LAMPIRAN
A Peta Penelitian Terdahulu yang relevan :
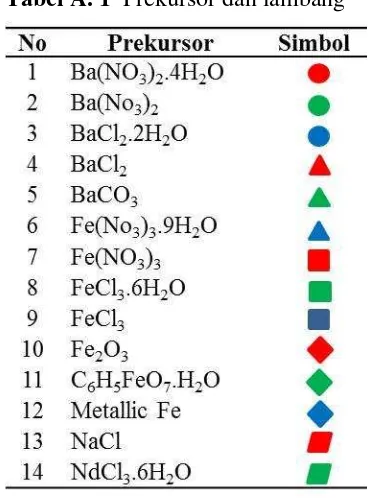
Tabel A. 1 Prekursor dan lambang
Tabel A. 2 Templat dan Lambang
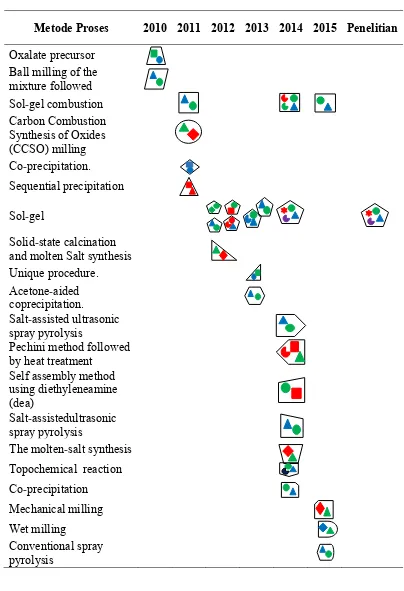
Tabel A. 4 Pemetaan penelitian terdahulu terkait penelitian disertasi yang dilakukan
Metode Proses 2010 2011 2012 2013 2014 2015 Penelitian
Oxalate precursor Ball milling of the mixture followed Sol-gel combustion Carbon Combustion Synthesis of Oxides (CCSO) milling Co-precipitation. Sequential precipitation
Sol-gel
Solid-state calcination and molten Salt synthesis Unique procedure. Acetone-aided coprecipitation.
Salt-assisted ultrasonic spray pyrolysis
Pechini method followed by heat treatment
Self assembly method using diethyleneamine (dea)
Salt-assistedultrasonic spray pyrolysis
The molten-salt synthesis Topochemical reaction Co-precipitation
Mechanical milling Wet milling
Conventional spray pyrolysis
B. Dasar penentuan temperatur kalsinasi Ba(Nd)xFe12O19
Selain fasa pembentukan kristal BaFe12O19 berdasarkan diagram fasa BaO-Fe2O3 (gambar 2.15) sebagai dasar penentuan temperatur kalsinasi digunakan juga hasil penelitian sebelumnya yang relevan seperti Tabel B.1 dan Gambar B.1 serta Gambar B.2 dibawah ini.
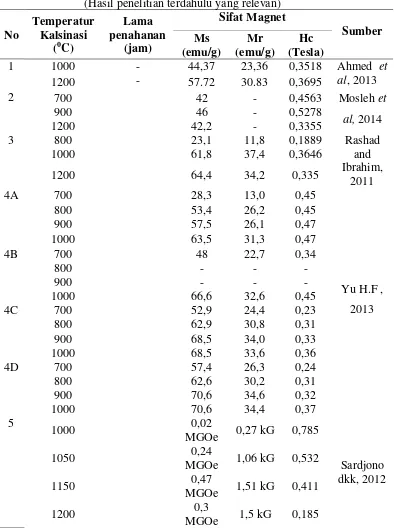
Tabel B.1 Sifat magnet BaFe12O19 berdasarkan temperatur kalsinasi (Hasil penelitian terdahulu yang relevan)
Berdasarkan Tabel B.1 di atas rata-rata nilai Hc maksimum pada temperatur kalsinasi 10000C.
Gambar B.1 Kurva Histerisis BaFe12O19 menunjukkan perubahan nilai Hc akibat perubahan suhu sintering, dan karena itu ukuran butir, untuk Bam, dan hubungan langsung antara Hc (Pullar, 2012)
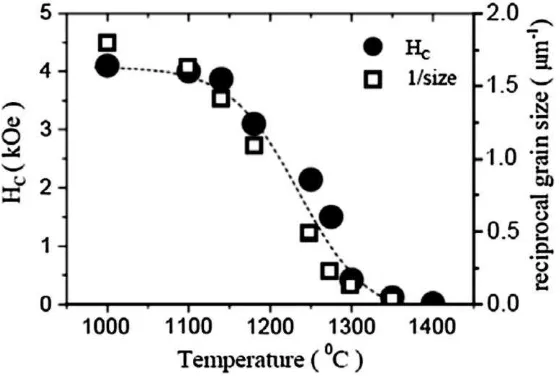
Gambar B.2 Perubahan nilai Hc dan ukuran butir akibat perubahan suhu sintering (Pullar, 2012)
C Pola difraksi sinar-x serbuk hasil sintesis Ba(Nd)xFe12O19
C.1 BaFe12O19
Angle d-value d-value Peak width Peak int Back. int Rel. int Signif. [ø2é] à1 [• ] à2 [• ] [ø2é] [counts] [counts] [%]
C.2 Ba(Nd)0,005Fe12O19
Angle d-value d-value Peak width Peak int Back. int Rel. int Signif. [ø2é] à1 [• ] à2 [• ] [ø2é] [counts] [counts] [%]
C.3 Ba(Nd)0,01Fe12O19
Angle d-value d-value Peak width Peak int Back. int Rel. int Signif. [ø2é] à1 [• ] à2 [• ] [ø2é] [counts] [counts] [%]
C.4 Ba(Nd)0,15Fe12O19
Angle d-value d-value Peak width Peak int Back. int Rel. int Signif.
D. Bentuk input program Espresso
& CONTROL ...
/
& SYSTEM ...
/
& ELECTRONS ...
/
& IONS ... /
ATOMIC_SPECIES Atom1 Mass1 Pseudo1 Atom2 Mass2 Pseudo2 ....
ATOMIC_POSITIONS units Atom1 x1 y1 z1
Atom2 x2 y2 z2 ....
K_POINTS options ...
Keterangan
&CONTROL variabel input yang mengendalikan fluks dari perhitungan dan jumlah I / O pada disk dan di layar, variabel umum mengendalikan run.
&ELECTRON variabel input elektron yang mengontrol algoritma digunakan untuk mencapai solusi konsisten diri dari persamaan KS untuk elektron..
&ION dibutuhkan ketika atom dipindahkan, diabaikan sebaliknya! variabel input yang mengontrol gerakan ion dalam molekul dinamika run atau relaksasi struktural
&CELL dibutuhkan ketika cell moves diabaikan sebaliknya variabel input yang mengendalikan bentuk evolusi dalam sel-bentuk variabel MD atau relaksasi struktural.
&EE dibutuhkan ketika biaya koreksi kepadatan kontra digunakan untuk memecahkan masalah dengan batas kondisi terbuka.
ATOMIC SPESIES, mana, massa dan pseudopotential digunakan untuk setiap spesies atom hadir dalam sistem.
K_POINTS dan bobot dari k-poin digunakan untuk integrasi BZ
Contoh input sistem molukuler N2
& CONTROL calculation = ’scf ’
restart_mode = ’from_scratch ’ pseudo_dir = ’./ pseudo ’ outdir = ’./tmp ’
/
& SYSTEM ibrav = 1 a = 15.0 ntyp = 1 nat = 2
ecutwfc = 30.0 nbnd = 8 /
& ELECTRONS
mixing_beta = 0.5 /
ATOMIC_SPECIES
N 14.00 N.pbe - kjpaw . UPF ATOMIC_POSITIONS angstrom
N 7.50000000 7.50000000 7.50000000 N 7.50000000 7.50000000 8.50000000 K_POINTS gamma
Penjelasan
&CONTROL
calculation = 'scf' melakukan perhitungan total energi melalui algoritma self consistent field. (perhitungan untuk satu konfigurasi atom).
restart_mode = 'from_scratch' melakukan perhitungan dari awal, Mulai dari dugaan awal untuk {ψi (~r )}
pseudo_dir = './pseudo' digunakan untuk menentukan path untuk file pseudopotential
outdir = './tmp' digunakan untuk menentukan direktori untuk file sementara yang digunakan oleh PWSCF. Dalam hal ini, sebuah direktori bernama tmp di bawah direktori saat ini sebagai out dir tersebut. Jika direktori ini tidak hadir, PWSCF akan menciptakannya di runtime.
/
&SYSTEM
Ibrav=1 bentuk crystal simple cubic
a = 15.0 digunakan untuk menentukan parameter kisi untuk kisi kubik sederhana yang kita gunakan. Di sini, diggunakan nilai 15.0 angstrom untuk parameter kisi.
nat = 2 bearti ada 2 buah atom pada sistem.
ecutwfc = 30.0 Energy cutoff for pseudo-potentials berarti menggunakan 30 Ry cutoff energi untuk ekspansi fungsi gelombang. Khas Nilai ecut wfcberkisar antara 20-100 Ry. Semakin besar nilai ecutwfc yang lebih akurat namun akan mengambil lebih umber daya komputasi. Di sini mengambil agak kecil nilai ecutwfc untuk mendapatkan hasil energi kinetik yang cepat cutoff (for planewaves). Nbnd = 8 menyertakan 8 band atau orbital dalam perhitungan untuk
molekul N2, ada 5 elektron valensi untuk setiap atom N. Jadi, ada 2 x 5 = 10 elektron dalam sistem, setiap band atau orbital akan ditempati oleh 2 elektron, jadi perlu setidaknya 5 band untuk perhitungan. Disini berarti ada 3 band kosong.
&ELECTRONS
electron_maxstep = 150 menetapkan SCF iterasi maksimum untuk 150. (jumlah iterasi) Nilai default adalah 60.
mixing_beta = 0.5 nilai parameter pencampuran. Nilainya berkisar 0,0-1,0. Nilai default adalah 0,7. Untuk sistem sederhana, nilai yang lebih besar akan memberikan lebih cepat konvergensi SCF.
/
&IONS
/
ATOMIC_SPECIES
N 14.00 N.pbe - kjpaw . UPF nana fiel referensi untuk atom N
ATOMIC_POSITIONS angstrom posisi atom dalam satuan angstrom
x y z
x y z
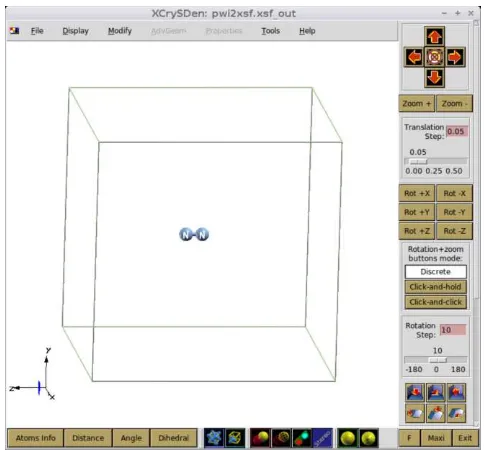
Contoh bentuk tampilan output (gambar molekul N2 hasil program) dapat dilihat pada Gambar 2.40 berikut.
Gambar 2.40 Tampilan Visual Xcrysden molekul N2 (Fathurrahman, 2015)
Hasil Output
bravais - lattice index = 1
lattice parameter ( alat ) = 28.3459 a.u.
unit - cell volume = 22775.6292 (a.u .)^3 number of atoms / cell = 2
number of atomic types = 1 number of electrons = 10.00 number of Kohn - Sham states = 8
kinetic - energy cutoff = 30.0000 Ry charge density cutoff = 120.0000 Ry convergence threshold = 1.0E -06
mixing beta = 0.5000
number of iterations used = 8 plain mixing
E Program Input dan Output Simulasi
Ba 137.32700 Ba.pbe-nsp-van.UPF O 15.99900 O.pbe-rrkjus.UPF Fe 55.84500 Fe.pbe-sp-van_ak.UPF
ATOMIC_POSITIONS {crystal}
O 0.157000000000000 0.843000000000000 0.052000000000000 O 0.157000000000000 0.843000000000000 0.448000000000000 O 0.686000000000000 0.843000000000000 0.448000000000000 O 0.503000000000000 0.006000000000000 0.149400000000000 O 0.503000000000000 0.006000000000000 0.350600000000000 O 0.497000000000000 0.503000000000000 0.649400000000000 O 0.006000000000000 0.503000000000000 0.649400000000000 O 0.497000000000000 0.503000000000000 0.850600000000000 O 0.006000000000000 0.503000000000000 0.850600000000000 O 0.497000000000000 0.994000000000000 0.850600000000000 O 0.497000000000000 0.994000000000000 0.649400000000000 O 0.994000000000000 0.497000000000000 0.149400000000000 O 0.503000000000000 0.497000000000000 0.149400000000000 O 0.503000000000000 0.497000000000000 0.350600000000000 O 0.994000000000000 0.497000000000000 0.350600000000000
K_POINTS automatic 8 8 2 0 0 0
CELL_PARAMETERS {alat}
0.866025403784439 -0.500000000000000 0.000000000000000 0.000000000000000 1.000000000000000 0.000000000000000 0.000000000000000 0.000000000000000 3.948895260583572
PENJELASAN
&CONTROL
calculation = 'relax'perintah untuk melakukan perhitungan optimasi geometri. Untuk optimasi geometri lebih sulit, membutuhkan waktu lebih dari50 langkah.
restart_mode = 'from_scratch'berarti bahwa kita ingin melakukan perhitungan dari awal, Mulai dari dugaan awal untuk{ψi (~r )}
pseudo_dir = './pseudo'digunakan untuk menentukan path untuk file pseudopotential
nstep = 1000 variabel baru untuk pengaturan jumlah yang agak besar ksederhana yang kita gunakan. Disini menggunakan nilai 5.92900 angstromu ntuk parameter kisi.
nat = 64 bearti ada 64 buah atom pada sistem.
ntyp = 3 berartibahwa hanya ada 3 jenisatom (yaitu : Barium, Fe dan Oksigen) dalam sistem (molekul BaFe12O19).
nspin = 2 tipe spin (spin-polarized (single-axis magnetization)).
ecutwfc = 30.0 Energy cutoff for pseudo-potentials berarti menggunakan 30Ry cut off energy untuk ekspansi fungsi gelombang. Khas Nilai ecutwfc berkisar antara 20-100 Ry. Semakin besar nilai ecutwfc yang lebih akurat namun akan mengambil lebih sumber daya komputasi. Di sini mengambil agak kecil nilai ecutwfc untuk mendapatkan hasil yang cepat.
starting_magnetization(3) = 0.5d0 simetri konfigurasi awal antara -1 dan 1 untuk menghitung nilai magnet
occupations = 'smearing' pekerjan smearing oleh beberapa fungsi
smearing = 'm-v'marzari-vanderbilt
degauss = 0.01 lebar semearing
/
&ELECTRONS
mixing_beta = 0.10,1 berarti bahwa kita tentukan nilai 0,1 untuk nilai pencampuran parameter. Nilainya berkisar 0,0-1,0. Nilai default adalah 0,7. Untuk sistem sederhana, nilai yang lebih besar akan memberikan lebih cepat konvergensi SCF. Untuk sistem lebih sulit kurangi nilai mixing_beta.
scf_must_converge = .false. /
/
ATOMIC_SPECIES
Ba 137.32700 Ba.pbe-nsp-van.UPF O 15.99900 O.pbe-rrkjus.UPF Fe 55.84500 Fe.pbe-sp-van_ak.UPF
ATOMIC_POSITIONS {crystal}
Na x y z
Output
Program PWSCF v.5.2.0 starts on 19Feb2016 at 14:54:12
This program is part of the open-source Quantum ESPRESSO suite for quantum simulation of materials; please cite
"P. Giannozzi et al., J. Phys.:Condens. Matter 21 395502 (2009); URL http://www.quantum-espresso.org",
in publications or presentations arising from this work. More details at http://www.quantum-espresso.org/quote
Parallel version (MPI), running on 4 processors R & G space division: proc/nbgrp/npool/nimage = 4 Waiting for input...
Reading input from standard input
Current dimensions of program PWSCF are:
Max number of different atomic species (ntypx) = 10 Max number of k-points (npk) = 40000
Max angular momentum in pseudopotentials (lmaxx) = 3 file O.pbe-rrkjus.UPF: wavefunction(s) 2S renormalized
Subspace diagonalization in iterative solution of the eigenvalue problem: a serial algorithm will be used
Parallelization info ---
sticks: dense smooth PW G-vecs: dense smooth PW Min 433 259 75 57471 26715 4047 Max 434 260 76 57484 26734 4051 Sum 1735 1039 301 229909 106901 16199
Generating pointlists ...
bravais-lattice index = 0
MD5 check sum: a011de417d3466d8e94df216395bc359 Pseudo is Ultrasoft + core correction, Zval = 10.0
Pseudo is Ultrasoft, Zval = 6.0
MD5 check sum: 874d5528bf087cea5d785f7b6a7bf583 Pseudo is Ultrasoft, Zval = 16.0
24 Sym. Ops., with inversion, found (12 have fractional translation)
k( 33) = ( 0.2886751 0.2500000 0.0000000), wk = 0.0937500 k( 34) = ( 0.2886751 0.2500000 -0.1266177), wk = 0.0937500 k( 35) = ( 0.3608439 0.3750000 0.0000000), wk = 0.0937500 k( 36) = ( 0.3608439 0.3750000 -0.1266177), wk = 0.0937500 k( 37) = ( 0.4330127 0.2500000 0.0000000), wk = 0.0468750 k( 38) = ( 0.4330127 0.2500000 -0.1266177), wk = 0.0468750 k( 39) = ( 0.5051815 0.3750000 0.0000000), wk = 0.0468750 k( 40) = ( 0.5051815 0.3750000 -0.1266177), wk = 0.0468750
Dense grid: 229909 G-vectors FFT dimensions: ( 54, 54, 200)
Smooth grid: 106901 G-vectors FFT dimensions: ( 40, 40, 160)
Largest allocated arrays est. size (Mb) dimensions
Kohn-Sham Wavefunctions 19.47 Mb ( 3366, 379) NL pseudopotentials 39.65 Mb ( 3366, 772) Each V/rho on FFT grid 4.45 Mb ( 145800, 2) Each G-vector array 0.44 Mb ( 57480) G-vector shells 0.07 Mb ( 9419) Largest temporary arrays est. size (Mb) dimensions Auxiliary wavefunctions 77.86 Mb ( 3366, 1516) Each subspace H/S matrix 35.07 Mb ( 1516, 1516) Each <psi_i|beta_j> matrix 4.46 Mb ( 772, 379) Arrays for rho mixing 17.80 Mb ( 145800, 8)
Check: negative/imaginary core charge= -0.000002 0.000000 Initial potential from superposition of free atoms
starting charge 619.96808, renormalised to 632.00000 Starting wfc are 490 randomized atomic wfcs
total cpu time spent up to now is 714.9 secs
Output (Gambar Struktur Molekul Ba2Fe24O38)
2 Ba2NdFe24O38
Input
&CONTROL calculation = 'relax'
O 0.495964538 0.504035462 0.346018649 O 1.008070924 0.504035462 0.346018649
K_POINTS automatic 8 8 2 0 0 0
CELL_PARAMETERS {alat}
0.866025403784439 -0.500000000000000 0.000000000000000 0.000000000000000 1.000000000000000 0.000000000000000 0.000000000000000 0.000000000000000 3.948895260583572
O 0.495964538 -0.008070924 0.153981351 O 0.495964538 -0.008070924 0.346018649 O 0.504035462 0.495964538 0.653981351 O -0.008070924 0.495964538 0.653981351 O 0.504035462 0.495964538 0.846018649 O -0.008070924 0.495964538 0.846018649 O 0.504035462 1.008070924 0.846018649 O 0.504035462 1.008070924 0.653981351 O 1.008070924 0.504035462 0.153981351 O 0.495964538 0.504035462 0.153981351 O 0.495964538 0.504035462 0.346018649 O 1.008070924 0.504035462 0.346018649
K_POINTS automatic 8 8 2 0 0 0
CELL_PARAMETERS {alat}
0.866025403784439 -0.500000000000000 0.000000000000000 0.000000000000000 1.000000000000000 0.000000000000000
0.0 00000 3.948895260583572
O 0.688426181 0.844213090 0.446009037 O 0.495964538 -0.008070924 0.153981351 O 0.495964538 -0.008070924 0.346018649 O 0.504035462 0.495964538 0.653981351 O -0.008070924 0.495964538 0.653981351 O 0.504035462 0.495964538 0.846018649 O -0.008070924 0.495964538 0.846018649 O 0.504035462 1.008070924 0.846018649 O 0.504035462 1.008070924 0.653981351 O 1.008070924 0.504035462 0.153981351 O 0.495964538 0.504035462 0.153981351 O 0.495964538 0.504035462 0.346018649 O 1.008070924 0.504035462 0.346018649
K_POINTS automatic 8 8 2 0 0 0
CELL_PARAMETERS {alat}
0.866025403784439 -0.500000000000000 0.000000000000000 0.000000000000000 1.000000000000000 0.000000000000000
0.0 00000 3.948895260583572
O 0.844213090 0.688426181 0.946009037 O 0.844213090 0.688426181 0.553990963 O 0.688426181 0.844213090 0.053990963 O 0.155786910 0.844213090 0.053990963 O 0.155786910 0.844213090 0.446009037 O 0.688426181 0.844213090 0.446009037 O 0.495964538 -0.008070924 0.153981351 O 0.495964538 -0.008070924 0.346018649 O 0.504035462 0.495964538 0.653981351 O -0.008070924 0.495964538 0.653981351 O 0.504035462 0.495964538 0.846018649 O -0.008070924 0.495964538 0.846018649 O 0.504035462 1.008070924 0.846018649 O 0.504035462 1.008070924 0.653981351 O 1.008070924 0.504035462 0.153981351 O 0.495964538 0.504035462 0.153981351 O 0.495964538 0.504035462 0.346018649 O 1.008070924 0.504035462 0.346018649
K_POINTS automatic 8 8 2 0 0 0
CELL_PARAMETERS {alat}
0.866025403784439 -0.500000000000000 0.000000000000000 0.000000000000000 1.000000000000000 0.000000000000000 0.000000000000000 0.000000000000000 3.948895260583572