Sampling:
The Basics
“S
ampling” is a key current Good Manufacturing Practice (cGMP) activity that impacts nearly every activity of manufacturing pharmaceutical prod-ucts. Sampling is used during the assessment of:• Raw materials, labeling, and components prior to release • Validation of equipment,
pro-cesses, systems, and products • Products during production • Finished products prior to release • Products during stability studies,
and
• Data before, during, and after pro-duction
The appropriate knowledge and application of cGMP requirements for sampling is critical to the development of a scientifically sound quality sys-tem.
In this article, a general discussion of cGMP requirements for sampling will be followed by targeted discussions for incoming materials and dosage forms. Where applicable, specific examples and experiences of the author are provided to address typical situ-ations that can arise.
The purpose of this article is not to provide a statistical tutorial on the mathematical principles of sampling plans, or to recom-mend definitive sampling plans to use in every circumstance. In-stead, the general principles and approaches that should be con-sidered for cGMP applications of sampling are presented and discussed. It is the author’s hope that this report will stimulate al-ternative approaches, introduce new considerations, and answer basic questions that create hurdles and issues in pharmaceutical manufacturing.
Though the final section of this article provides a listing of sev-eral important and useful resources on sampling that may answer specific questions and concerns, the ultimate reference on
acceptance sampling is Juran’s Quality Control Handbook. This exhaustive resource should be viewed as a “must-have” for every Quality Assurance (QA) professional.Juran’s Quality Control Handbook1includes sections on sampling risks, implementation of
acceptance sampling programs, attributes versus variables, relia-bility sampling, bulk sampling, and the definitive statistical basis for all aspects of sampling programs. No sampling plan should be developed without some regard for the approaches and consider-ations discussed by Juran, et at.1
cGMP Requirements for Sampling
Before we look at the specific cGMP requirements for sampling, let’s look at what a sample is and means from a general perspec-tive. According to the American Heritage Dictionary, a sample (or sampling) as it might relate to cGMPs is defined as:
In short, a sample is a portion that accurately represents the population from which the characteristics of the population can be determined.
The cGMPs mention samples, sampling plans, or sampling methods repeatedly. When reviewed overall, there are four themes that occur throughout these references:
❶Sampling plans and methods must be written and defined ❷Samples must be representative of the population
❸Samples or sampling plans must be based on appropriate
statistical criteria, and
❹Samples must be properly identified and handled
Let’s examine each of these overall requirements in more detail.
❶Sampling Plans and Methods Must be Written and Defined
As with all cGMP requirements, sampling plans and meth-ods must be predetermined and written. The most com-mon approaches to written methods for sampling are:
• Develop a single Standard Operating Procedure (SOP) that details the plan to use with predetermined inspection levels, sampling sizes, and acceptance limits – then, any individual requirement for sampling will simply refer to the sample plan SOP.
• Develop a specific SOP detailing the sampling plan for use with each individual type of material – for example, an SOP will be written individually for incoming packag-ing components, raw materials, labelpackag-ing, etc.
Each approach has advantages, but the key consideration is that you must pre-determine the specific sampling plan to be used for any type of material to be tested.
❷Samples Must be Representative of the Population
• Segregation that occurs during blending, transport, or handling
• Variability occurring during the manufacturing process, especially if the process is prolonged (such as during campaigns to manufacture packaging components) • Part-to-part variability, due to differences in manufacturing
components (such as bottles formed on equipment with multiple heads)
• Changes in operators during manufacturing
• A variety of other factors that impact production consis-tency
Let’s face it… though process validation is a key element in the pharmaceutical manufacturing process, it is not always consid-ered by vendors producing raw materials, packaging components, excipients, or labeling. Thus, the importance of an appropriate sampling plan is heightened for materials supplied by others.
❸Samples or Sampling Plans Must be Based on Appropriate
Statistical Criteria
The use of an appropriate statistically-based sampling plan is important to ensure our sample is truly representative of the popula-tion. In other words, a solid sampling plan based on statistical crite-ria can provide additional confidence that the sample, or specimen, on which we base accept/reject decisions, will provide the “true” answer regarding the quality of the material.
The term “statistics” often creates the impression that the sam-pling plan must be complex, and use extensive statistical tables, formula, and calculations. Though in some cases, it is appropriate to utilize and perform more complex data manipulations (for exam-ple, with Design Of Experiments [DOE] studies), sampling plans for routine uses can and should be simple and easy to use.
Standards Institute (ASQ/ANSI) Z1.4 sampling plans does not ensure that the plan is statistically-based. Some sampling plans derived from these widely used programs would actually allow acceptance of some lots with critical defects. Each sampling plan must be developed to consider the specific attributes being meas-ured, and the risks associated with accepting a defective lot.
❹Samples Must be Properly Identified and Handled
Finally, cGMPs mention, in several locations, the need to proper-ly identify and handle samples. Despite the relative simplicity of this requirement, most firms routinely fail product or material lots, or undergo Out-of-Specification (OOS) investigations due to either improper sample identification or poor handling of samples prior to testing. Any testing program and sampling plan must include appropriate requirements for labeling and handling.
Now that we have discussed general cGMP requirements for samples or sampling, we will look in more detail at some
approaches for sampling plans for specific materials and product dosage forms. In the following pages, approaches for sampling plans will be discussed for:
• Incoming Packaging Components • Incoming Raw Materials
• Labeling Materials
• Non-sterile Liquid Products • Sterile Products
• Creams, Suspensions, and Emulsions • Powder Blends
• Tablets, Capsules, and Other Solid Dosage Forms
First, let’s look at a few basic concepts related to sampling and sampling plans.
Basics of Sampling and Sampling Plans
pharmaceutical product types:
■What is a Sampling Plan?
A sampling plan is a written approach to collecting and testing samples to ascertain material conformance to quality require-ments. Included in the plan will be:
• Sample size – The number of samples taken (or quantity) must be specified in the written plan. This will eliminate sam-pler discretion, and better ensure an appropriate and ade-quate sample.
• Method of sampling – The exact manner in which samples are to be taken, and the sample location must be included. • Tests or assessments – The testing, inspection, or
assess-ment required will be specified in the plan. Because the sam-pling (and testing) plan is pre-determined and written, the tests conducted will be directed toward determination of con-formance to requirements.
• Criteria for acceptance/rejection – Specific criteria for deter-mining whether the material is acceptable or fails require-ments must be specified. Again, unless this is pre-deter-mined, the tendency to compromise for borderline situations will arise.
■Must Sampling be Random or Specified?
In general, samples taken for pharmaceutical purposes are cho-sen randomly. Random sampling means that the samples are chocho-sen without regard for appearance, ease of sampling, location, etc. Random samples must be truly random with the method for selec-tion specified. However, there are situaselec-tions (such as for determi-nation of blend uniformity – see section below) in which the exact location of sampling is specified. In each case, the plan for selecting samples must be specified in the sampling plan.
■What is Sampling Bias?
occurred. This bias can lead to acceptance of non-conforming lots or rejection of conforming lots. Thus, caution must be exercised when designing a sampling plan to ensure that bias is not intro-duced. The use of sampling devices, such as a sample thief, often introduces biases that cannot be predicted or easily eliminated. Thus, care must be used to either eliminate the bias or identify its impact.
■What is Meant by Batch Homogeneity and Batch Uniformity?
Batch homogeneity means that each increment or portion of a batch is visually or analytically the same as all others. Batch uni-formity means that the trend of results, not necessarily all individ-ual results, is similar through all portions of the batch. Though these terms are not the same, they are similar and used nearly interchangeably.
Concepts of Sampling Risk
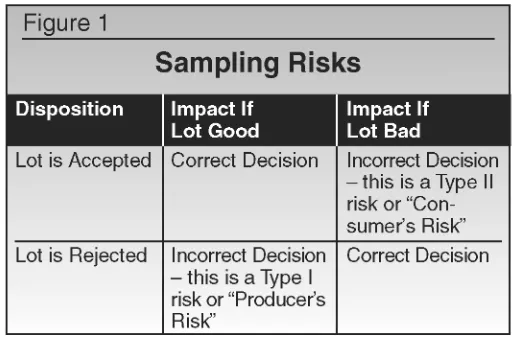
Any discussion of sampling and sampling plans must also con-sider the elements of risk associated with any inspection involving less than 100% of the population. Two types of risks are inherent in any acceptance sampling plan. These risks are illustrated in Figure 1.
actually would have met acceptance criteria if 100% of the units would have been evaluated. In actual practice, this “risk” is mini-mized because most pharmaceutical manufacturers utilize a retest-ing process to fully investigate results that are initially OOS.
However, this process of investigating OOS results is time consum-ing and adds expense.
A Type II risk is called a “Consumer’s Risk” because the impact would be that the ultimate consumer would actually receive a defective or nonconforming product. This risk is certainly of greater concern, both because of the potential for harm, but also because an acceptable test result is rarely questioned – in other words, there is no “back-up” opportunity for recognition of a con-sumer’s risk that exists for a producer’s risk (i.e., retesting).
Neither type of risk can be totally eliminated. Thus, the sam-pling plan for pharmaceutical products must include a risk assess-ment of the potential for making an incorrect disposition decision and the likely outcome if incorrect decisions are made. For evalu-ating product attributes (items classified as either good versus defective, such as tablet appearance), this risk is usually deter-mined by a management decision or corporate philosophy, and is expressed as Acceptable Quality Level (AQL). AQL is the maxi-mum average percent defective that is acceptable for the product being evaluated.
So, let’s see how this risk assessment might be expressed as an AQL by looking at two examples dealing with labels:
❶Correct label print text – because the “risk” associated with
accepting as-good labels that are defective is significant (both to the consumer, and because this would be a direct cGMP violation), the AQL for this type of defect would be very low. The lowest AQL typically defined in sampling plans is 0.01, meaning that a larger sample would be required to detect even a low level of defects. No defects would be toler-ated in the sample population. Thus, the consumer’s risk is significantly minimized.
❷Correct label print color – because the “risk” associated with
be higher. In this case, the firm may choose an AQL of 6.5, which would require a smaller sample needed to assess the population, and more tolerance for random defects.
For product variable characteristics (parameters that are meas-ured, such as analytical test results on products), this risk assess-ment is usually alleviated by the overall process design, process validation, and analytical method validation combined to provide an acceptable level of confidence in product released. For exam-ple, if a process is incapable of consistently yielding product with drug active results 90-110% (the product specification range), the consumer’s risk increases. Or, if the analytical method has a method variability of + or -3%, can you have confidence releasing a batch with a final drug active result of 91%? Thus, this risk is at least partially alleviated, in this case, by establishing a release specification outside the “danger zone.” In other words, in the case of a release range of 90-110% and an analytical variability of + or -3%, the actual release specification may be established as 94-106%. Thus, even if the method is biased by 3%, with a result of 94%, the actual result (91%) would still be acceptable.
So, the selection of any sampling plan for a pharmaceutical product must be a considered decision. The plan must consider the products, processes, test/inspection methods, as well as, the potential producer’s and consumer’s risks the firm is willing to accept. It is generally inappropriate to merely select a “typical” sam-pling plan without a proper consideration of these factors.
Incoming Packaging Components
A sampling plan for incoming packaging components must consider many factors:
• The criticality of the component as it relates to ultimate prod-uct quality (i.e., primary packaging components versus sec-ondary packaging)
• Typical lot sizes
• Reactivity of the component with product • Potential for “fatal” defects
• Potential for foreign material contamination, and • Other specific factors relating to material use
In other words, any plan for sampling and testing packaging components must utilize knowledge of the material usage, along with the potential risks posed by defects in the components.
Most sampling plans for packaging components classify defects into three categories:
❶Critical Defects – these are defects that would almost certainly
impart risks to product utility or compliance to specifications. Examples of critical defects include:
• Foreign bottles mixed with the lot • Incorrect materials of construction
• Defects with glass containers that would likely result in glass contamination of the product
• Holes or other defects that would result in leaking containers or jeopardize product sterility
❷Major Defects – these are defects that, in sufficient quantity or
under certain conditions, could render product unacceptable or unusable (i.e., issues during use). Examples of major defects include:
• Bottles out-of-round or with uneven molding or form
• Chips, bubbles, cracks, or other defects that may result in pro-duction or product quality issues
• Failed dimensions (i.e., height, diameter, etc. – dimensions that would not necessarily result in critical product defects, such as failed sterility)
• Incorrect color that is not cosmetic-only • Foreign material contamination
❸Minor Defects – these are defects that may result in minor
concerns. Examples of minor defects include:
• Color variation
• Dirt on exterior containers
• Minor extraneous plastic in molded bottles or caps • Other defects that are cosmetic-only
The sampling plan should identify the number of samples required for each defect type (it is possible that the number of samples exam-ined will be different for each defect type), testing or inspection per-formed, and number of defects above which the lot would be rejected or require further inspection.
So, the question that must be answered in developing a sampling plan is how to ensure that the sample is representative of the entire lot, and that the plan is statistically-based. The best approach for ensuring representative sampling is to remove samples uniformly from across the batch. For example, if the plan requires a sample size of 80 units, these units should be taken from all points in the lot.
NOTE: When removing samples from a batch, it is important to identify the location of samples taken. One approach often used is to place a sticker with a statement, such as, “This Case Sampled By/Date __________” on each case or unit from which a sample was removed.
How can you best satisfy the cGMP requirement that a sampling plan is statistically-based? Answering this question can be especially difficult if a variety of packaging components with various uses in dosage forms are received within the firm. For example, is it possible to establish a statistically-based plan – a simple one – for a firm that produces liquids, powders, capsules, and creams encompassing >100 product formulations and another >250 packaging configurations?
The example plan outlined above (AAFES Plan) has the advantage that it is simple, is statistically-based, and provides an opportunity to conduct additional inspection for initial fail-ures. However, from a practical standpoint, the continuing inspections scenario requires that an additional sample be taken from the lot, unless the maximum possible sample is taken initially. Either way, the need for continuing inspection is a productivity concern.
Many other potential sampling plans that would meet cGMP requirements could be selected. The key to developing any sampling plan is to be able to answer the question that the Food and Drug Administration (FDA) investigator might ask:
“What justification can you provide to ensure that this sam-pling plan will disqualify any lot that could result in defective finished product?”
One additional key to sampling plans for incoming packaging components is to consider the potential for remediating or cor-recting lot concerns identified during inspection. In other words, oftentimes, a lot that fails incoming inspection is desperately needed for production. Can you design a procedure that allows for inspection/rework of the nonconforming lot, then release the lot? The answer is, “Yes, under certain circumstances.” If the defect that resulted in failure of the lot can be inspected, and eas-ily detected and removed, it may be possible to result in a
release disposition after inspection. This process must be speci-fied in SOPs, and documentation must exist to demonstrate that the lot was acceptable after rework/inspection.
Incoming Raw Materials
in Figure 4, the plan makes “apparent” statistical sense.
Note: Some raw materials, such as bagged ingredients, will con-tain more than 500 units. When lots of materials are greater than 500 units, an alternate sampling plan should be considered.
In each case above, the sampling plan must specify that all por-tions of the batch, beginning, middle, and end, must be sampled. Even in the worst-case scenario above, the batch can be well repre-sented in the final sample.
It must also be pointed out that some materials and product requirements specify that more rigorous sampling regimens be fol-lowed. For example, if a material is microbiologically sensitive and/or potentially non-homogeneous, it may be prudent to sample every container regardless of lot size. Knowledge of the material and the process must be included in the risk assessment associated with selecting a sampling plan.
• Does the “mixing” of individual samples into a composite really produce a homogeneous, thus, representative, sample?
• Does the mixing process actually degrade or harm the sample? For example, if the sample is tested for particle size distribution, the rigors of mixing can destroy the “real” condition of the mate-rial.
• Can a non-homogeneous lot actually “pass” incoming testing using a composite, though it may have locations of high or low results?
Despite these concerns, the use of a composite sample to repre-sent the entire lot is routinely used. And, unless the questions (i.e., concerns) noted in the questions above are properly considered, FDA will likely raise questions about the plan.
Several precautions must be taken to protect samples of raw materials:
• Temperature sensitivity must be considered – some samples must be stored in cool conditions
• Other factors that could impact material quality must be consid-ered, such as moisture and light
• As discussed earlier, proper sample identification is always required
In summary, the simple “square root of n plus one” sampling plan is usually acceptable for raw materials. Though not statistically signif-icant from a mathematical viewpoint, this plan does provide for a representative sample, and is easy to implement and use. Many of the problems and rejections associated with incoming materials are often traced to poor sampling technique or sample handling. Pro-visions for proper technique is a must for sampling SOPs.
Labeling Materials
• Label printing process
• Confidence in the label manufacturer
• Label control and verification procedures in place in your firm, and
• Level of technology available for label inspection and control
Note: For the sake of this discussion, labeling materials are considered unit labels, packaging instructional inserts, unit car-tons with labeling information, and other printed materials that contain product usage, dosage, or warning information.
Let’s look at each of these in more detail.
Label Printing Process
The printing process and procedures in place at the label man-ufacturer are significant in developing a sampling and inspection process. The key aspects of the printing process that must be considered include:
How are printed labels cut? How many replicates of each label are printed for each “shot?”
For label manufacturers that print multiple “shots” of each label, the sampling and inspection plan for incoming labels must ensure that all representatives of the “shot” be inspected. A “shot” would be a multi-lane printing system that prints multiple versions of the same label (i.e., four different print mats, for example) that are then cut and combined. In these cases, it is important that the sampling plan include an inspection of consecutive labels that represent all members of the “shot.”
every separate printing event is represented in the inspection.
How does the printer handle splices in label rolls? What con-trols exist?
Several recalls occur each year when label manufacturers improperly splice the end of one label roll to the beginning of the next. These splices must be properly handled and controlled to avoid splice errors. Label manufacturers must ensure proper cGMP practice, such as line clearance and double-checks, to avoid these concerns. Incoming sampling plans should include an inspection of these splices, or at least some minimal number of them. Ideally, label manufacturers will avoid entirely, or at least minimize, the number of these splices required.
Is it possible for labeling to change after approval? What comput-er systems are used, and what controls exist to ensure that the label approved will be the label printed and delivered?
The label manufacturer’s processes and procedures for con-trolling changes to master labels is critical. Unless the label manu-facturer has superior controls for ensuring that the approved label is the printed label, the incoming sampling plan and inspection must include a thorough review of each printed label lot against the master label.
Confidence in the Label Manufacturer
Of equal importance to understanding the label printing process is a basic confidence or trust in the label manufacturer. As trust and experience increase, the incoming sampling and inspection plan can be reduced. Key questions that must be answer-ed for label manufacturers include:
Do adequate controls exist to ensure segregation of each labeling type and lot?
detail label segregation. On-site audits are necessary to ascertain this. The audit should assess how labels are controlled after print-ing, during storage, and at shipment. Ideally, labeling should have a strict chain of custody throughout the manufacturing and han-dling process.
Are the individuals involved in the manufacturing and control of the label manufacturing process experienced in pharmaceutical operations, controls, and procedures? Do employees receive training in cGMP?
At this point in time, most label manufacturers for pharmaceuti-cal companies are well-versed in cGMP requirements, and have incorporated these concepts into their own operation. An on-site audit should assess cGMP essentials, such as the presence of adequate procedures, training, documented production records, material segregation and controlled access, routine in-process inspection, and strict control of “masters.” Failure to have good con-trols in any of these areas would be an item of concern for a poten-tial pharmaceutical customer.
Is the firm innovative and progressive in developing controls to ensure printed label quality?
Today, the use of automated manufacturing and inspection equipment has become commonplace, and even essential to remain competitive with respect to label quality and productivity. A proactive approach to improvement and enhanced controls points directly toward a high and consistent level of quality. The higher the demonstrated quality, the less incoming inspection is potentially required.
Does the label manufacturer have a solid history in providing high quality materials?
consistent-ly that they have systems and procedures in place to control both label manufacturing and segregation? Any effort to reduce or min-imize incoming inspection should consider the historical results of the supplier.
Label Control and Verification Procedures in Place in Your Firm
In addition to knowledge and confidence in the label manufac-turer, the controls and verification systems in place at your firm also play a role in the sampling plan for incoming labeling. Key questions that must be answered include:
Do you have 100% label verification systems in place? Have these systems been validated and proven reliable? Has redun-dancy been designed into these systems?
In today’s pharmaceutical manufacturing world, all manufacturers should have systems to 100% verify label correctness. However, do you have confidence that these systems always function properly? The more rigorous the validation, the more confidence you might have in these systems. Thus, the level of incoming inspection may be reduced. Likewise, redundancy in inspection or verification systems can provide justification for less rigorous incoming inspection.
Does your firm use many different labels of similar size and shape, or a relatively small number?
One key question you should ask about your own operations is, “How great is the actual risk for using an incorrect label? ”If your number of labeling materials is low, or if each label is clearly of a dif-ferent size, shape, or color, the risk for failure might be relatively low. In these cases, less incoming inspection may be warranted. The reverse would, of course, also be true.
Can you devise a scenario in which the incorrect label delivered by the label manufacturer could be used and undetected?
approved, and used? Would this scenario require multiple systems failures, or be essentially impossible? If so, less inspection rigor is warranted.
Level of Technology Available for Label Inspection and Control
Finally, the type and level of inspection technology available to your label inspection group may have an impact on the sampling and inspection plan you develop. For example:
Do you use an optical comparator or similar “intelligent” instru-mentation to verify all incoming labeling prior to release?
The use of “intelligent” instrumentation to inspect incoming labeling may provide significant confidence that incorrect labeling could not be used. Care must be exercised, however, to not use technology as an excuse to minimize incoming inspection without merit.
Are all labels electronically inspected and verified “off-line” prior to release and use?
One approach used by many pharmaceutical firms is to con-duct 100% inspection prior to release. This approach, though seemingly overkill to some, does provide assurance that the cor-rect label was received and released, and provides added redun-dancy with on-line verification systems.
Do you have other systems in place for 100% inspection of labeling prior to use to circumvent the need for sampling and inspection?
Again, the greater the level of inspection and verification in place, the less incoming inspection is needed.
misla-beling has been one of the top ten causes for pharmaceutical product recalls in each of the last ten years. Despite the fact that cGMP requirements for labeling and label control have remained essentially unchanged since issuance in 1978, firms still struggle with the basic concepts of development, inspection, use, control, release, and segregation of labeling.
Non-Sterile Liquid Products
Sampling of liquid products – those that are true solutions, not suspensions, emulsions, or creams – are among those with fewest special concerns. If the product is a true solution (all mate-rials in solution – no possibility of separation under normal condi-tions – each sample is, by definition, the same as every other sample), no special concerns for sampling usually exist. Certainly, proper handling of samples after collection is important. As with all products and all sampling approaches, exercise proper sampling plan design and execution to ensure that results obtained repre-sent the product evaluated.
Sterile Products
Several special sampling concerns exist for sterile products. These products require additional controls for the production envi-ronment, product protection, and product handling to ensure that sterility is assured. With these products, product and environmen-tal protection has been engineered into the process. For example, protection from environmental contamination is controlled by the use of filtration to remove essentially all particulates, control of air-flow and direction, sterilization of equipment, containers, potential product contact surfaces, and rigorous controls to prevent “peo-ple” contamination. Sampling plans for these products typically are used to assess the proper function of these systems. The follow-ing aspects of the process are usually sampled and monitored:
• Air
• Equipment sterilization • Personnel
These systems are sampled via environmental monitoring plans and procedures. Sampling plans are designed and executed to comprehensively test each aspect of these systems. For exam-ple, air is tested for both non-viable and viable (microorganisms) contamination in the manufacturing environment. Samples are taken from the hands and clothing of personnel to ensure that systems and personnel practices are properly followed.
It is not the intent of this article to discuss these sampling pro-grams in detail. In short, however, the typical cGMP requirements discussed in the introductory section of this report (statistically valid, written, representative, sample identification, etc.) are also required for each of these sampling efforts.
Creams, Suspensions, and Emulsions
Creams, suspensions, emulsions, and other products that are prone to separation or settling pose special concerns for sampling and testing. During each process step in which separation or set-tling could occur, comprehensive sampling and testing must be performed to ensure that the process is performing as designed. Validation studies will usually provide the documentation needed to prove the process, and establish the critical process parameters that must be achieved in order to produce conforming product. Routine sampling and testing during commercial production should merely verify that expected processing parameters are being
achieved.
Each process must be evaluated to determine the proper vali-dation and ongoing verification sampling and testing approach. You must determine this for each individual product. A detailed discussion on aspects of this determination will not be reviewed in this article because each product is unique. However, the follow-ing represents some of the author’s experience relatfollow-ing to sam-pling and testing these products:
■Sampling Techniques
mixing or agitation conditions. To remove a sample, the mixing or agitation should cease when sampling open containers (to ensure safety of the sampler). Samples must be removed quickly when safe to ensure the sample is representative, and that settling has not occurred. In addition, samples from open containers should not necessarily be removed only from the upper few inches of the prod-uct batch. Validation studies should identify from what portion of the batch the sample should be taken.
Even when sampling closed systems (i.e., samples taken from a sampling port or other device, and not taken directly from an open container) requires precautions. In most of these cases, it is impor-tant to purge the sampling port before removing the sample for analysis. Discharging a small portion of material prior to removing the sample can avoid samples that are not representative.
■Handling of Samples
It is imperative that in-process samples for these products be properly handled. Handling must ensure that degradation of the sample integrity does not occur. If the intent of the sample is to test the exact attributes of the product at that point, it may be necessary to avoid further mixing of the sample. For these products, a sample that completely fills the sample container to a point that no space exists will usually protect sample integrity. Without head-space, inadvertent mixing is unlikely.
However, samples taken to analyze the integrity of the sample mixture may require further mixing prior to analysis. In these cases, collecting the sample in a container that has adequate headspace to allow further agitation prior to analysis may be required.
The key point here is that you must consider the purpose of the samples taken. What attributes are you attempting to analyze? It is important that you “pre-determine” this purpose, and design a sam-pling approach and sample handling “protocol” to ensure that the sample taken will yield the information required.
■Validation of Mix Failures
data are available that define the length of downtime of mixing sys-tems without affecting the mixture (or emulsion, etc.), the batch may be in jeopardy. Thus, it may be useful during validation to intentionally cease mixing at key points in the process, sample at various time points, and test the product to determine the length of
time the mixing
Can cream, emulsion, and suspension samples be composited and tested? In most cases, composite samples can be used to assess the overall quality of the product. Individual samples can be collected, then combined to produce an overall sample reflecting the quality of the batch. However, you must use caution with these products to ensure that the mixing of individual samples does not introduce additional variability in the process that can mask prod-uct issues. You should validate the sample compositing process. This can occur by analyzing the individual samples, then compar-ing results (or average results from all individuals) to the composite sample result. A failure to yield statistically comparable results can indict the compositing process.
In short, if you produce emulsions, creams, or suspensions, you already know that these products pose special concerns. The sam-pling plan designed to either validate the processes for these prod-ucts, or to assess ongoing production, must consider these special concerns and greater effort to “pre-determine” the purpose of each set of samples is warranted.
Powder Blends
Perhaps the area of sampling that has caused the most contro-versy is how sampling is applied to powder blends. In other words, what sampling plan is required to demonstrate, in a consistent,
Despite all controls, procedures, and
high-quality attitude, the most important
reproducible way, homogeneity of the powder blend prior to further processing? Let’s explore some key considerations.
Following publication of FDA’s Guidance for Industry, ANDAs: Blend Uniformity Analysis (August 3, 1999), significant industry concerns were raised regarding FDA’s requirements for demon-strating blend uniformity when United States Pharmacopeia (USP) content uniformity testing is required on the product. Some of these concerns included:
• Current limitations in sampling technology – specifically, the use of sample thief technology has been proven prone to error and inconsistency
• Powder segregation of samples after sampling
• Weighing errors can occur during preparation of blend sam-ples for analysis
• Difficulty in proving that the blender sample plan will be truly representative – demonstrating that all worst-case locations are included in the sample, and
• Blender sampling fails to consider segregation that can occur during discharge, storage, and transport prior to final pro-cessing
Despite these concerns, the current cGMP requirement in 21 CFR 211.110(a)(3) states:
“…control procedures shall include… adequacy of mixing to assure uniformity and homogeneity.”
This requirement, thus, applies to development, process vali-dation, and post-validation commercial batches for solid oral drug products.
uni-formity by sampling at the discharge stage, or from final in-process blend storage units (i.e., drums, totes, etc.). In all cases, firms uniformly utilize unit-dose sampling sizes to fulfill Barr Decision and FDA current expectations.
Because of the numerous issues mentioned above with thief sampling of oral drug blends, dealing with blend sample failures is an ongoing concern. In other words, the uncertainty of blend sam-pling technology and approaches often lead to blend analysis ures. So, we face the age-old question, “How do I know these fail-ing results are real? What additional testfail-ing is needed to overturn these failing results?” Some firms universally interpret blend sam-ple failures as “true failures,” and reject any study exhibiting failing results. Other firms attempt to discredit failing results through an investigation that can involve OOS investigations, resampling, retesting, utilization of scientific rationale, or a combination of these. The risk of this approach to overturning failing blend results can be a delayed regulatory approval or FDA inspection issues.
As a result of these issues and industry concerns, the Product Quality Research Institute (PQRI) Blend Uniformity Working Group developed an approach to demonstrating blend uniformity by com-bining blend testing and compendial dosage unit testing. This approach (stratified sampling) postulates that the analysis of dosage units (i.e., finished tablets) can supplement or provide sta-tistical evidence that a failing blend result was due to poor sam-pling or handling technique. This approach has not been officially “accepted” by FDA, though several members of FDA served on the PQRI expert committee that developed the recommendation. De-spite this lack of official acceptance, several firms have utilized strat-ified sampling to justify acceptance of failing blend data.
For a full copy of the stratified sampling approach and rationale supporting it, reference the PQRI web site (listed in the final sec-tion of this document).
Tablets, Capsules, and Other
Solid Dosage Forms
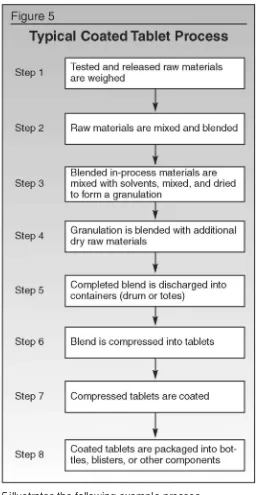
Figure 5illustrates the following example process.
Detailed approaches for sampling of materials up through step five are discussed earlier in this article. Following are additional considerations for the finishing steps involved in tablet production:
■Compression into Tablets
There are several key sampling/testing considerations regard-ing the tablet compression process:
• Start-up: It is critical that initial tablets produced be carefully sampled and tested to ensure that equipment set-up is cor-rect, granulation was produced as expected, and the combi-nation of equipment and product are functioning well to-gether. During the initial period of production, additional samples should be taken and tested for conformance to physical requirements (analytical conformance should have been assessed and proven during validation). During start-up of routine, commercial production, samples should be reviewed from each set of compression tooling. In other words, you should remove at least two consecutive cycles of tablets, and carefully inspect (and document) physical ap-pearance, dimensions, and functionality. Inspection should include:
i. Appearance – complete tablets, no capping, no chips, and complete and correct embossing (if performed)
ii. Dimensions – tablet thickness, weight, and other critical parameters should be measured
iii. Functionality – tablet hardness, friability, etc. should be checked
The combination of this initial early inspection is essential to a successful compression run.
• During the run: Samples should be taken at regular intervals during the batch to ensure that tablets continue to conform to requirements
adjustment of the press that could alter physical dimensions or characteristics of the tablets
• At the end of the run: Often, the end of the batch is the time when nonconforming tablets frequently are produced – thus, it may be prudent to remove and analyze additional samples near the end of the run
• Validation: Extensive sampling and testing must occur during validation studies that include analytical testing, in addition to physical inspection. Because many granulation formulas are sensitive to de-blending or segregation, analytical verification of blend homogeneity must be verified at all stages of the batch, with special attention given to start-up, after adjust-ments, when the compression hopper is depleted or nearing depletion, and at the end of the batch.
■Tablet Coating
The tablet coating process must be carefully sampled and eval-uated to ensure proper application of coating. It is imperative that the process be monitored throughout, with additional sampling and evaluation after adjustments or during times deemed critical, such as at the beginning of the process and near the end.
■Tablet Packaging
Sampling and analysis of solid dosage forms should occur at various points during the packaging process. Two key aspects of packaging quality are usually considered:
❶Tablet Integrity ❷Fill Accuracy
Product should be sampled and inspected at the filler and after each critical process (blistering, for example) to ensure that tablet damage is not occurring.
In summary, sampling of all aspects of the tablet production process is important to ensure at all stages that tablet integrity con-forms to requirements. Each process must be carefully evaluated, usually via review of a process flow chart prior to validation, to ascertain critical process steps, or points at which defects could occur. After identifying these points, a sampling plan must be developed to prove both during validation and routine production that tablets meet all requirements.
Guide to References on Sampling Plans
A recent search on the Yahoo internet web site for “sampling plans” revealed 617,000 sites that include some reference to sam-pling or samsam-pling plans. In addition, hundreds of reference books have been written that are either dedicated to sampling or sampling plans. So, the availability of information on this subject is abundant. What are the best locations to find needed information that will assist those in the pharmaceutical industry? Though the author does not claim to have knowledge of all or even most of the best information sources, the following are good resources for additional help or information on this subject:
Web Sites
Several Internet web sites provide helpful information that can aid the pharmaceutical practitioner develop an appropriate sampling plan:
❶www.aafes.com/qa/docs/supqap-statistical_
sampling_plans_home_page.htm
This site is the Army and Air Force Exchange Service (AAFES) site. In this site, you will find the details presented briefly in the sec-tion on “Incoming Packaging Components.” On this site, you can locate the AAFES approach using continuing inspection criteria based on the MIL-STD 105E or ASQ/ANSI Z1.4 sampling plans. This site is easy to follow, easy to read, and a good site for the sta-tistical novice to locate and learn more about these plans.
❷www.samplingplans.com
Characteristics [OC] Curves, control charts, etc.), and can provide valuable information on getting started on sampling plans. This site provides excellent examples of sampling plans, challenge questions, and an extensive glossary of terms and definitions.
❸www.cqeweb.com
This site provides a wealth of information on many subjects that comprise the body of knowledge for the American Society for Quality (ASQ) Certified Quality Engineer Certification process. One chapter deals specifically with the topic, “Sampling.” This chapter provides another excellent review of the basic and more detailed statistics that serve as the basis for many sampling plans. It includes a very good glossary of terms with excellent graphics and examples.
❹www.iso.ch
This is the web site for the International Organization for
Standardization (ISO). This is an extensive web site detailing stan-dardized approaches for many quality topics, including sampling and sample plans. Any effort to develop sampling plans for products for global marketing should consider the procedures already developed and in use by the ISO.
❺www.pqri.org
This is the Product Quality Research Institute (PQRI) site that includes the complete Blend Uniformity Working Group report, “The use of stratified sampling of blend and dosage units to demonstrate adequacy of mix for powder blends.” In addition to this report, the let-ter provided to FDA introducing this report and blend uniformity data used to support the recommendations are available.
❻Many other pharmaceutical organizations have valuable
infor-mation on web sites that provide further details and discussions on sampling:
• www.ivthome.com
The Institute of Validation Technology(IVT) includes many articles, papers, and additional resources to aid in the develop-ment of sampling plans or approaches to cGMP compliance. • www.pda.org
• www.ispe.org
The Society for Life Science Professionals (formerly Institute for Pharmaceutical Engineers) provides excellent technical and industry information regarding aspects of cGMP compliance. • www.gmp1st.com
The GMP Institute provides a wealth of helpful cGMP compli-ance information.
• www.asq.org
The American Society for Quality is one of the largest and old-est organizations dedicated to providing information for profes-sionals involved in quality.
This discussion could never identify all of the excellent organiza-tions providing information on cGMP compliance and, specifically, sampling and sampling plans. If the above web sites cannot provide the information required, query other web site search engines to find the specific information desired.
Textbook Reference
Several classic textbooks provide extensive information on the development and use of sampling plans for quality assurance pur-poses. However, the ultimate resource is Juran’s Quality Control Handbook (A. Blanton Godfrey and Joseph M. Juran (coeditors-in-chief), 5thEdition, McGraw-Hill, 1999). is the universal reference book
for information about quality control sampling plans. All quality practi-tioners should have, and refer often to, this excellent resource. ❏
About the Author
Eldon Henson is a frequent contributor to Institute of Validation Technologypublications. He has authored IVT’s Auditing
and M.A. degrees in microbiology from Southern Illinois University-Carbondale, and has worked in various quality and manufacturing roles at Abbott Laboratories, Novartis, Boehringer-Ingelheim, Sigma-Aldrich, and is currently Director, Quality
Assurance at KV Pharmaceutical in St. Louis, MO. Henson can be reached at [email protected].