawal test. Antinociceptive thresholds mu-opioid receptor subtypes, mu , mu , or a novel mu-1 2 were determined by use of an automated tail-flick unit
opioid receptor in the mouse paw-withdrawal test. (BM kiki, Tokyo). Mice were adapted to the testing
environment for at least 1 h before any stimulation. Each animal was restrained with a soft cloth to reduce visual
2. Materials and methods stimuli and the radiant heat source was positioned under the glass floor directly beneath the hind paw. The reaction
2.1. Animals time to remove the paw from the source of noxious radiant
heat was measured. The intensity of the light beam was Adult male ddY mice weighing 22–25 g were housed in adjusted so that baseline reaction time was 2–4 s. The light a light- and temperature-controlled room (lights on 09:00– beam was focused on the same plantar spot of the hind 21:00 h; 248C) and had free access to food and water. The paw in all animals. To prevent tissue damage trials were experiments were performed with the approval of the terminated automatically if the mouse did not lift the paw Committee of Animal Experiments in Tohoku Pharma- within 10 s. The average threshold of the control response
ceutical University. before injections of compounds was determined by two
consecutive measurements each separated by 10 min. No
2.2. Injection procedure animal was used more than once. To prevent experimenter
bias, observers were uninformed of the dose of the The procedure for intrathecal (i.t.) injections was endomorphins, DAMGO and 3-methoxynaltrexone being adapted from the method of Hylden and Wilcox [11] with injected, and were uninformed of whether 3-methoxynal-a const3-methoxynal-ant injection volume of 2 ml / mouse. For i.t. trexone, naloxonazine orb-funaltrexamine was given as a administration, a 29-gauge needle connected to a Hamilton pretreatment. After determination of pre-drug values, microsyringe was inserted directly between L5 and L6, and animals were injected. Antinociceptive activity for each the drug was administered at a rate of 2ml / 10 s. animal was calculated with the following equation and represented as percent of maximum possible effect (%
2.3. Drugs MPE)5(P22P1 / 102P1)3100, where P1 and P2 are
2.5. Data analysis and statistics
Statistical significance of the data was estimated with a mixed two-factor analysis of variance (ANOVA) followed by Dunnett’s multiple comparison test. A level of prob-ability of 0.05 or less was accepted as significant. The ED50 values and their 95% confidence limits (95% CL) for the antinociceptive effect of the endomorphins and DAMGO were computed according to the method of Litchfield and Wilcoxon [14] using Programs 11 and 47 of the Pharmacological calculations system of Tallarida and Murray [37].
3. Results
3.1. Potency and time course of i.t. injection of
endomorphin-1 and -2
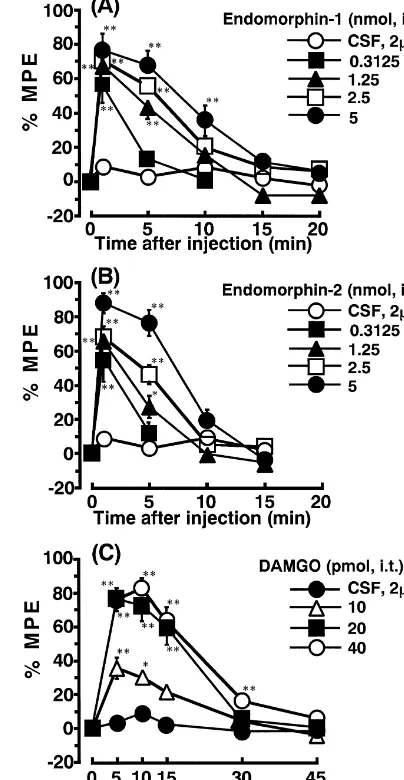
The time course of antinociceptive activity for i.t. endomorphin-1, -2 and DAMGO is shown in Fig. 1. Groups of 10 mice were tested for antinociception at 1, 5, 10, 15, 20, 30 and 45 min. Endomorphin-1 and -2 at 1 min post-injection produced dose-dependent antinociception with ED50 values of 0.14 (0.06–0.32) and 0.24 (0.12– 0.49) nmol, respectively (figure not shown).
Endomorphin-1 and -2 after 5 min of i.t. injection induced antinociception with ED50 values of 1.9 (0.59– 6.11) and 2.6 (1.51–4.47) nmol, respectively (Fig. 1A,B). The two peptides had equal potency as seen from ED50
values. The ED50 value for i.t. DAMGO was 14.0 (8.52– 22.99) pmol at the 5–10 min peak time of antinociception (Fig. 1C).
Fig. 1. Time course of effects of i.t. administered endomorphin-1 (A),
3.2. Antagonistic effects of s.c.b-funaltrexamine and
endomorphin-2 (B) and DAMGO (C) in the mouse paw-withdrawal test.
naloxonazine on antinociception induced by
Antinociception was expressed as percent of maximum possible effect (%
endomorphins MPE)5(post-drug responsive latency2pre-drug responsive latency / 102
pre-drug responsive latency)3100. Each data point represents the
Groups of 10 mice received eitherb-funaltrexamine (40 mean6S.E.M. for 10 mice. **P,0.01, *P,0.05, compared to the respective value in the CSF-control group.
mg / kg, s.c.) or naloxonazine (35 mg / kg, s.c.) 24 h before i.t. injection of endomorphins or DAMGO. Pretreatment of
b-funaltrexamine markedly decreased the antinociceptive DAMGO-induced antinociception were 48 (39.91–57.73) responses in both endomorphins and DAMGO treatment mg / kg, 12 (9.65–14.93) mg / kg and 60 (51.58–69.79) groups compared with non-pretreatment groups, confirm- mg / kg, respectively.
ing the involvement of mu-opioid receptors in the response
of the three opioid peptides. Endomorphin-1- and DAM- 3.3. Effects of pretreatment with i.t. injected GO-induced antinociception were insensitive to nalox- naloxonazine on spinal endomorphin-induced
onazine (35 mg / kg, s.c.), whereas endomorphin-2 re- antinociception
sponses were completely antagonized by a lower dose
(23.3 mg / kg,s.c.) of the mu -opioid receptor antagonist.1 The antagonistic effects of naloxonazine on antinocicep-Different dose responses for naloxonazine against a fixed tion induced by equipotent doses of i.t. endomorphins and
dose of endomorphins (5 nmol) or DAMGO (20 pmol) DAMGO were examined. Groups of mice received graded
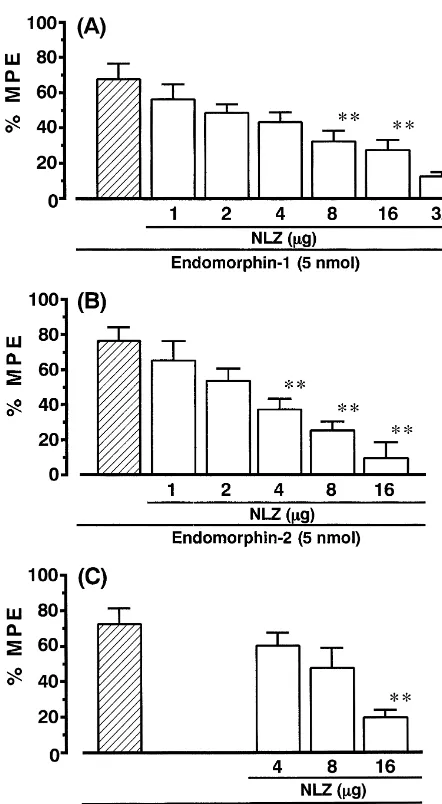
an-Fig. 2. Effects of naloxonazine and b-funaltrexamine on i.t. endomorphin-1-, endomorphin-2- and DAMGO-induced antinociception in the mouse paw-withdrawal test. Graded doses of naloxonazine (NLZ) andb-funaltrexamine (b-FNA, 40 mg / kg) were administered s.c. 24 h before i.t. administration of endomorphin-1, endomorphin-2 and DAMGO. Antinociceptive effects were measured 5 min after i.t. administration of the endomorphins or 10 min after i.t. DAMGO. Each column represents the mean S.E.M. for 10 mice. **P,0.01 compared to each agonist alone.
tinociception of endomorphin-1. Higher doses (8 or 16mg) endomorphin-2 shifted 3-fold to the right (Fig. 4C). of naloxonazine significantly decreased endomorphin-1- However, 3-methoxynaltrexone (2.5 ng) in combination
and DAMGO-induced antinociception, but the antagonism with endomorphin-1 and DAMGO was ineffective at
was not complete. Pretreatment with naloxonazine at a attenuating the responses to i.t. administration of both dose of 32 mg completely antagonized antinociception endomorphin-1 and DAMGO.
induced by endomorphin-1 (Fig. 3). Endomorphin-2-in-duced antinociception was completely antagonized by
pretreatment with a dose of 16 mg of naloxonazine, 4. Discussion
indicating that the endomorphin-2 response was more
sensitive to i.t. naloxonazine than endomorphin-1. ID50 Both endomorphin-1 and -2 induced dose-dependent values for i.t. naloxonazine on endomorphin-1, antinociception after spinal administration in the paw-endomorphin-2 and DAMGO-induced antinociception withdrawal test. The peak effects of endomorphins-induced were 10.0 (4.24–23.57)mg, 3.8 (2.33–6.21) mg and 10.5 antinociception occurred rapidly, within 1 min of the i.t.
(6.70–16.46) mg, respectively. injection, and disappeared at 10–15 min after injection.
The present results of i.t. injected endomorphins are in 3.4. Inhibition of endomorphin-induced antinociception agreement with those of Stone et al. [36] and Sakurada et
by3-methoxynaltrexone al. [31] who reported that the antinociceptive effect of the
endomorphins is short-lasting and is absent 15 min follow-The selective morphine-6-glucuronide antagonist, 3- ing i.t. injection as assayed by the tail-flick test and the methoxynaltrexone (0.25 mg / kg, s.c.), which blocked tail-pressure test. Endomorphins are small peptides that endomorphin-2-antinociception at a dose that was inactive consist of only four amino acids, making them vulnerable against DAMGO did not affect endomorphin-1-induced to rapid degradation by peptidases. Dipeptidyl peptidase antinociception (Fig. 4A). Pretreatment with 0.25 mg / kg IV is a membrane-bound serine proteinase proposed to be of 3-methoxynaltrexone markedly decreased 60% of the involved in the inactivation of endomorphins [35]. antinociceptive response to i.t. endomorphin-2 without We used a variety of doses of i.t. and s.c. naloxonazine influencing endomorphin-1- and DAMGO-induced an- and 3-methoxynaltrexone to determine sensitivity to
an-tinociception (Fig. 4A). tagonists of mu opioid subtypes involved in the
an-At 5 and 10 min after injection, the response to i.t. tinociceptive responses to the endomorphins and DAMGO. endomorphin-2 was markedly decreased by coadministra- b-Funaltrexamine irreversibly antagonizes both mu - and1
tion of 2.5 ng of 3-methoxynaltrexone (Fig. 4C). The ED50 mu -opioid receptors [27] and inhibits both supraspinal2
Fig. 4. Effects of 3-methoxynaltrexone on the antinociceptive dose– response curves of endomorphin-1, endomorphin-2 and DAMGO. (A) Groups of mice were administered either endomorphin-1 (5 nmol), Fig. 3. Effects of naloxonazine on endomorphin-1, endomorphin-2 and
endomorphin-2 (5 nmol) or DAMGO (20 pmol) and the indicated dose of DAMGO-induced antinociception in the mouse paw-withdrawal test.
3-methoxynaltrexone. 3-methoxynaltrexone was administered 25 min Groups of mice received the indicated dose of naloxonazine in
combina-before i.t. endomorphins and 20 min combina-before i.t. DAMGO. Antinociceptive tion with endomorphin-1 (5 nmol, i.t.), endomorphin-2 (5 nmol, i.t.) and
effects were measured 5 min after i.t. endomorphins and 10 min after i.t. DAMGO (20 pmol, i.t.). Graded doses of naloxonazine (NLZ, white
DAMGO. (B,C) Groups of mice were administered the indicated doses of column) were administered i.t. 24 h before i.t. administration of the three
endomorphin-1 (B) or -2 (C) alone or in combination with a fixed dose of peptides. **P,0.01 compared to each agonist alone.
3-methoxynaltrexone (2.5 ng i.t.). **P,0.01, *P,0.05 compared to each agonist alone.
raspinal antinociception [9,13,25,26]. Recent behavioral pharmacological studies suggest the presence of mu -1
receptors sensitive to naloxonazine in spinal sites as completely blocked by pretreatment with a reasonable dose assayed with the formalin, hot-plate, tail-pressure and tail- of 35 mg / kg s.c. naloxonazine to obtain a relative mu1
flick tests [31–33]. Autoradiographic studies show that selectivity in the paw-withdrawal test. Moreover, mu - and mu -opioid receptor subtypes are localized in1 2 endomorphin-2 was significantly antagonized by pretreat-spinal and suprapretreat-spinal structures involved in the modula- ment with a low dose of naloxonazine (10.37 mg / kg s.c.),
tion of nociception [19]. an irreversible mu -opioid receptor antagonist, suggesting1
It should be noted that a reasonable naloxonazine dose that endomorphin-2 may be a highly selective agonist at to obtain a relative mu selectivity in mice would be 351 the mu -opioid receptor subtype of mu-opioid subtypes1
mg / kg s.c.) significantly attenuated DAMGO-induced endomorphin-2 acts through a site distinct from antinociception (Fig. 2), indicating that at high doses endomorphin-1 and DAMGO. The discovery that the naloxonazine loses much of its selectivity mu1 opioid antinociception induced by morphine-6b-glucuronide,
receptors. heroin, and 6-acetylmorphine (a heroin metabolite) can be
Intrathecal naloxonazine did not block the antinocicep- antagonized by 3-methoxynaltrexone at doses which are tive effects of DAMGO and endomorphin-1 at a dose of 4 inactive against morphine has proven to be a means to mg which significantly antagonized endomorphin-2-in- distinguish the different antinociceptive mechanisms of duced antinociception. DAMGO and endomorphin-1 are alkaloids within the mu-opioid receptor agonist family extremely insensitive to antagonism by pretreatment with [2,39]. The present study is the first to show that 3-i.t. naloxonazine judging from the ID50 values of nalox- methoxynaltrexone can also distinguish the actions of onazine. These results suggest that a reasonable dose of i.t. different peptide mu agonists. 3-methoxynaltrexone selec-administered naloxonazine to get a relative mu selectivity1 tively blocked the antinociception of endomorphin-2 far
in mice would be 4mg / mouse. more effectively than that of endomorphin-1 or DAMGO.
As seen from the ID50 values of naloxonazine, Thus, based on antagonism by 3-methoxynaltrexone,
endomorphin-2 was relatively sensitive to naloxonazine endomorphin-2, but not endomorphin-1 or DAMGO, has whereas endomorphin-1 was relatively insensitive to an- behavioral and pharmacological similarity to morphine-6b -tagonism by both i.t. and s.c. pretreatment with nalox- glucuronide and heroin.
onazine. This indicates that endomorphin-1 can act as a A single gene encoding the mouse mu-opioid receptor
predominantly mu -opioid2 receptor agonist and (MOR-1) has been cloned [28,38] and seven splice
var-endomorphin-2 as a mu -opioid receptor agonist in the1 iants have been identified [1,21,22,42]. Morphine produces current test paradigm. As in our previous report with the antinociception by activating mu-opioid receptors encoded tail-pressure test, naloxonazine (35 mg / kg s.c.) did not by the MOR-1 gene. In antisense mapping studies [29,30], completely abolish the action of i.t. endomorphin-2. The the antisense oligodeoxynucleotides targeting exon 1 of reason for this difference may be related to different MOR-1 effectively reduce the antinociceptive effect
pro-nociceptive assays. duced by morphine without affecting the action of
An alternative explanation for the differential effective- morphine-6b-glucuronide, an extremely potent mu-opioid ness of the mu antagonists against the various agonists is receptor agonist. By contrast, antisense probes targeting that endomorphin-2 has lower efficacy than endomorphin-1 exons 2 and 3 reduce the antinociceptive effect produced or DAMGO. Irreversible antagonists have long been used by morphine-6b-glucuronide, but not that by morphine to estimate efficacy [3], and lower efficacy agonists are even though both opioids belong to the mu-opioid family. more susceptible than higher efficacy agonists to a reduc- Indeed, i.c.v. morphine-6b-glucuronide produces an-tion in maximal effects by irreversible antagonists [12]. tinociceptive effects in a MOR-1 exon 1 knockout mouse Thus, theb-funaltrexamine and naloxonazine results could in which morphine and DAMGO are inactive [34]. reflect different receptor subtypes or different efficacies. More recently, five new MOR-1 exons (exon 6, 7, 8, 9 The lack of a selective irreversible antagonist for subtypes and 10) have been identified, and antisense mapping of other than the mu receptor limits the ability to distinguish these exons revealed that they were all involved in between these possibilities. Studies with GTP-g-S binding morphine antinociception without altering antinociception assays, however, have failed to detect significant differ- of morphine-6b-glucuronide [21,22]. Thus, the antisense
ences in efficacy between endomorphin-1 and experiments support the idea of distinct sites, but the
endomorphin-2 [8,10]. newly discovered exons are not a component of the
Reversible antagonists, by contrast, have provided evi- receptor responsible for morphine-6b-glucuronide
an-dence that different agonists act through different re- tinociception [21,22]. Both endomorphin-1 and
ceptors. Evidence for the presence of separate mu and delta endomorphin-2 bind with high affinity to each of the new receptors in the vas deferens, for example, included the splice variants of mu receptor [21,22].
demonstration that naloxone antagonized the effects of The original division of mu - and mu -receptor subtypes1 2
morphine more potently than those of enkephalin [15]. In was characterized primarily on the basis of pharmaco-the present study, endomorphin-2 was more susceptible logical studies using naloxonazine. Similarly, the more
than DAMGO and endomorphin-1 to antagonism by the recent use of 3-methoxynaltrexone revealed binding
reversible antagonist, 3-methoxynaltrexone. Indeed, as characteristics of morphine-6b-glucuronide and heroin that shown in Fig. 4C, 2.5 ng of the antagonist produced a were distinct from morphine, suggesting a possible novel
Endomorphin-2 is an endogenous opioid in primary sensory afferent
of antagonism shown by these compounds. Regardless, the
fibers, Peptides 19 (1998) 1783–1789.
present results demonstrate that endomorphin-2 interacts
[18] S. Martin-Schild, A.A. Gerall, A.J. Kastin, J.E. Zadina, Differential
with mu receptors in a manner that is similar to morphine- distribution of endomorphin 1- and endomorphin 2-like immuno-6b-glucuronide and pharmacologically distinguishable reactivities in the CNS of the rodent, J. Comp. Neurol. 405 (1999)
from morphine as well as other peptides, including 450–471.
[19] A.S. Moskowitz, R.R. Goodman, Autoradiographic distribution of
DAMGO and the closely related peptide endomorphin-1.
mu and mu opioid binding in the mouse central nervous system,1 2
Thus, the endomorphins can serve a unique role in
Brain Res. 360 (1985) 117–129.
understanding the complex agonist–receptor interactions at [20] S.L. Nishimura, L.D. Rech, G.W. Pasternak, Biochemical
characteri-3
the mu receptor and its potential isoforms, subtypes, and zation of high-affinity H-opioid binding. Further evidence for Mu
1
physical states. sites, Mol. Pharmacol. 25 (1984) 29–37.
[21] Y.-X. Pan, J. Xu, E. Bolan, C. Abbadie, A. Chang, A. Zuckerman, G. Rossi, G.W. Pasternak, Identification and characterization of three new alternatively spliced m-opioid receptor isoforms, Mol. Phar-macol. 56 (1999) 396–403.
References
[22] Y.-X. Pan, J. Xu, E. Bolan, A. Chang, L. Mahurter, G. Rossi, G.W. Pasternak, Isolation and expression of a novel alternatively spliced [1] L.A. Bare, E. Mansson, D. Yang, Expression of two variants of the mu-opioid receptor isoform, MOR-1F, FEBS Lett. 466 (2000) 337–
human mu-opioid receptor mRNA in SK-N-SH cells and human 340.
brain, FEBS Lett. 354 (1994) 213–216. [23] G.W. Pasternak, Pharmacological mechanisms of opioid analgesics, [2] G.P. Brown, K. Yang, M.A. King, G.C. Rossi, L. Leventhal, A. Clin. Neuropharmacol. 16 (1993) 1–18.
Chang, G.W. Pasternak, 3-Methoxynaltrexone, a selective heroin / [24] G.W. Pasternak, P.L. Wood, Multiple mu opiate receptors, Life Sci. morphine-6b-glucuronide antagonist, FEBS Lett. 412 (1997) 35–38. 38 (1986) 1888–1898.
[3] R.F. Furchgott, P. Bursztyn, Comparison of dissociation constants [25] D. Paul, R.J. Bodnar, M.A. Gistrak, G.W. Pasternak, Different m and of relative efficacies of selected agonists acting on parasympa- receptor subtypes mediate spinal and supraspinal analgesia in mice, thetic receptors, Ann. NY Acad. Sci. 144 (1967) 882–899. Eur. J. Pharmacol. 168 (1989) 307–314.
[4] I.E. Goldberg, G.C. Rossi, S.R. Letchworth, J.P. Mathis, J. Ryan- [26] C.G. Pick, D. Paul, G.W. Pasternak, Comparison of naloxonazine Moro, L. Leventhal, W. Su, D. Emmel, E.A. Bolan, G.W. Pasternak, andb-funaltrexamine antagonism of mu 1 and mu 2 opioid actions, Pharmacological characterization of endomorphin-1 and Life Sci. 48 (1991) 2005–2011.
endomorphin-2 in mouse brain, J. Pharmacol. Exp. Ther. 286 (1998) [27] L.D. Recht, G.W. Pasternak, Effects of b-funaltrexamine on radio-1007–1013. labeled opioid binding, Eur. J. Pharmacol. 230 (1987) 341–344. [5] R.R. Goodman, G.W. Pasternak, Visualization of mu-1 opiate [28] T. Reisine, G.I. Bell, Molecular biology of opioid receptors, Trends
receptors in rat brain by using a computerized autoradiographic Neurosci. 16 (1993) 506–510.
subtraction technique, Proc. Natl. Acad. Sci. USA 82 (1985) 6667– [29] G.C. Rossi, Y.-X. Pan, G.P. Brown, G.W. Pasternak, Antisense
6671. mapping the MOR-1 opioid receptor: evidence for alternative
[6] P. Hackler, J.E. Zadina, L.J. Ge, Isolation of relatively large amounts splicing and a novel morphine-6b-glucuronide receptor, FEBS Lett. of endomorphin-1 and edomorphin-2 from human brain cortex, 369 (1995) 192–196.
Peptides 18 (1997) 1635–1639. [30] G.C. Rossi, L. Leventhal, Y.-X. Pan, J. Cole, W. Su, R.J. Bondnar, [8] L.M. Harrison, A.J. Kastin, J.E. Zadina, Differential effects of G.W. Pasternak, Antisense mapping of MOR-1 in rats: Distinguish-endomorphin-1 endomorphin-2, and Tyr-W-MIF-1 on activation of ing between morphine and morphine-6b-glucuronide antinocicep-G-proteins in SH-SY5Y human neuroblastoma membranes, Peptides tion, J. Pharmacol. Exp. Ther. 281 (1997) 109–114.
19 (1998) 749–753. [31] S. Sakurada, J.E. Zadina, A.B. Kastin, S. Katsuyama, T. Fujimura, [9] J.S. Heyman, C.L. Williams, T.F. Burks, H.I. Mosberg, F. Porreca, K. Murayama, M. Yuki, H. Ueda, T. Sakurada, Differential in-Dissociation of opioid antinociception and central gastrointestinal volvement of m-opioid receptor subtypes in endomorphin-1- and propulsion in the mouse; studies with naloxonazine, J. Pharmacol. -2-induced antinociception, Eur. J. Pharmacol. 372 (1999) 25–30. Exp. Ther. 245 (1988) 238–243. [32] S. Sakurada, S. Takeda, T. Sato, T. Hayashi, M. Yuki, M. Kutsuwa, [10] K. Hosohata, T.H. Burkey, J. Alfaro-Lopez, E. Varga, V.J. Hruby, K. Tan-no, C. Sakurada, K. Kisara, T. Sakurada, Selective antagon-W.R. Roeske, H.I. Yamamura, Endomorphin-1 and endomorphin-2 ism by naloxonazine of antinociception by Tyr-D-Arg-Phe-b-Ala, a are partial agonists at the human m-opioid receptor, Eur. J. Phar- novel dermorphin analogue with high affinity atm-opioid receptors, macol. 346 (1998) 111–114. Eur. J. Pharmacol. 395 (2000) 107–112.
[11] J.L.K. Hylden, G.L. Wilcox, Intrathecal morphine in mice: a new [33] T. Sato, S. Sakurada, N. Takahashi, T. Sakurada, K. Tan-No, K. technique, Eur. J. Pharmacol. 67 (1980) 313–316. Wako, K. Kisara, Contribution of spinal m1-opioid receptors to [12] T.P. Kenakin, Efficacy in drug receptor theory: outdated concept or morphine-induced antinociception, Eur. J. Pharmacol. 369 (1999)
under-valued tool?, Trends Pharmacol. Sci. 20 (1999) 400–404. 183–187.
[13] G.S.F. Ling, R. Simantov, J.A. Clark, G.W. Pasternak, Naloxonazine [34] A.G. Schuller, M.A. King, J. Zhang, E. Bolan, Y.-X. Pan, D.J. action in vivo, Eur. J. Pharmacol. 129 (1986) 33–38. Morgan, A. Chang, M.E. Czick, E.M. Unterwald, G.W. Pasternak, [14] J.T. Litchfield, F. Wilcoxon, A simplified method of evaluating J.E. Pintar, Retention of heroin and morphine-6b-glucuronide dose-effect experiments, J. Pharmacol. Exp. Ther. 96 (1949) 99– analgesia in a new line of mice lacking exon 1 of MOR-1, Nature
113. Neurosci. 2 (1999) 151–156.
[15] J.A. Lord, A.A. Waterfield, J. Hughes, H.W. Kosterliz, Endogenous [35] R. Shane, S. Wilk, R.J. Bodnar, Modulation of endomorphin-2-opioid peptides: multiple agonists and receptors, Nature 267 (1977) induced analgesia by dipeptidyl peptidase IV, Brain Res. 815 (1999)
495–499. 278–286.
[37] R.J. Tallarida, R.B. Murray, Manual of Pharmacological Calcula- and enkephalin binding sites in the central nervous system, Proc. tions with Computer Programs, Springer, New York, 1987. Natl. Acad. Sci. USA 78 (1981) 6181–6185.
[38] G.R. Uhl, S. Childers, G.W. Pasternak, An opiate-receptor gene [41] J.E. Zadina, L. Hackler, L.J. Ge, A.J. Kastin, A potent and selective family reunion, Trends Neurosci. 17 (1994) 89–93. endogenous agonist for the m1-opiate receptor, Nature 386 (1997) [39] J.R. Walker, M. King, E. Izzo, G.F. Koob, G.W. Pasternak, Antagon- 499–502.