Journal of Life Sciences
Volume 6, Number 9, September 2012 (Serial Number 53)
David Publishing Company www.davidpublishing.com
Publication Information
Journal of Life Sciences is published monthly in hard copy (ISSN 1934-7391) and online (ISSN 1934-7405) by David Publishing Company located at 9460 TELSTAR AVE SUITE 5, EL MONTE, CA 91731, USA.
Aims and Scope
Journal of Life Sciences, a monthly professional academic journal, covers all sorts of researches on molecular biology, microbiology, botany, zoology, genetics, bioengineering, ecology, cytology, biochemistry, and biophysics, as well as other issues related to life sciences.
Editorial Board Members
Dr. Stefan Hershberger (USA), Dr. Suiyun Chen (China), Dr. Farzana Perveen (Pakistan), Dr. Francisco Torrens (Spain), Dr. Filipa João (Portugal), Dr. Masahiro Yoshida (Japan), Dr. Reyhan Erdogan (Turkey), Dr. Grzegorz Żurek (Poland), Dr. Ali Izadpanah (Canada), Dr. Barbara Wiewióra (Poland), Dr. Valery Lyubimov (Russia), Dr. Amanda de Moraes Narcizo (Brasil), Dr. Marinus Frederik Willem te Pas (The Netherlands), Dr. Anthony Luke Byrne (Australia), Dr. Xingjun Li (China), Dr. Stefania Staibano (Italy), Dr. Wenle Xia (USA), Hamed Khalilvandi-Behroozyar (Iran).
Manuscripts and correspondence are invited for publication. You can submit your papers via Web Submission, or E-mail to [email protected] or [email protected]. Submission guidelines and Web Submission system are available at http://www.davidpublishing.com.
Editorial Office
9460 TELSTAR AVE SUITE 5, EL MONTE, CA 91731, USA Tel: 1-323-9847526, Fax: 1-323-9847374
E-mail:[email protected], [email protected]
Copyright©2012 by David Publishing Company and individual contributors. All rights reserved. David Publishing Company holds the exclusive copyright of all the contents of this journal. In accordance with the international convention, no part of this journal may be reproduced or transmitted by any media or publishing organs (including various websites) without the written permission of the copyright holder. Otherwise, any conduct would be considered as the violation of the copyright. The contents of this journal are available for any citation. However, all the citations should be clearly indicated with the title of this journal, serial number and the name of the author.
Abstracted / Indexed in
Database of EBSCO, Massachusetts, USA Chemical Abstracts Service (CAS), USA Cambridge Scientific Abstracts (CSA), USA
Chinese Database of CEPS, American Federal Computer Library center (OCLC), USA Ulrich’s Periodicals Directory, USA
Chinese Scientific Journals Database, VIP Corporation, Chongqing, China
Subscription Information
Price (per year): Print $520, Online $360, Print and Online $680.
David Publishing Company
9460 TELSTAR AVE SUITE 5, EL MONTE, CA 91731, USA Tel: 1-323-9847526, 323-410-1082; Fax: 1-323-9847374 E-mail: [email protected]
David Publishing Company www.davidpublishing.com DAV ID P UBL ISH IN G
J LS
Journal of Life Sciences
Volume 6, Number 9, September 2012 (Serial Number 53)
Contents
Microbiology and Biochemistry
961 Application of Molecular Genetic Methods in Anthropological and Paleodemographic Studies of Fragmentary and Damaged Skeletal Material from Rescue Excavations
Katerina Boberova, Eva Drozdova and Kristyna Pizova
970 A Comparative Study of Soluble Protein Extractions of Populus deltoides × (Trichocarpa ×
Deltoides) for 2-DE
Joke Dupae, Ann Cuypers, Jean-Paul Noben, Jana Boulet, Nele Weyens, Karen Verstraelen and Jaco Vangronsveld
980 Xanthophyll Cycle and Its Relative Enzymes
Xirui Xiong, Xuefei Wang and Ming’an Liao
985 Isolation and Identification of Cardenolide Compounds of Gomphocarpus sinaicus and Their Fungicidal Activity Against Soil Borne and Post Harvest Fungi
Moustafa A. Abbassy, Ezzat A. Kadous, EL-Sayed A.M. Abd-Allah and Gehan I.Kh. Marei
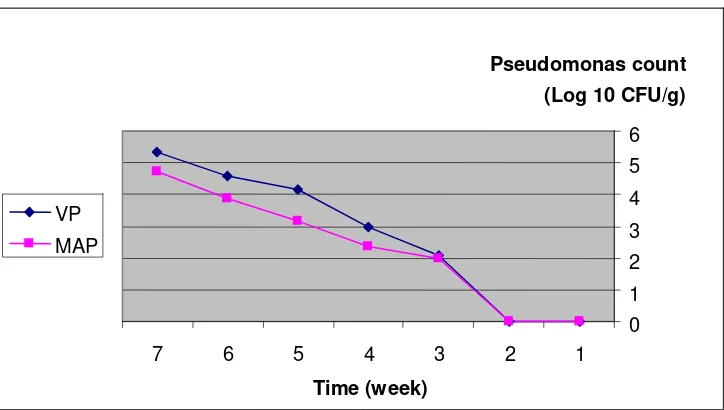
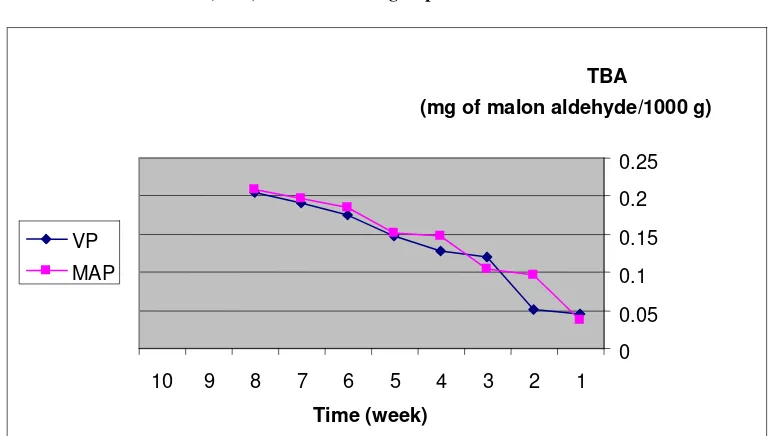
995 Microbial Aspects in Comparing Modified Atmosphere Packaging and Vacuum Packaging on
Shelf Life of Fresh Bull Meat
Abbas Najafpourkhadem, Lamiya Rokouie, Firouz Oghabi, Naser Valaie and Ali Shafighi
Botany and Zoology
1003 Effect of Different Organic Nutrient Sources and Two NPK Rates on the Performance and Nutrient Contents of A Newly Released Cassava Variety
Samson Adeola Odedina, Joy Nwakaego Odedina, Stephen Olusola Ojeniyi and Funmi Akinlana
1008 Nutritional Profile of Untraditional Colored Wheat Grains and Bread Making Utilisation
Nursery and Field Conditions
Niemat Abdalla Saleem, Khalil Ayoub Adam Mohamed and Mohamed El Nour
1025 Feeding Enrichment and Leopard Pacing in ZOO Dvůr Králové, a.s.
Ivana Gardiánová, Ivona Svobodová and Petra Stašáková
Interdisciplinary Researches
1028 Health Impact Assessment of Air Pollution in Some Regions in Albania
Mirela Lika (Çekani), Anjeza Çoku and Erida Nelaj
1034 The Mulching Effect of the Olives Mill Wastewaters on the Soil Cohesion Improvement and Wind Erosion Reduction in the Southern Tunisian Arid Zones
Mounir Abichou, Mohamed Labiadh, Nagwa Elnwishy and Hanane Abichou
1041 Predicting Potential Distribution of Gaur (Bos gaurus) in Tadoba-Andhari Tiger Reserve, Central India
Ambica Paliwal and Vinod Bihari Mathur
1050 Management Guidelines for Calabrian Pine Reforestations Carried Out in Southern Italy in the 1950s-70s
Silvano Avolio, Vincenzo Bernardini, Erica Clerici and Matteo Tomaiuolo
1057 Adaptability of Permanent Grassland to Drought
Ľuboš Vozár, Ján Jančovič, Peter Kovár and Slávka Bačová
1061 The Effect of Utilisation on the Floristic Composition of Meadow Communities
Anna Kryszak, Jan Kryszak, Agnieszka Strychalska and Agnieszka Klarzynska
1068 Biotechnological Strategies for Phytoremediating Triazinic Herbicides in the Humid Pampa (Argentine)
Application of Molecular Genetic Methods in
Anthropological and Paleodemographic Studies
of Fragmentary and Damaged Skeletal Material
from Rescue Excavations
Katerina Boberova, Eva Drozdova and Kristyna Pizova
Laboratory of Biological and Molecular Anthropology, Institute of Experimental Biology, Faculty of Science, Masaryk University,
Brno 611 37, Czech Republic
Received: March 27, 2012 / Accepted: April 27, 2012 / Published: September 30, 2012.
Abstract: The purpose of this study was to demonstrate the application of molecular genetic methods in anthropology and paleodemography in case where the examined bone material is damaged and fragmented, and where the skeletal remains of children are investigated. The application of traditional anthropological methods is limited, especially if sex determination of children and fragmentary skeletal remains is detected. Sex typing genetic markers (SRY, amelogenin) were used for sex determination of children and undetermined fragmentary skeletal remains from the burial site Pohansko, south outer precincts (Czech Republic). This is an approximately 1,200 year-old burial site (the Great Moravian period), which was excavated during rescue excavations. After the genetic analysis, sex was determined in more than half of the investigated samples. The results of the genetic analysis were used for completion of demographic data of this archaeological site. The results of sex determination of several samples were independently verified by the Institute of Criminology in Prague (Czech Republic), using the PowerPlex® ESX 17 System (Promega). This study
showed the suitability of modern molecular genetic methods to skeletal anthropology and paleodemographic analyses.
Key words: Sex determination, aDNA, amelogenin, SRY, PowerPlex® ESX 17 System, Pohansko.
1. Introduction
Sex determination is one of the most basic anthropological characteristics important for any reconstruction of the demographic structure of past populations. A large representation of subadult skeletons or fragmentary skeletal remains of unknown sex is a frequent problem in demographic analysis of historic populations because it leads to inaccuracy of results. The presence of fragmentary skeletal material is often associated with rescue archaeological research where there is a lack of time for careful excavation and it is necessary to use heavy equipment to uncover the findings, which can damage skeletal material. In cases
Corresponding author: Katerina Boberova, M.Sc., Mgr.,
research fields: molecular anthropology, physical anthropology. E-mail: [email protected].
where the skull or pelvis is damaged or missing, sex determination is difficult and often impossible. Sex assessment of subadult skeletal remains is unreliable because the sexually dimorphic characters are not yet developed. Anthropological methods of sex determination are limited in such cases [1, 2], and genetic methods can be a suitable alternative.
homology at the amelogenin locus [3]. Using specific amelogenin PCR primers, different base pair (bp) fragments are amplifiable from the X and Y chromosomes. Primer pairs used for sex identification are mainly those described by Sullivan et al. [4, 5], exhibiting a length dimorphism for the X and Y chromosomes (106 and 112 bp). The presence of a single band indicates that the sample came from a female, while two bands identify the sample’s source as male. An advantage of this approach is that the X chromosome product itself plays a role as a positive control [3].
The amelogenin marker has been successfully employed in sex determination of skeletal remains from different archaeological sites [6-11]. However, in some cases authors have reported observation of allelic drop-out, a frequent problem with amplification of degraded aDNA, potentially leading to incorrect sex determination [7, 8, 12, 13]. Due to the relatively frequent occurrence of this phenomenon, it is suitable to use an additional genetic sex marker; for example, SRY [14]. The SRY gene codes for the testes determining factor and is located on the short arm of the Y chromosome (Yp11.3). The occurrence of a single 93 bp amplicon of the SRY gene would distinguish authentic male DNA from a female DNA sample [14]. The SRY marker has been successfully used for sex typing of skeletal materials [15-17], often combined with the amelogenin marker [18-23].
The objective of our study was to demonstrate the application of genetic methods in anthropological and paleodemographic research when fragmentary adult and child skeletal remains are analyzed. The Pohansko archaeological site, situated in the south-east part of the Czech Republic near the town of Breclav, was the source of the samples in this study. Archeological excavations at this site uncovered the foundations of a fortified settlement, church, burial grounds and other buildings. One of the settlement units at Pohansko is the south outer precincts. It is dated approximately to the 9th-10th century, and was discovered during the
period of 1960-1962 [24]. During rescue excavation in the years 1960, 1962, 1975-1979, and 1991-1994, groups of graves and isolated graves were excavated amongst the numerous settlement objects [25-27]. A total of 205 graves were explored [28]. The majority of them contained the skeletons of children and fragmentary skeletal remains of unknown sex. Molecular sex identification of children and undetermined skeletal remains was performed by PCR amplification of amelogenin and SRY markers. For verification, the results of genetic sex determination of three samples were subsequently proved using the PowerPlex® ESX 17 System (Promega). An anthropological and demographic study of the population from Pohansko has been published [24], but it was partially distorted due to the large number of individuals without known sex. Application of molecular genetic methods in anthropology offers a solution to this limitation and helps to improve our knowledge about historic populations.
2. Materials and Methods
The most serious problem when working with degraded aDNA from skeletal material is contamination [30]. It is therefore necessary to adopt many stringent precautions against contamination. To ensure the highest possible reliability of the present work, the guidelines for aDNA analysis suggested by Cooper and Poinar [31] and Pääbo et al. [32] were followed as closely as possible.
Our laboratory is divided into three different laboratory rooms so that aDNA extraction, PCR amplification, and analysis of the PCR products are performed separately. The laboratory rooms, workspaces and instruments were UV irradiated (254 nm) prior to use, and were cleaned with denatured ethanol and bleach solutions to destroy extraneous DNA molecules after each work step. Different sets of pipettes were used for aDNA extraction, PCR amplification, and analyses of PCR products. Laboratory overalls with hood and face masks and disposable gloves were worn to decrease the risk of contamination with recent DNA. Separate personal protective equipment was used for each laboratory room. Only materials and chemicals of DNA-free quality or autoclaved were used, and all consumables were further UV irradiated for at least 20 minutes.
During the preparation of each sample, the bone fragments were cleaned with household bleach (NaOCl) and 96% ethanol. To remove contaminants from the bone sample surface, we ground away 1-2 mm of the bone with micro tool (DREMEL® StylusTM Lithium-Ion). After grinding, the bone fragments were UV irradiated for 10 minutes on each side to eliminate contaminant DNA. Dried samples were powdered in a mortar or bone mill under sterile conditions. The bone powder was decalcified using 0.5 M EDTA (pH 8.0) on rotator at 4 °C overnight. The supernatant was discarded and the decalcification step was repeated (4 °C overnight). A new portion of 0.5 M EDTA was added and the sample was decalcified at 4 °C for a week. After decalcification, the supernatant was stored in a sterile tube. DNA was extracted from the
supernatant using QIAamp® DNA Mini Kit (Qiagen®) according to the manufacturer’s protocol. DNA extraction was performed in a laminar flow box. A minimum of 3 independent extractions per sample were performed. During both the isolation and PCR steps, negative controls were performed to identify possible contamination by recent DNA.
For molecular sex determination, the SRY marker and the amelogenin marker were chosen because of their relative small size. For amplification of the SRY marker, the protocol introduced by Cunha et al. [18] was adopted. The amelogenin marker was amplified using the slightly modified protocol introduced by Hummel [13]. The PCR protocol for both markers included initial denaturation at 94 °C for 3 min, followed by 20 cycles of 93 °C/45 s, 60 °C/1 min, and 72 °C/1 min; and 35 cycles of 93 °C/45 s, 66 °C/1 min, and 72 °C/1 min. Final extension at 72 °C was kept for 3 min. To avoid false negative and false positive results, each DNA sample was amplified at least three times, separately for the SRY and amelogenin markers. PCR products were separated using 3% agarose (SRY) or 4.4% and 5% MetaPhor® Agarose (amelogenin) gel electrophoresis, and visualised by ethidium bromide staining with UV illumination.
Three bone samples were selected randomly and sent to the Institute of Criminology in Prague (Czech Republic) for independent sex determination. DNA was extracted from bone powder using the MinElute PCR Purification Kit (Qiagen®) according to the protocol described by Yang et al. [33] and Anderung et al. [34]. The PowerPlex® ESX 17 System (Promega) amplification kit was used to obtain the genetic profiles of the samples. Detection of PCR products using capillary electrophoresis was carried out using an ABI PRISM® 3130xl Genetic Analyzer (Applied
Biosystems). Data analysis was performed with GeneMapper® ID-X software, version 1.2.
3. Results and Discussion
application of aDNA analysis from unidentified bone fragments and child skeletal remains in anthropology and paleodemography. Analysis of authentic DNA from historical bone material is problematic because the preservation of DNA molecules depends on many circumstances including environmental conditions, age and preservation of bones, and handling of bones during and after excavation [35, 36]. DNA from ancient bones is fragmented and degraded by hydrolysis and oxidation over time, which complicates its analysis [37, 38]. Moreover, contamination with modern DNA is a serious problem, and many precautions against contamination must be adopted [39].
We analysed the skeletal remains of a population from Pohansko, the south outer precincts. A total of 86 bone specimens of child skeletal remains and 34 bone specimens of unidentified skeletal remains were investigated for genetic sex determination (bone samples could not be removed from 3 skeletons, 1 child and 2 unidentified, because of their poor state of preservation). In general, the skeletal remains were badly preserved, which likely influenced the quality and quantity of authentic DNA of the samples (data not shown).
Using the two common sex markers, SRY and amelogenin, sex was determined in 66 samples (55%): 31 male and 35 female. Only the concordant results of both sex markers were considered as valid. Results are summarized in Table 1. In 38 cases, the results of the SRY and amelogenin marker analyses were not concordant; in 15 cases only either amelogenin or SRY was revealed; and in one sample the analysis yielded no markers. All these data were excluded from the overall evaluation. The failure in SRY and amelogenin locus detection may have been due to a high rate of aDNA fragmentation. The skeletal remains from Pohansko were treated with standard anthropological methods and kept in deposit for approximately 20-40 years, which could have caused the degradation of aDNA. According to Pruvost et al. [35], freshly excavated
Table 1 The results of genetic sex determination (only valid data shown)a.
Grave No. Age at deathb Genetic sex determination
(Table 1 continued)
Grave No. Age at deathb Genetic sex determination
136 6 y M
aAbbreviations: nd, not determined; M, male; F, female; y, years;
m, months. bAge determination was adopted from Drozdova
[24].
bones are better for aDNA amplification than bones from collections because ancient DNA can degrade quickly after bones are removed from the preserving conditions of their original setting.
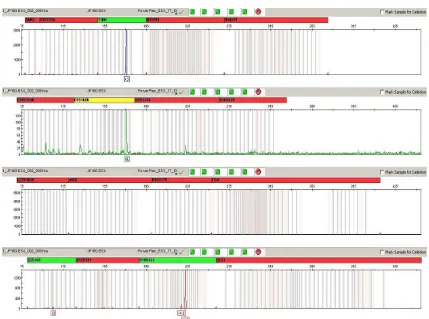
For verification of the concordant results, 3 bone samples (from graves 103, 155 and 160) were sent for evaluation to the Institute of Criminology in Prague. The skeletal remains from grave 103 belong to an adult individual (age not determined). The bones were fragmented and in most cases unidentified. Genetic analysis was negative for SRY, and only the X allele of amelogenin was present. Sex determination as female was confirmed by PCR amplification using the PowerPlex® ESX 17 System (Promega). The genetic profile was incomplete, but it was clearly visible that only the X allele of amelogenin was detected (Fig. 1).
The skeleton from grave 155 belongs to an 18-month-old child and was badly preserved. Genetic analysis was positive for SRY, and the Y allele of amelogenin was revealed, so the sex was determined as
male. As in the case of the skeletal remains from grave 103, the genetic profile was not complete because of low concentration and degradation of DNA, but X and Y amelogenin alleles were detected (Fig. 2). The sex of the infant remains from grave 155 was therefore confirmed as male.
The fragmentary skeletal remains from grave 160 probably belong to an adult individual (age not determined). By genetic analysis, the sex was determined as male (SRY positive, Y amelogenin allele present). Unfortunately, using the PowerPlex® ESX 17 System amplification kit, the genetic profile was of poor quality and amelogenin loci were not detected (Fig. 3). This may be attributed to ongoing degradation of authentic DNA of the sample (investigations in our laboratory and the laboratory of the Institute of Criminalogy were time-separated by more than 1 year). In this case, the results of sex determination with SRY and amelogenin markers failed to verify independently.
The success rate of sex determination (55%) based on the combination of SRY and amelogenin markers in the present study are comparable to other aDNA studies. In a similar study, Vanharova and Drozdova [23] determined the gender in 4,000 year-old samples up to 40% (21 of 53 samples). Zink et al. [20] identified the gender of 2,500-5,000 year-old samples by analysis of SRY and amelogenin genes with a success rate of 43.9% (18 out of 41 cases).
Fig. 1 Electropherogram of DNA typing results for sample 103 using PowerPlex® ESX 17 System (Promega) amplification kit.
Fig. 3 Electropherogram of DNA typing results for sample 160 using PowerPlex® ESX 17 System (Promega) amplification kit.
A) Sex determ ination based on anthropological analysis only
Males; 27
Females; 40
Children; 87 Unidentified individuals; 36
Fig. 4 The sex structure of individuals from Pohansko, south outer precincts, before genetic analysis.
B) Sex determ ination based on concordance in both genetic m arkers
Males ; 30
Females; 55
Unidentified adults; 18 Boys; 28
Girls; 20 Unidentified children; 39
Fig. 5 The sex structure of individuals from Pohansko, south outer precincts, after genetic analysis.
remains, we identified 3 males and 15 females. The sex identification of the remaining 16 individuals was not successful. Figs. 4 and 5 compare the number of adult males, adult females, children without known sex and unidentified individuals before and after genetic sex assessment.
4. Conclusion
Damaged and fragmentary bones constitute a significant portion of skeletal material from rescue excavations. The traditional anthropological approach to demographic analysis is limited if poorly preserved
B) sex determination based on concor dance in both genetic Markers
ancient bones are investigated, especially when sex determination is part of the analysis. In the present study, we demonstrate the utility of molecular genetic methods, specifically the application of genetic sex markers, in the analysis of fragmentary ancient skeletal remains. The SRY and amelogenin sex markers were used for genetic sex determination of fragmentary and child skeletal remains from rescue excavations at the Pohansko archaeological site. Sex was genetically determined in more than half of the investigated cases. Moreover, the results of sex determination of some skeletal remains were independently verified by the Institute of Criminology in Prague. The results of this study allowed the demographic profile of the Pohansko population to be completed with more accurate knowledge about the mortality of this population.
Acknowledgments
This research was supported by the Ministry of Education (Project No. MSM 0021622427), Czech Republic. We thank the Institute of Criminology in Prague for their analysis of the bone fragments from the Pohansko archaeological site. The authors thank Kristina Kovarik, MSc. for language corrections.
References
[1] H. Schutkowski, Sex determination of infant and juvenile skeletons: Morphognostic features, American Journal of Physical Anthropology 90 (2) (1993) 199-205.
[2] M. Faerman, G.K. Bar-Gal, D. Filon, C.L. Greenblatt, L. Stager, A. Oppenheim, et al., Determining the sex of infanticide victims from the Late Roman era through ancient DNA analysis, Journal of Archaeological Science 25 (1998) 861-865.
[3] J.M. Butler, Forensic DNA Typing, Biology, Technology, and Genetics of STR Markers, 2nd ed., Elsevier Academic Press, Amsterdam, Boston, Heidelberg, London, New York, Oxford, Paris, San Diego, San Francisco, Singapore, Sydney, Tokyo, 2005.
[4] K.M. Sullivan, A. Mannucci, C.P. Kimpton, P. Gill, A rapid and quantitative DNA sex test: fluorescence-based PCR analysis of X-Y homologous gene amelogenin, BioTechniques 15 (4) (1993) 636-641.
[5] A. Mannucci, K.M. Sullivan, P.L. Ivanov, P. Gill, Forensic application of a rapid and quantitative DNA sex
test by amplification of the X-Y homologous gene amelogenin, International Journal of Legal Medicine 106 (4) (1994) 190-193.
[6] C. Lassen, S. Hummel, B. Herrmann, Molekulare Geschlechtsbestimmung an Skelettresten früh- und neugeborener Individuen des Gräberfeldes Aegerten, Schweiz, Anthropologischer Anzeiger 55 (2) (1997) 183-191.
[7] E. Meyer, M. Wiese, H. Bruchhaus, M. Claussen, A. Klein, Extraction and amplification of authentic DNA from ancient human remains, Forensic Science International 113 (2000) 87-90.
[8] D. Schmidt, S. Hummel, B. Herrmann, Brief communication: multiplex X/Y-PCR improves sex identification in aDNA analysis, American Journal of Physical Anthropology 121 (2003) 337-341.
[9] M. Cipollaro, G. Di Bernardo, G. Galano, U. Galderisi, F. Guarino, F. Angelini, et al., Ancient DNA in human bone remains from Pompeii archaeological site, Biochemical and Biophysical Research Communications 247 (3) (1998) 901-904.
[10] T. Waldron, G.M. Taylor, D. Rudling, Sexing of Romano-British baby burials from the Beddingham and Bignor villas, Sussex Archaeological Collections 137 (1999) 71-79.
[11] M. Faerman, D. Filon, G. Kahila, C.L. Greenblatt, P. Smith, A. Oppenheim, Sex identification of archaeological human remains based on amplification of the X and Y amelogenin alleles, Gene 167 (1995) 327-332.
[12] M.A. Jobling, P. Gill, Encoded evidence: DNA in forensic analysis, Nature Reviews Genetics 5 (2004) 739-751. [13] S. Hummel, Ancient DNA Typing: Methods, Strategies
and Applications, Springer Verlag, Berlin, Heidelberg, 2003.
[14] F.R. Santos, A. Pandya, C. Tyler-Smith, Reliability of DNA-based sex tests, Nature Genetics 18 (1998) 103. [15] R. Palmirotta, F. Verginelli, G. Di Tota, P. Battista, A.
Cama, S. Caramiello, et al., Use of a multiplex polymerase chain reaction assay in the sex typing of DNA extracted from archaeological bone, International Journal of Osteoarchaeology 7 (1997) 605-609.
[16] L. Luptakova, A. Babelova, R. Omelka, B. Kolena, M. Vondrakova, M. Bauerova, Sex determination of early medieval individuals through nested PCR using a new primer set in the SRY gene, Forensic Science International 207 (2011) 1-5.
[18] E. Cunha, M.L. Fily, I. Clisson, A.L. Santos, A.M. Silva, C. Umbelino, et al., Children at the convent: comparing historical data, morphology and DNA extracted from ancient tissues for sex diagnosis at Santa Clara-a-Velha (Coimbra, Portugal), Journal of Archaeological Science 27 (2000) 949-952.
[19] A.R. Zink, W. Grabner, A.G. Nerlich, Molecular identification of human tuberculosis in recent and historic bone tissue samples: The role of molecular techniques for the study of historic tuberculosis, American Journal of Physical Anthropology 126 (2005) 32-47.
[20] A. Zink, C.J. Haas, U. Reischl, U. Szeimies, A.G. Nerlich, Molecular analysis of skeletal tuberculosis in an ancient Egyptian population, Journal of Medical Microbiology 50 (2001) 355-366.
[21] E. Cappellini, B. Chiarelli, L. Sineo, A. Casoli, A. Di Gioia, C. Vernesi, et al., Biomolecular study of the human remains from tomb 5859 in the Etruscan necropolis of Monterozzi, Tarquinia (Viterbro, Italy), Journal of Archaeological Science 31 (2004) 603-612.
[22] D. Caramelli, C. Lalueza-Fox, C. Capelli, M. Lari, M.L. Sampietro, E. Gigli, et al., Genetic analysis of the skeletal remains attributed to Francesco Petrarca, Forensic Science International 173 (2007) 36-40.
[23] M. Vanharova, E. Drozdova, Sex determination of skeletal remains of 4000 years old children and juveniles from Hostice 1 za Hanou (Czech Republic) by ancient DNA analysis, Anthropological Review 71 (2008) 63-70. [24] E. Drozdova, Breclav-Pohansko VI: Slovansti Obyvatele
Velkomoravskeho Hradiska Pohansko u Breclavi (Demograficka a Antropometricka Studie), Masarykova univerzita, Brno, 2005.
[25] J. Vignatiova, Breclav-Pohansko II. Slovanske Osidleni Jizniho Predhradi, Masarykova univerzita, Brno, 1992. [26] E. Drozdova, J. Benes, Breclav-Pohansko: South outer
precincts, Anthropological study, Anthropologie 35 (3) (1997) 263-275.
[27] J. Vignatiova, E. Klanicova, Predbezna zprava o vysledcich archeologickeho vyzkumu na jiznim predhradi Pohanska u Breclavi v letech 1991-1994, in: Z. Merinsky (Ed.), Konference Pohansko 1999. 40 Let od Zahajeni Vyzkumu Slovanskeho Hradiska Breclav-Pohansko. Archaeologia Mediaevalis Moravice et Silesiana 1/2000, ÚAM FF MU, Brno, 2001, pp. 21-30.
[28] R. Prichystalova, Detske hroby zjuzneho predhradia
velkomoravskeho hradiska na Pohansku pri Breclavi, Studijne Zvesti Archeologickeho Ustavu SAV 42 (2007) 163-181.
[29] C. Keyser-Tracqui, B. Ludes, Methods for the study of ancient DNA, in: A. Carracedo (Ed.), Methods in Molecular Biology: Forensic DNA Typing Protocols 297, Humana Press, Totowa, New Jersey, 2005, pp. 253-264.
[30] M.L. Sampietro, M.T.P. Gilbert, O. Lao, D. Caramelli, M. Lari, J. Bertranpetit, et al., Tracking down human contamination in ancient human teeth, Molecular Biology and Evolution 23 (9) (2006) 1801-1807.
[31] A. Cooper, H.N. Poinar, Ancient DNA: Do it right or not at all, Science 289 (5482) (2000) 1139.
[32] S. Pääbo, H. Poinar, D. Serre, V. Jaenicke-Després, J. Hebler, N. Rohland, et al., Genetic analyses from ancient DNA, Annual Review of Genetics 38 (2004) 645-679. [33] D.Y. Yang, B. Eng, J.S. Waye, J.C. Dudar, S.R. Saunders,
Technical note: Improved DNA extraction from ancient bones using silica-based spin columns, American Journal of Physical Anthropology 105 (1998) 539-543.
[34] C. Anderung, P. Persson, A. Bouwman, R. Elburg, A. Götherström, Fishing for ancient DNA, Forensic Science International: Genetics 2 (2008) 104-107.
[35] M. Pruvost, R. Schwarz, V.B. Correia, S. Champlot, S. Braguier, N. Morel, et al., Freshly excavated fossil bones are best for amplification of ancient DNA, Proceedings of the National Academy of Sciences of The United States of America 104 (3) (2007) 739-744.
[36] J. Burger, S. Hummel, B. Herrmann, W. Henke, DNA preservation: A microsatellite-DNA study on ancient skeletal remains, Electrophoresis 20 (1999) 1722-1728.
[37] S. Pääbo, Ancient DNA: Extraction, characterization, molecular cloning, and enzymatic amplification, Proceedings of the National Academy of Sciences of the United States of America 86 (1989) 1939-1943.
[38] E. Willerslev, A. Cooper, Ancient DNA, Proceedings of the Royal Society B: Biological Sciences 272 (2005) 3-16. [39] M.T.P. Gilbert, H.J. Bandelt, M. Hofreiter, I. Barnes,
Assessing ancient DNA studies, Trends in Ecology and Evolution 20 (10) (2005) 541-544.
A Comparative Study of Soluble Protein Extractions of
Populus deltoides
× (
Trichocarpa
×
Deltoides
) for 2-DE
Joke Dupae1, Ann Cuypers1, Jean-Paul Noben2, Jana Boulet1, Nele Weyens1, Karen Verstraelen1 and Jaco Vangronsveld1
1. Centre for Environmental Sciences, Hasselt University, Agoralaan Building D, B-3590 Diepenbeek, Belgium
2. Biomedical Institute, Hasselt University, Agoralaan, Building A, 3590 Diepenbeek, Belgium
Received: November 21, 2011 / Accepted: April 27, 2012 / Published: September 30, 2012.
Abstract: Background:The disclosure of the poplar genome strengthens its position as well-established model organism. Populus has been subject of several proteome studies, but up to date no comparative study was performed on the extraction method of soluble proteins for this species. The extraction is the most critical step in two-dimensional gel electrophoresis and each extraction method has its advantages, disadvantages and limitations. Therefore protein extraction methods should be optimized for each tissue before starting an experimental setup. In prospect of future DIGE (Differential Gel electrophoresis) experiments for the investigation of the effects of cadmium and inoculation with plant growth promoting bacteria at the proteome level, the aim of this study was to optimize an extraction method for soluble proteins of poplar leaves and roots. Results:The acetone-phenol extraction method was found to be the most suited, rendering a high spot number and low background interference. During further optimization, several critical steps in the extraction method were revealed. Conclusion:Aiming to optimize the extraction of soluble leaf and root proteins of Populus deltoides × (trichocarpa × deltoides) compatible with DIGE analysis, a protocol rendering high reproducibility, low background interference and a high spot numberwas established, however no novel insights were acquired.
Key words: Proteomics, two-dimensional gel electrophoresis, soluble proteins, protein extraction, Populus.
1. Introduction
Due to their low protein content and high protease activity, proteomic analysis of plant tissues appears to be particularly challenging. Moreover, other compounds such as phenols, terpenes, organic acids, pigments, oxidative enzymes, lipids, etc. can interfere with two dimensional gel electrophoresis (2-DE) and can cause vertical or horizontal streaking, smearing or reduction in the number of distinctly resolved spots as indicated by Carpentier et al., Saravanan et al. and Wang et al. [1-3]. Protein extraction is therefore a crucial step in two- dimensional gel electrophoresis.
Populus is a well-established model organism to
elucidate the biological function unique to trees, as
Corresponding author: Joke Dupae, Ph.D. candidate, research fields: environmental biology, proteomics. E-mail: [email protected].
proposed by Plomion et al. [4]. The genome of
Populus trichocarpa has been sequenced by Tuskan et
al. [5], paving the way for poplar proteomics. Proteome studies on poplar have been published by Bohler et al. and Kieffer et al. [6-8], but up to date no comparative study on protein extraction of poplar has been released. As protein extraction is critical in 2-DE and since every extraction method has its limitations, advantages and disadvantages as stated by Carpentier et al. [1], it is essential to find an optimal extraction protocol before starting an experimental set-up. Therefore the aim of this study was to optimize an extraction method for soluble proteins of poplar leaves and roots.
were selected for comparison: (1) a combination of acetone and phenol; a slightly changed protocol based on Carpentier et al. [1], (2) a combination of TCA/acetone and phenol; this protocol was proposed by Wang et al. [9] to be universal, rapid and especially suited for recalcitrant plant tissues. And finally, (3) a TCA/DTT/acetone extraction; a protocol already applied on Populus tremula L. × P. alba L. (Populus ×
Canescens (Aiton) Smith)—clone INRA 717-1-B4
and Populus tremula L. leaves described by Bohler et
al. and Kieffer et al. [6-8]. By comparing these protocols and by further improving the spot pattern, an optimized extraction protocol for soluble leaf and root proteins of Populus deltoides × (trichocarpa ×
deltoides) was established, however no fundamentally
novel insights were acquired. In prospect of future experiments, buffer compatibility was checked for DIGE analysis.
2. Materials and Methods
2.1 Plant Material
Cuttings (30 cm, with an average shoot height of 10 cm) of Populus deltoides × (trichocarpa × deltoides) were grown on sand in 4l pots during 10 weeks. They were watered three times a week with 1/2 strength Hoagland’s solution [10], until they had sufficient expanded leaves. Of each plant three fully expanded leaves, developed after transfer to the pot system, were harvested. To minimize biological variation, methods were evaluated “within leaf”, meaning that every possible combination of two out of three extraction methods was performed on separate leaf halves derived from same leaf. This design allowed for comparing the three methods within one plant. For each combination three biological replicates were performed using three plants in total. In future experiments poplar cuttings will be exposed to different treatments and proteins obtains from roots and leaves will be compared. Therefore, highest reproducible protocol was tested for its applicability on roots as well.
2.2 Extraction Methods
At first we compared the differences between extraction of soluble proteins within the same leaf during 1 h and overnight for each extraction method. Subsequently we compared the three methods to each other in a loop design and one phenol based method, described by Sarma et al. [12], was included afterwards.
2.2.1 TCA/Acetone-Phenol Extraction
Prior to grinding in liquid nitrogen, the primary and secondary veins of the leaves were removed. The extraction was performed according to Wang et al. [9] (tested on bamboo (Bambusa vulgaris), grape (Vitis
vinifera), iris (Iris pseudacorus), olive (Olea europea),
lemon (Citrus limonum), pine (Pinus nigra), redwood
(Sequoia sempervirens), sugarcane (Saccharum
officinarum), and tobacco (Nicotiana tabacum)) with
some modifications. Because of the much higher amount of starting material, we used 10% TCA/acetone; 80% methanol-0.1M ammonium acetate and 80% acetone at 10 mL per gram ground tissue, a phenol-Tris buffer (a pH of 8.0 is required for optimal results, as described by Thiellement et al. [11]), was used instead of a phenol-SDS buffer and our samples were allowed to precipitate during 1 h or overnight at – 20 °C. The dry pellet was finally resuspended in resuspension buffer (7 M urea, 2 M thio-urea, 4% chaps and 30 mM Tris) (incubation at 18 °C, 1,200 rpm, 2 h (Eppendorf mixer)), centrifuged (70,000 g, 90 min, 18 °C) and stored at – 80 °C.
2.2.2 Acetone-Phenol Extraction
This protocol is based on the work of Carpentier et al. [1] (tested on banana (Musa spp.), apple (Malus
domestica) and potato (Solanum tuberosum)) with
and 10 volumes of extraction buffer (50 mM Tris pH 8.5, 25 mM Na2EDTA, 100 mM KCl, 30% w/v sucrose, 2% -mercapto-ethanol and 0.4 mM PMSF) were added per gram FW. Ten minutes later, the same volume of phenol was added and the solution was mixed thoroughly. After centrifugation (8,000 rpm, 5 min, 4 °C) the phenolic phase was collected, 5 volumes of ammonium acetate in proportion to the volume of collected phenol, were added and the sample was allowed to precipitate at – 20 °C for 1 h or overnight. After centrifugation (10,000 g, 10 min, 4 °C) the pellet was washed three times with ammonium acetate and once with acetone/DTT (0.2% w/v). Finally, the pellets were resuspended in resuspension buffer as described in 2.2.1.
2.2.3 TCA/DTT/Acetone Extraction
This protocol is based on the work of Bohler et al. [6] (tested on poplar ([Populus tremula L. × P. alba L.
(Populus × Canescens (Aiton) Smith)]—clone INRA
717-1-B4)) without modifications. As mentioned above, the same handlings prior to grinding were performed and the samples were allowed to precipitate for 1 h or overnight at – 20 °C. The final pellet was treated as described in 2.2.1.
2.2.4 A Phenol Extraction Protocol Rendering High Resolution and Reproducibility of 2-DE
The extraction was carried out as described by Sarma et al. [12] (tested on soybean) with some modifications. They proposed a protocol based on phenol extraction of soluble proteins, rendering a high resolution of 2-DE gels and a high reproducibility. Protease inhibitors were excluded from the extraction buffer since no problems with proteases were observed in the previous extraction methods. The final pellet was treated as described in 2.2.1.
2.3 RuBisCO Interference
Addition of ampholine to the resuspension buffer caused a position shift of the RuBisCO Large Subunit (LS) in the first dimension, resulting in lower interference of RuBisCO LS in the second dimension
gel, an observation first described by Espagne et al. [13]. Comparing the buffers used, adding 0.5% ampholine pH 4-7 resulted in a lower RuBisCO resolubilization. In order to control the RuBisCO in poplar leaf extracts, 1.25% (v/v) IPG 4-7 NL buffer (GE Healthcare) was added to the resuspension buffer, samples were mixed (1,200 rpm) for 2 h at 18 °C and finally centrifuged (70,000 g, 90 min, 18 °C) to remove the unsolubilized proteins.
2.4 Protein Quantification
The protein concentration of each sample was determined using the RC DC protein assay kit II (BIORAD, California) using BSA (1.54 mg/mL) as the standard. This protein assay is an improved version of the Lowry assay [14], modified to be reducing agent compatible (RC) and detergent compatible (DC). Color development is achieved by a two step reaction: (1) a reaction between proteins and copper in an alkaline medium followed by (2) a reduction of Folin reagent by copper-treated proteins [14]. The Microfuge Tube Assay Protocol, provided by BIORAD, was used with a repetition of step 4 and 5. Leaf samples and root samples were 1/2 diluted prior to quantification and all solutions were brought to 27 °C prior to use.
2.5 2-DE
500 V for 3 h, gradient step of 1,000 V for 3 h, constant step of 1,000 V for 3 h, gradient step of 8,000 V for 3 h and finally a constant step of 8,000 V for 7 h at 20 °C with a maximum current setting of 50 µA/strip. On the paper wicks at the negative electrode, 150 µL DeStreak rehydration solution (GE Healthcare) was added to reduce streaking. After the IEF, the IPG strips were equilibrated at 18 °C for 15 min in equilibration buffer (75 mM Tris pH 8.8, 6 M urea, 30% v/v glycerol, 2% w/v SDS and a trace of bromophenol blue) supplemented with 1% w/v DTT. A second equilibration step of 15 min with the same equilibration buffer, now containing 2.5% w/v iodoacetamide, was carried out in the dark afterwards. The first equilibration step provides a completely reduced state of unalkylated, denaturated proteins. Whereas, in the second step, iodoacetamide alkylates thiol groups in order to prevent reoxidation during electrophoresis. On top, the Immobiline DryStrips are saturated with the SDS buffer system, required for the second dimension separation. The IPG strips were then sealed on top of 200 mm × 260 mm × 1 mm, 12.5% polyacrylamide gels with 0.5% agarose in SDS running buffer. The SDS-PAGE step was performed at 15 °C in Ettan Dalt II tanks (GE Healthcare) at 0.5 W pergel for 20 h.
2.6 Staining
To visualize the protein spots, silver staining (according to Shevchenko [15]) or Gel Code Blue staining (according to the manufacturer guidelines; Thermo Scientific) were performed. After staining, the gels were scanned using a flatbed scanner (CanoScan 4400F, Canon) at highest resolution. Comparing the extraction methods and testing the highest reproducible protocol on root samples, 100 µg soluble protein was loaded onto silver stained gels. For the comparison of RuBisCO focusing and the reproducibility test, Gel Code Blue staining was used (50 or 300 µg proteins, see results). Staining the gels with Gel Code Blue gives an idea about
reproducibility of DIGE gels (Bohler S., personal comments).
2.7 Image Analysis
Gel analysis was performed using ImageMaster 2D Platinum 5.0. For quantitative comparison, two parameters were used: the total spot number and the ratio of the number of automatically detected spots to the number of spots after manual verification. The last parameter is included to quantify background interference; the closer the ratio is to 1, the less background interference in the gel. Further on, this ratio will be referred to as the “background ratio”. Values are the means ± standard deviation.
2.8 Statistical Analysis
Statistical analysis of all data was performed using SAS 9.1. All tests included 3 biological replicates. Since all data had a normal distribution, a student’s
t-test was performed to determine statistically significant differences between the groups. The significance level was set at α = 0.05.
2.9 Spot Picking and Identification
histidine were set as fixed and variable modifications, respectively. Maximally one missed cleavage was allowed. Additional information (e.g. peptide sequence, charge state of each peptide) will be provided in supplementary data.
Resulting peptide identifications were accepted if they could be established at greater than 95.0% probability as specified by the Peptide Prophet algorithm described by Keller et al. [17] within Scaffold version 2_05_02 (Proteome Software Inc., Portland, OR). Protein identifications were accepted if they could be established at greater than 99.9% probability and contained at least 2 identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm within Scaffold according to Nesvizhskii et al. [18]. To assign a function, the accepted protein identifications were searched for homology against NCBI nonredundant database
(Populus; taxid3689). For additional information
concerning the identification we refer to the supplementary data.
3. Results and Discussion
In order to find an optimal protocol for the extraction of soluble proteins from poplar leaves, a comparison was made between three extraction methods: (1) a combination of acetone and phenol, (2) a combination of TCA/acetone and phenol and (3) a TCA/DTT/acetone extraction. At first, the extraction time was optimized. Samples precipitated overnight resulted in better spot patterns and less background interference than samples precipitated during one hour (data not shown). Subsequently, the three extraction methods were compared within the leaves in a loop design. Since the total spot number was discernibly lower after the TCA/acetone-phenol extraction, no further analysis was performed at this stage and the TCA/acetone-phenol extraction was excluded from further comparison (Fig. 1).
A new comparison between acetone-phenol and TCA/DTT/acetone was made and subjected to further analysis (Fig. 2). However the TCA/DTT/acetone
Fig. 1 Comparison of the three extraction methods within the poplar leaves in a loop design; silver staining. Extraction with TCA/acetone-phenol clearly rendered fewer spots and an inferior spot pattern. Therefore it was excluded from further analysis.
extraction (average total spot number: 606 ± 41; average background ratio: 0.759 ± 0.07) was used on poplar before, the acetone-phenol extraction appeared to be the best with an average total spot number of 983 ± 53 and an average background ratio of 0.905 ± 0.02. Furthermore, there was a significant statistical difference between both extraction methods for the spot number (P = 0.0004) and the background ratio (P= 0.0258). Leaves of Populus tremula L. × P. alba
L. (Populus × canescens (Aiton) Smith)—clone INRA
717-1-B4 and of Populus tremula L., used by Bohler et al. [6] and Kieffer et al. [7, 8] respectively, are morphologically different (e.g. much thinner) compared to our poplar clone (Bohler S., personal comments). Therefore the observed difference in protein extraction capacity between these leaf types and ours, could be explained by the difference in leaf composition and structure.
Sarma et al. [12] reported that they developed a phenol extraction that renders a high resolution and reproducibility of 2-DE. Using this extraction method, a similar spot pattern was obtained compared to the acetone-phenol extraction (Fig. 3). No significant difference was found in the average background ratio (P = 0.7673), but the average total spot number was significantly higher with the acetone-phenol extraction (P = 0.0313).
Based on the former results, acetone-phenol extraction was chosen as a proper extraction method for poplar leaves and further tests were performed in order to optimize the spot pattern. As suggested by
Thiellement et al. [11], the pH of the phenol should always be at 8.0 to prevent nucleotide interference. In preliminary experiments, we observed that buffers made only one day in advance (determined in the manuscript as “fresh buffers”) render clearer gels (data not shown). Additionally, calculating the volumes of all solutions in proportion to the fresh weight of the starting material, enhances the spot pattern (data not shown). Adding ampholine pH 4-7 to the resuspension buffer, Espagne et al. [13] reported a position shift of the RuBisCO large subunit (LS). However, adding ampholine pH 3-10, led to a selective focusing of RuBisCO LS to its correct pI. They suggested that the different mobility shift resulted from the poor RuBisCO solubilization in the 4-7 ampholine pH range. Besides the position shift of the RuBisCO LS, distinctly separated spots in the RuBisCO LS region were obtained. Moreover, some of these spots had never been identified in the plant proteome before. These results raised the question whether addition of IPG 4-7 NL buffer (GE Healthcare) to our resuspension buffer, could result in a better focused RuBisCO LS region since less RuBisCO LS is solubilized. Therefore, proteins were resuspended in the presence of 1.25% (v/v) IPG 4-7 NL buffer (GE Healthcare) and centrifuged at 70,000 g during 90 min prior to loading. First, testing the Gel Code Blue staining, 50 µg proteins was loaded, resulting in a better focused RuBisCO LS spot (Fig. 4a). The protein amount was increased up to 300 µg to obtain the same spot pattern as on silver stained gels.
Fig. 4 Focusing of RuBisCO LS region. Gel Code Blue. A: 1.25% (v/v) IPG 4-7 NL buffer + 50 µg proteins loaded; B: 1.25% (v/v) IPG 4-7 NL buffer + 300 µg proteins loaded; C: no IPG 4-7 NL buffer + 300 µg loaded. By adding 1.25% (v/v) IPG 4-7 NL buffer the RuBisCO LS spot is clearly better focused.
Loading 300 µg proteins, the RuBisCO spot is less focused but still better than without IPG 4-7 NL buffer (Fig. 4b, c).
The presence of RuBisCO LS often interferes with the identification of spots with the same molecular mass or the same isoelectric point. To determine the RuBisCO LS interference, 4 spots (indicated by
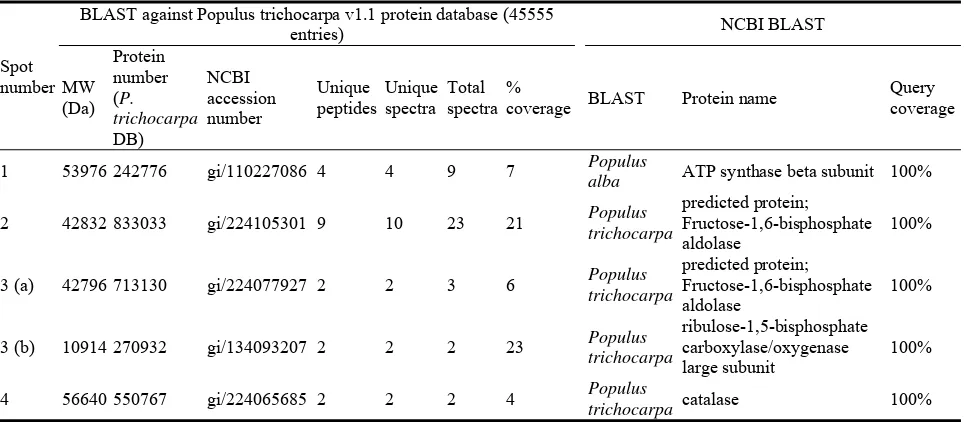
numbered arrows on Fig. 5) were picked for LCQ analysis. 3 spots could be identified (Table 1) as (1) an ATP synthase beta subunit, (2) a fructose 1.6 bisphosphate aldolase and (3) a catalase. The same aldolase was identified in spot 4, but here, it was comigrating with RuBisCO LS. Spot location was inconsistent with the MW of each of both proteins separately, but its occurrence was reproducible from
gel to gel. The reason for this is not known. Taken together and as judged from a limited number of spots analyzed, it seems that only minor background interference might be expected coming from RuBisCO LS in planned physiological studies in poplar using the protocol as described above.
Hydrolysis of acrylamide could occur during overnight isoelectric focusing, resulting in poor focused spot patterns and sometimes even “gaps” in
Table 1 Protein identification to determine RuBisCO interference.
Spot number
BLAST against Populus trichocarpa v1.1 protein database (45555
entries) NCBI BLAST
MW
coverage BLAST Protein name
Query coverage
1 53976 242776 gi/110227086 4 4 9 7 Populus
alba ATP synthase beta subunit 100%
2 42832 833033 gi/224105301 9 10 23 21 Populus
the basic region. Adding 150 µL DeStreak rehydration solution on the negative paper wick, hereby preventing depletion of urea and thiourea and thus diminishing hydrolysis of acrylamide according to Kask et al. [19], resulted in a better spot pattern (data not shown) but could not prevent the “gaps” in the basic region (Verstraelen K, personal comments).
Differences in the average spot number were observed for the acetone-phenol extraction between the different comparisons, giving doubt to the reproducibility of the extraction protocol. However,
these differences could be due to the silver staining procedure, considering its low reproducibility as described by Görg et al. [20]. To test the reproducibility of the acetone-phenol extraction, gels were stained with Gel Code Blue, which is less sensitive but more reproducible than silver staining, stated by Görg et al. [20]. Analysis of 4 gels (biological replicates) resulted in an average total spot number of 208 ± 2 and high matching percentages, suggesting a high reproducibility of the extraction method (Fig. 5, Table 2).
Fig. 5 Acetone-phenol extraction of leaf samples. Gels were stained with Gel Code Blue and 300 µg proteins were loaded. After matching, a high reproducibility could be observed (Table 1). The reference gel is shown with the spot pairs; red circles represent spots belonging to the reference gel (L12), blue circles represent spots belonging to gel L10. Numbered arrows indicate the spots that were picked for LCQ analysis.
Table 2 Matching results of the gel from the reproducibility test.
GelName1 GelName2 Number of matches Percent matches
L9 L12 198 95.0
L10 L12 200 94.8 L11 L12 194 93.3 L12 was set as reference gel. # Matches: the number of spots that could be matched between the two specified gels (GelName1 and 2), Percent Matches: percentage of spots of GelName1 that could be matched to the reference gel (GelName2).
After optimizing the extraction method on leaf samples, the same protocol was used for root samples. This rendered very clear spot patterns with an average total spot number of 1611 ± 121 and an average background ratio of 1.03 ± 0.07 in Fig. 6.
4. Conclusion
According to the data presented above, the acetone-phenol extraction seems to be the highest reproducible extraction method for soluble proteins of poplar leaves, yielding a high spot number and low background interference. Applying the extraction method on root samples rendered very clear spot patterns as well. Further optimization revealed several critical steps: (1) pH of the phenol should always be at 8.0 to assure that nucleotides will be in the buffer phase and not in the phenol-rich phase, in accordance to Thiellement et al. [11], (2) the extraction buffer should always be made fresh and (3) the volumes of buffers, phenol, … added should be carefully calculated in proportion to the fresh weight of the starting material.
Some more tests made to improve the quality of the spot pattern, revealed that applying 150 µL DeStreak rehydration solution (GE Healthcare) on the paper wick of the negative electrode increases the quality of the spot pattern. On top, addition of 1.25% (v/v) IPG 4-7 NL buffer (GE Healthcare) to the buffer prior to resuspension results in a better focused RuBisCO LS region. It should be mentioned however that, when performing DIGE, ampholytes will compete with proteins to bind the fluorophores, therefore IPG buffer may not be added prior to DIGE labeling. Scientists focusing on the RuBisCO region could use this method combined with other commercial dyes which are sensitive and can be used quantitatively.
Acknowledgments
Research funded by the institute for the Promotion of Innovation through Science and Technology in
Flanders (IWT-Vlaanderen) for JD and NW, by the Fund for Scientific Research Flanders (FWO-Vlaanderen), Ph.D. grant for JB and by a Ph.D. grant for KV from a BOF (Bijzonder onderzoeksfonds) project of Hasselt University, BOF06N01. The UHasselt Methusalem project 08M03. The authors are grateful to Erik Royackers from the Biomedical Research Institute of Hasselt University for the identification of proteins by mass spectrometry and acknowledge the DoEJoint Genome Institute and Poplar Genome Consortium for providing the Populus
genome sequence.
References
[1] S.C. Carpentier, E. Witters, P. Deckers, R. Swennen, B. Panis, Preparation of protein extracts from recalcitrant plant tissues: An evaluation of different methods for two-dimensional gel electrophoresis analysis, Proteomics 5 (2005) 2497-2507.
[2] R.S. Saravanan, J.K.C. Rose, A critical evaluation of sample extraction techniques for enhanced proteomis analysis of recalcitrant plant tissues, Proteomics 4 (2004) 2522-2532.
[3] W. Wang, M. Scali, R. Vignani, A. Spadafora, E. Sensi, S. Mazzuca, et al., Protein extractuon for two-dimensional electrophoresis from olive leaf, a plant tissue containing high levels of interfering compounds, Electrophoresis 24 (2003) 2369-2375.
[4] C. Plomion, C. Lalanne, S. Claverol, H. Meddour, A. Kohler, M.B. Bogeat-Triboulot, et al., Mapping the proteome of poplar and application to the discovery of drought-stress responsive proteins, Proteomics 6 (2006) 6509-6527.
[5] G.A. Tuskan, S. DiFazio, S. Jansson, J. Bohlmann, I. Grigoriev, U. Hellsten, et al., The genome of black cottonwood, Populus trichocarpa (Torr. & Gray), Science 313 (2006) 596-1604.
[6] S. Bohler, M. Bagard, M. Oufir, S. Planchon, L. Hoffmann, Y. Jolivet, et al., A DIGE analysis of developing poplar leaves subjected to ozone reveals major changes in carbon metabolism, Proteomics 7 (2007) 1584-1599.
[7] P. Kieffer, J. Dommes, L. Hoffmann, J.F. Hausman, J. Renaut, Quantitative changes in protein expression of cadmium-exposed poplar plants, Proteomics 8 (2008) 2514-2530.
L. Hoffmann, et al., Combining proteomics and metabolite analyses to unravel cadmium stress-response in poplar leaves, Journal of Proteome Research 8 (1) (2009) 400-417.
[9] W. Wang, R. Vignani, M. Scali, M. Cresti, A universal and rapid protocol for protein extraction from recalcitrant plant tissues for proteomic analysis, Electrophoresis 27 (2006) 2782-2786.
[10] D.R. Hoagland, D.I. Arnon, The water culture method for growing plants without soil, California Agricultural Experiment Station Bulletin 347 (1938) 36-39.
[11] H. Thiellement, M. Zivy, C. Darmeval, Plant Proteomics: Methods and Protocols, Humana Press Inc., New Jersey, 2006, pp. 9-14.
[12] A.D. Sarma, N.W. Oehrle, D.W. Emerich, Plant protein isolation and stabilization for enhanced resolution of two-dimensional polyacrylamide gel electrophoresis, Analytical Biochemistry 379 (2008) 192-195.
[13] C. Espagne, A. Martinez, B. Valot, T. Meinnel, C. Giglione, Alternarive and effective proteomic analysis in Arabidopsis, Proteomics 7 (2007) 3788-3799.
[14] Lowry, N.J. Rosebrough, A.L. Farr, R.J. Randall, Protein measurement with the folin phenol reagent, Journal of
Biological Chemistry 193 (1951) 265-275.
[15] A. Shevchenko, M. Wilm, O. Vorm, M. Mann, Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels, Analytical Chemistry 68 (1996) 850-858.
[16] D. Dumont, J.P. Noben, J. Raus, P. Stinissen, J. Robben, Proteomic analysis of cerebrospinal fluid from multiple sclerosis patients, Proteomics 4 (2004) 2117-2124.
[17] A. Keller, A.I. Nesvizhskii, E. Kolker, R. Aebersold Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search, Analytical Chemistry 74 (2002) 5383-5392. [18] A.I. Nesvizhskii, A. Keller, E. Kolker, R. Aebersold, A
statistical model for identifying proteins by tandem mass spectrometry, Analytical Chemistry 75 (2003) 4646-4658.
[19] L. Kask, K. Larsson, B. Bjellqvist, Elimination of basic gaps at high pH values in 2-DE, Proteomics 9 (2009) 5558-5561.
Xanthophyll Cycle and Its Relative Enzymes
Xirui Xiong, Xuefei Wang and Ming’an Liao
College of Horticulture, Sichuan Agricultural University, Ya’An City, SiChuan 625014, China
Received: February 17, 2012 / Accepted: July 12, 2012 / Published: September 30, 2012.
Abstract: Light is a fundamental source of energy but is also potentially harmful to organisms. Plants have evolved a variety of regulatory mechanisms to respond to the naturally varying light conditions. Xanthophyll cycle is now recognized as a key regulator and photoprotective mechanism found ubiquitously in plants. Xanthophyll cycle has multiple functions, such as thermal dissipation, protection against oxidative stress caused by light, modulation of the structure of thylakoid membrane, involving in blue light signal transduction and regulating the synthesis of ABA (Abscisic acid). VDE (Violaxanth de-epoxidase) and ZE (zeaxanth epoxidase), are involved in xanthophyll cycle. This paper outlined the functions of xanthophylls cycle and its relative enzymes.
Key word: Xanthophyll cycle, function, VDE, ZE.
1. Introduction
Light was a fundamental source of energy but is also potentially harmful to organisms [1]. Plants had evolved a variety of regulatory mechanisms to respond to the naturally varying light conditions [2]. Xanthophyll cycle is now recognized as a key regulator and photoprotective mechanism found ubiquitously in plants [3, 4]. Xanthophyll cycle, also known as violaxanthin cycle, is that violaxanthin (V) transforms into anthers yellow mass (A) and zeaxanthin (Z) occuring de-epoxidation reaction in the bright light, and that Z re-transforms into V occuring epoxidation in low light or dark [5, 6].
Xanthophyll cycle was wide spread in higher plants, ferns, mosses and some algae, located in the thylakoid membrane. There was a difference that the violaxanthin cycle was in higher plants, and that the dinoflagellates flavin silicon cycle was in some algae, but the two loops were both formed Z or A [7]. There was neither violaxanthin cycle nor dinoflagellates flavin silicon cycle in Cyanobacteria, in which
β-carotene become into Z in bright light [8]. We outlined the functions of xanthophylls cycle and its
Corresponding author: Ming-An Liao, Ph.D., professor, research field: plant physiology. E-mail: [email protected].
relative enzymes.
2. Functions of Xanthophyll Cycle
In 1962, Yamamoto et al. [9] firstly discovered the presence of xanthophyll cycle in higher plants. Untill 1987, Demmig et al. [10] found that Z might be related with the dissipation of excess light energy, then xanthophyll cycle became attractive. Xanthophyll cycle had multiple functions, such as thermal dissipation, protection against oxidative stress caused by light, modulation of the structure of thylakoid membrane, involving in blue light signal transduction and regulating the synthesis of ABA.
2.1 Thermal Dissipation
[16]. The mutant of lack of effective thermal dissipation performed more sensitive to strong light, especially in strong light with other stress factors. Numerous evidences showed that in photosynthetic apparatus of higher plants, the amount of Z and A was related to the extent of thermal dissipation, measured as NPQ [17, 18]. NPQ was inhibited by 80 % or more when leaves were fed with DTT, an inhibitor of VDE [17-19]. So far, most of the experiments support the view that thermal dissipation quenching was dependent on xanthophyll cycle.
In normal photosynthesis, thylakoid lumen pH should be maintained at 5.5 or so. And generally speaking, the thermal dissipation of the main limiting factor was the formation of Z.
2.2 Protection Against Lipid Peroxidation
The chlorophyll of thylakoid membrane was a type of photosensitive material. Havaux et al. [20] and Sarry [21] showed when Z production was inhibited by DTT, light stress would strongly enhance lipid degradation, and caused the peroxidation of unsaturated fatty acids in the thylakoid membrane. It was thus suggested that xanthophyll cycle might protect lipids from oxidative stress. In fact, some other experiments had shown that lutein and Z could quench the singlet state oxygen [22, 23].
2.3 Modulation of the Structure of the Thylakoid Membranes
Yamamoto et al. [24] suggested that xanthophylls cycle components may affect the physical properties of the thylakoid membrane. In 1991, Gruszeski and Strzalka [25] observed that VDE could cause the change of the fluidity of the thylakoid membrane. Havaux and Gruszeski [26] showed that after high light treatment, the decrease of the thylakoid membrane fluidity was closely related to the increase of Z. They also showed that Z-induced decrease of the membrane fluidity could enhance the stability of the thylakoid membrane under conditions of strong light and high
temperature, thereby could increase the tolerance of photosynthetic apparatus against light and temperature [22]. However, the PSⅡstability decreased in an A. thaliana mutant with high Z and no V [27]. VDE enzyme existed in the etiolated Phaseolus vulgaris leaves, the activity of which was inhibited by DTT. This indicated VDE enzyme appeared before the thylakoid and photosynthetic activity did.
2.4 Involving in Blue Light Signal Transduction and Regulating the Synthesis of ABA
Xanthophyll cycle was involved in blue signal light transduction in plants and Z was considered to be a photoreceptor. In the dark, the cotton coleoptile accumulating V but lacking of Z was not bent when induced by blue light pulse. But the degree of bending of the one containing different levels of Z was positively correlated with the increase of Z when induced by blue light pulse. The formation of Z and the side of openness of the thylakoid membrane matrix caused by blue light pulse, which was associated with the concentration of Z, were inhibited by DTT.
Somebody proposed Z was an intermediate pathway of the synthesis of ABA. So regulating the synthesis of ABA may be another function of xanthophyll cycle. In excessive light energy, VDE maybe reduce the synthesis of ABA. ZE was involved both in xanthophyll cycle and the synthesis of ABA precursors [16].
3. Enzymes Involved in Xanthophyll Cycle
In xanthophyll cycle, VDE converted V into Z via intermediate A, whereas ZE catalyzed the backward reaction [11]. VDE was the key enzyme in xanthophyll cycle and ZE was essential.
3.1 Violaxanthin De-Epoxidase
lettuce and found its molecular mass to be 43 KD with an isoelectric point of 5.4. They reported the amino acid composition of VDE, and considered acidic amino acid Asp (containing Asn) and Glu (containing Gln) held the largest proprortion, which may be the reason that VDE was acidic. Bugos and Yamamoto [29] had cloned the cDNA of VDE in lettuce and expressed it in E.coli. In VDE protein, there were three interesting domains: (1) a cysteine-rich region which was most likely to be the inhibitory site of dithiothreitol (DTT); (2) a lipocalin signature, probably the binding site of violaxanthin; and (3) a highly charged domain where 47% of the amino acid residues contain charged side chain, possible involved in its binding to the thylakoid membrane [1].
The activity of VDE was strictly regulated by pH of thethylakoid lumen. VDE was mobile with the lumen pH above 7.0, and became tightly bound to the membrane at pH below 6.5 [30]. When pH decreased to 6.3, the de-epoxidation started, and the optimum pH of V de-epoxidation was 5.8 in isolated pea chloroplast [9]. Ascorbate was a co-substrate of VDE [24] and the activity of VDE was regulated by ascorbate concentration [31, 32], as well as by its availability [33]. In addition, the de-epoxidation was also affected by the amount of VDE protein and V availability [32]. An early finding showed that MGDG (monogalactosyl diacylglycerol) played an important role in VDE activity. Its activity was also affected by the ratio of MGDG to V [34]. In recent study, MGDG was found to be four times more efficient than DGDG (digalactosyl diacylglycerol) and up to thirty-eight times than other lipids in the reaction of VDE [28]. Further, low concentration DTT could intensively inhibit the VDE activity [8, 32], while high concentration DTT inhibited the activity of ascorbate peroxidase [35]. So low concentration of DTT treatment was usually used for studying the function of xanthophyll cycle [19].
So far, the amino acid composition of VDE had been known and the cDNA sequences encoding VDE have been cloned. The activity of VDE can not be directly
measured in vivo and new experimental equipment or methods will be needed. It may be effective way to study VDE via using different mutants or efficiently regulating the gene expression of VDE.
3.2 Zeaxanthin Epoxidase
ZE was a dual-function monooxygenase, located in the thylakoid membrane substrate surface, and can convert Z to A and V. The ZE catalyzing reaction required the presence of the co-substrate NADPH, oxygen and another cofactor FAD [7]. Its optimum pH was 7.0, close to the pH of the stroma. The enzyme had not yet been isolated from plant tissues. Burbidge et al. [36] had obtained the DNA sequence of cDNA of ZE and found its molecular mass to be 72.5 KD with an isoelectric point of 7.28.
ZE was known far less than VDE, so it was also essential to study ZE. The use of the mutants may help to further illustrate the Z-dependent quenching mechanism.