www.elsevier.com/locate/ibmb
Phylogenetic analysis of Phlebotomus species belonging to the
subgenus Larroussius (Diptera, Psychodidae) by ITS2 rDNA
sequences
Trentina Di Muccio
a, Marino Marinucci
a, Liliana Frusteri
a, Michele Maroli
a, Bernard
Pesson
b, Marina Gramiccia
a,*aLaboratorio di Parassitologia, Istituto Superiore di Sanita`, Viale Regina Elena 299, 00161 Rome, Italy bLaboratoire de Parasitologie, Faculte´ de Pharmacie, Universite´ Louis Pasteur, Strasbourg I, France
Received 5 August 1999; received in revised form 28 December 1999; accepted 4 January 2000
Abstract
In the genealogy of Phlebotomus (Diptera: Psychodidae), morphological analyses have indicated that the subgenus Larroussius is a monophyletic group which is most closely related to the subgenera Transphlebotomus and Adlerius. We conducted a phylogen-etic analysis of the relationships among six representative species of the subgenus Larroussius and one species representatitive of the Phlebotomus subgenus, assessing sequences of the Second Internal Transcribed Spacer (ITS2) of the ribosomal RNA (rRNA). Three of the species (P. perniciosus, P. ariasi and P. perfiliewi perfiliewi) were collected in different parts of the Mediterranean area. The trees estimated from parsimony and neighbour-joining analyses supported the monophyly of the Larroussius subgenus inferred from the morphological analysis. According to our data, P. ariasi may be a sister group to the rest of the Larroussius subgenus, although additional sequence data are needed to confirm this observation. Our results suggest that P. perniciosus and P.
longicuspis are distinct species, in spite of the fact that there are only slight morphological differences. The strict congruence
between the phylogeny of the Larroussius subgenus inferred from the ITS2 sequences and that based on morphological studies further confirmed the ability of the spacer sequence to identify recently-derived affiliations. 2000 Elsevier Science Ltd. All rights reserved.
Keywords: Phlebotomus; Larroussius; Leishmania infantum; rDNA; ITS2; Phylogeny
1. Introduction
Phlebotomine sandflies (Diptera: Psychodidae, Phlebotominae) include well-known vectors of patho-gens and parasites responsible for human diseases and animal leishmaniases in many regions of the world (listed in Killick-Kendrick, 1990). Those phlebotomines that represent vectors belong to two genera:
Phle-botomus (Old World) and Lutzomyia (New World). The
most widely accepted classification of the Phlebotomi-nae is that of Lewis et al. (1977), which is based on morphological adult characters and on traditional sys-tematic opinions (i.e., regardless of phylogenetic
affili-* Corresponding author. Tel.: +39-06-49903015; fax: + 39-06-49387065.
E-mail address: [email protected] (M. Gramiccia).
0965-1748/00/$ - see front matter2000 Elsevier Science Ltd. All rights reserved. PII: S 0 9 6 5 - 1 7 4 8 ( 0 0 ) 0 0 0 1 2 - 6
ations among taxa). The over-importance given to sev-eral characters of adults in terms of their discriminatory value has led to the creation of a few large and possibly polyphyletic genera, namely, Lutzomyia, Brumptomyia and Warileya (nearctic genera), and Phlebotomus and
Sergentomyia (palearctic genera). To develop a more
According to Seccombe et al. (1993), the genus
Phle-botomus, which includes all of the proven vectors of
human leishmaniases in the Old World (Killick-Kend-rick, 1990), presents few taxonomic problems at the level of genus and appears to be monophyletic on mor-phological grounds. However, a recent study has pro-vided evidence that the genus Phlebotomus is not mono-phyletic, though the study was based on molecular data (Depaquit et al., 1998). Furthermore, Rispail and Le´ger (1998a) have recently revised the definition of morpho-logical character states.
The Phlebotomus subgenus Larroussius, which includes all of the proven vectors of Leishmania
infantum (Killick-Kendrick, 1990), is readily defined by
the development in females of a long digitiform exten-sion of the spermathecal “neck” (Rispail, 1990). The tax-onomy of Larroussius seems to be satisfactory and, despite the existence of a few synonymisations, the status of the taxa appears neither controversial nor con-fused. The subgenus Larroussius is closely related to the subgenera Transphlebotomus and Adlerius, and the pro-liferation of species within Larroussius and Adlerius is probably recent (Rispail and Le´ger, 1991; Rispail and Le´ger, 1998b). Nonetheless, relatively little is known about the evolution and phylogenetic relationships of the
Larroussius vectors of Leishmania infantum; Esseghir et
al. (1997) evaluated the phylogenetic affiliations of P.
perniciosus, P. tobbi, P. perfiliewi perfiliewi and P. ari-asi, yet this study was conducted on mitochondrial
haplotypes of Phlebotomus vectors of Leishmania major. In the present study, we analysed evolutionary affili-ations inferred from ribosomal DNA (rDNA) Second Internal Transcribed Spacer (ITS2) sequences of the
Lar-roussius vectors of Leishmania infantum. In recent years,
the study of rDNA ITS2 sequences has provided a foun-dation for developing diagnostic assays to identify mem-bers of taxa of medically important complexes (Collins and Paskewitz, 1996) and has provided useful infor-mation on recently-derived phylogenies in insects, parti-cularly mosquitoes (Severini et al., 1996; Miller et al., 1996; Marinucci et al., 1999).
The phylogenetic analysis in our study indicated that all of the morphologically defined taxa are valid species and that the subgenus Larroussius is a phylogenetically coherent unity, as suggested by prior allozyme studies (Pesson et al., 1991) and numerical analyses of morpho-logical characters (Rispail and Le´ger, 1991).
2. Materials and methods
2.1. Insects
The nomenclature of Seccombe et al. (1993) was used for the taxonomic designation of the species studied. A total of seven Phlebotomus species were studied. The
following six species were from wild material:
Phle-botomus (Larroussius) ariasi Tonnoir, 1921; P. (La.) neglectus Tonnoir, 1921; P. (La.) perfiliewi perfiliewi
Parrot, 1930; P. (La.) tobbi Adler and Theodor, 1930;
P. (La.) perniciosus Newstead, 1911 and P. (La.) long-icuspis Nitzulescu, 1930. The remaining species, P.
(Phlebotomus) papatasi Scopoli, 1786, was from col-lected material. The psychodid sandfly Lutzomyia
shan-noni Dyar, 1929, was used as reference; the rDNA ITS2
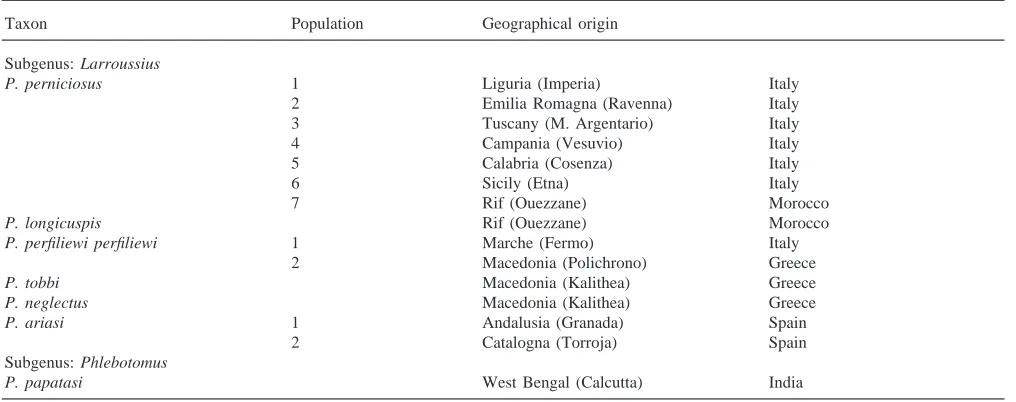
of this species was previously published by Miller et al. (1997). The species and populations used in this study and their geographical origin are provided in Table 1e.
2.2. DNA extraction, polymerase chain reaction and sequencing
Genomic DNA was extracted from single adult males of each species or population. The rDNA ITS2 region was amplified by polymerase chain reaction (PCR) using primers based on conserved sequences of the 5.8S and 28S coding regions (Collins and Paskewitz, 1996). PCRs and cloning of the PCR products followed procedures published in detail elsewhere (Collins and Paskewitz, 1996; Severini et al., 1996).
Clones were sequenced in both forward and reverse direction by the dideoxy method using the T7 Sequen-cing Kit and [α35S]dATP (Amersham Pharmacia
Biotech Italy, Milan). Compression zones were resolved with Deaza G/A T7 Sequencing mixes (Pharmacia Biotech). The sequences have been deposited in the EMBL under the accession numbers: AF205523-AF205527, AF207711 and AF205529.
The homologous sequence of L. shannoni (accession number U48382) was published previously by Miller et al. (1997).
To assess any intra-individual spacer variability, 3–5 clones from a single specimen of each species or popu-lation were sequenced. To evaluate intra-sample spacer variation, cloned DNA from 3–5 individuals of each species or population sample was sequenced.
2.3. Phylogenetic analysis
Table 1
Phlebotomus species and their geographical origin
Taxon Population Geographical origin
Subgenus: Larroussius
P. perniciosus 1 Liguria (Imperia) Italy
2 Emilia Romagna (Ravenna) Italy
3 Tuscany (M. Argentario) Italy
4 Campania (Vesuvio) Italy
5 Calabria (Cosenza) Italy
6 Sicily (Etna) Italy
7 Rif (Ouezzane) Morocco
P. longicuspis Rif (Ouezzane) Morocco
P. perfiliewi perfiliewi 1 Marche (Fermo) Italy
2 Macedonia (Polichrono) Greece
P. tobbi Macedonia (Kalithea) Greece
P. neglectus Macedonia (Kalithea) Greece
P. ariasi 1 Andalusia (Granada) Spain
2 Catalogna (Torroja) Spain
Subgenus: Phlebotomus
P. papatasi West Bengal (Calcutta) India
unweighted. The sequences were also analysed using neighbour-joining (NJ) (Saitou and Nei, 1987) with Kimura’s two-parameter correction (Kimura, 1980), as implemented in the TREECON for Windows v. 1.3b (Van de Peer and De Wachter, 1994); gaps were excluded from the analysis. Bootstrapping (Felsenstein, 1985) was performed using both parsimony (1000 replicates) and NJ (1000 replicates). The trees were rooted using the ITS2 sequence of L. shannoni as the outgroup.
3. Results
The sequenced fragments of rDNA from the seven phlebotomine species ranged from 388 base pairs (bp) in P. perniciosus to 430 bp in P. ariasi. All the phleboto-mine taxa exaphleboto-mined possessed a split 5.8S rRNA, con-sisting of a mature 5.8S rRNA and a 2S rRNA separated by a short transcribed spacer. These findings strongly support Shimada’s hypothesis (1992) that the lack of a transcribed spacer in the 5.8S rRNA (i.e., the presence of a continuous 5.8S rRNA) is a synapomorphy defining the Culicidae in the Nematocera genealogy (Miller et al., 1997). The presumptive boundaries of the split 5.8S and 28S genes were deduced from comparisons of align-ments with the sequence of L. shannoni (Miller et al., 1997), whereby the initial 105 (P. tobbi) to 116 (P.
neglectus) bases were putatively split 5.8S rDNA and
the last 39 bases were putative 28S rDNA. The 5.8S, 2S and 28S coding regions were well conserved and nearly identical to those reported for L. shannoni. Higher pro-pensity for variation (mostly indels) occurred in the short intervening sequence separating the 2S region from the rest of the coding region (data not shown).
By contrast, the ITS2 fragment appeared as a mosaic of variable and conserved regions. The presence of con-served blocks of nucleotides is probably due to the spacer function in ribosomal RNA maturation. No intra-individual spacer variability was detected, and only neg-ligible (,0.25%) levels of intra-species spacer variation were found. The length of the sequenced ITS2 fragments ranged from 241 bp in P. longicuspis to 291 bp in P.
ariasi. The overall base composition was as follows: A:
39.0% (35.7–41.5%), T: 37.7% (34.4–40.2%), C: 10.5% (8.2–12.1%) and G: 12.8% (11.0–13.9%). A bias toward A and T (76.7% of total) was consistent with the base composition of ITS2 sequences of L. shannoni (80%) and other Nematocerous taxa (Miller et al., 1997).
The alignment of the ITS2 sequences resulted in a total of 313 characters, including gaps (Fig. 1). Of the 313 characters, 204 (65.2%) were variable and 143 (45.7%) parsimony informative.
Spacer pairwise sequence differences among species of the subgenus Larroussius (here and in the following estimates, all insertions/deletions were removed from alignments) ranged from 4.5% between the two closely related taxa P. perniciosus and P. longicuspis to 34.3% between P. ariasi and P. tobbi, which was similar to the 33.6% difference between P. ariasi and P. perniciosus. The intra-species spacer variation within the Larroussius taxa is currently being studied: preliminary ITS2-based data in P. ariasi and P. perfiliewi perfiliewi were not conclusive due to inadequate geographic representa-tiveness. More consistent data were obtained in P.
per-niciosus populations (the same populations in Table 1):
Fig. 1. Sequence alignment of the ITS2 regions of rDNA of the Phlebotomus taxa and Lutzomyia shannoni. The ITS2 sequence of Lutzomyia
shannoni is from Miller et al. (1997). Nucleotide sequences identical to P. perniciosus are indicated by dots. Insertions and deletions are denoted
by a hyphen (-). Taxa are abbreviated as in Table 2.
phylogenetic analyses. Pairwise nucleotide differences and Kimura’s two-parameter distances are provided in Table 2.
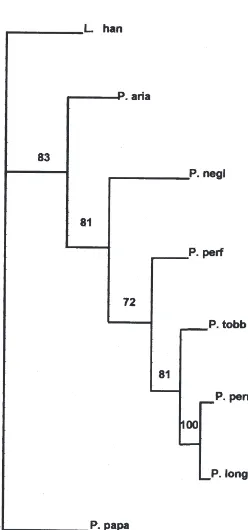
Parsimony analysis identified a single tree of 357 steps (Fig. 2). All of the relationships inferred in the parsi-mony-tree were supported in at least 72% of the boots-trap replications. A tree based on the distance analysis is shown in Fig. 3, with the levels of significance derived from bootstrapping analysis roughly corresponding to those given by the parsimony analysis. Both distance and parsimony trees showed identical nested topologies.
4. Discussion
Table 2
Kimura’s two-parameter distances (×100) for ITS2 rDNA sequences (above diagonal) and pairwise nucleotide differences (below diagonal) among the taxa included in this study. Taxa are abbreviated as the first letter of the genus and first four letters of the species name (e.g., Lutzomyia
shannoni=L. shan)
1 2 3 4 5 6 7 8
1.P.pern 4.78 19.77 77.61 51.87 28.44 39.89 79.36
2.P.long 18.31 70.37 52.96 29.67 38.21 87.58
3.P.tobb 76.49 53.40 30.20 41.57 75.84
4.P.papa 75.36 101.68 103.46 86.60
5.P.aria 52.23 45.18 72.08
6.P.perf 44.67 70.57
7.P.negl 81.49
8.L.shan
11 39 99 80 50 66 96
37 95 82 52 65 100
102 85 52 69 89
114 119 127 100
83 79 93
78 94
101
Fig. 2. Phylogenetic tree based on rDNA ITS2 sequence data and generated by maximum parsimony method. The tree was rooted with
Lutzomyia shannoni. Tree length: 357 steps, Consistency Index (C.I.)
of 0.810, Homoplasy Index (H.I.) of 0.190, C.I. excluding uniformative characters of 0.760, H.I. excluding uninformative characters of 0.240, Retention Index of 0.699 and Rescaled Consistency Index (R.C.) of 0.566. The tree is the 50% majority rule consensus of 1000 bootstrap replicates with the branch-and-bound search option. The numbers above the branches are bootstrap values given in percentages. Taxa are abbreviated as in Table 2.
Fig. 3. Phylogenetic tree based on rDNA ITS2 sequence data. The tree was constructed using neighbour-joining with Kimura’s two-para-meter distances (scale bar) and rooted with Lutzomyia shannoni. The numbers above the branches are bootstrap percentages (1000 replications) for clades supported above the 50% level. Taxa are abbreviated as in Table 2.
In our analysis, the ITS2 sequence from the subgenus
Phlebotomus (represented here by P. papatasi) was
placed as the sister group to a clade including all of the ITS2 sequences from the Larroussius taxa examined. Thus, even with the few available data, the monophyly of the Larroussius subgenus was confirmed, in concord-ance with the morphologically and molecularly based phylogenies of Rispail and Le´ger (1991, 1998b) and Esseghir et al. (1997) and Esseghir et al. (1997).
Based on morphological evidence, P. longicuspis, P.
perniciosus and P. tobbi are closely related species. In
a somewhat greater distinction was observed comparing
P. longicuspis and P. perniciosus to P. tobbi (i.e., 0.15
and 0.16 substitutions/site, respectively). In a large majority of tree reconstructions, P. tobbi was inferred as the sister group to the [P. longicuspis, P. perniciosus] clade. The inferred arrangement of these taxa was con-sistent with the species phylogeny based on morphologi-cal and biochemimorphologi-cal evidence and geographimorphologi-cal distri-bution. The western and southern palearctic species P.
longicuspis and P. perniciosus are very similar in
mor-phological characters, and Collantes and Martı´nez-Ort-ega (1997) proposed their synonymy on the basis of a biometric study. However, in Morocco, analysis of iso-enzyme patterns has revealed distinct intermediate mor-photypes for each of the two species (Benadbennbi et al., 1999). These data imply that the two species must have diverged only recently, yet their corresponding ITS2 are clearly distinct in terms of sequence and for the presence of indels that are not shared between them (Fig. 1). P. tobbi occupies the eastern Mediterranean area (from Croatia to Israel, Iran and Azerbaydzhan). Between the P. longicuspis-P. perniciosus and P. tobbi lineages, the percentage of variable base positions aver-ages 16%; this likely represents the level of differen-tiation of ITS2 sequences, which began to differentiate after the evolutionary split from a common ancestor of eastern and western populations, subsequently diverging into separate species. In our tree reconstructions, the P.
perfiliewi ITS2 lineage constitutes the sister group of a
clade that includes the [P. longicuspis, P. perniciosus] clade and the P. tobbi lineage. These genealogical prox-imities are similar to the phylogeny inferred from mito-chondrial data by Esseghir et al. (1997) and seem to be consistent with morphological evidence. Within P.
per-filiewi, three different subspecies have been described,
namely, P. p. perfiliewi, P. p. transcaucasicus and P. p.
galilaeus. Phylogenetic inferences in our study were
based exclusively on sequence data from populations from the distribution area of P. p. perfiliewi. Whereas Esseghir et al. (1997) distinguished two mitochondrial lineages in this subspecies, we did not observe signifi-cant differences in ITS2 sequences when comparing eastern populations (Macedonia, Greece) with western populations (Marche, Italy).
Both trees (Figs. 2 and 3) placed P. ariasi as the sister group to the assemblage consisting of the remaining
Lar-roussius taxa and they show a more distant relationship
to P. neglectus than that suggested by morphological evidence. Additional data will be needed to confirm our observation and to more precisely determine the phylo-genetic position of P. neglectus.
Recently, Depaquit et al. (1998) used parsimony analysis of sequences from 28S D2 rDNA to re-investi-gate affiliations among major phlebotomine lineages. The results were, in part, found to be in contrast with traditional phlebotomine phylogeny based on
mor-phology. In particular, the subgenus Larroussius
(represented by P. perniciosus and P. neglectus) was placed as the sister group to the [Lutzomyia,
Sergento-myia] clade, whereas the Phlebotomus subgenera Phle-botomus and ParaphlePhle-botomus were placed together in
a separate clade. As pointed out by these authors, defini-tively rejecting the hypothesis of a monophyletic
Phle-botomus would require additional data.
In summary, the ITS2 molecular data produced informative phylogeny within the Larroussius subgenus. Furthermore, except for the P. neglectus sequence, the ITS2 phylogeny obtained by maximum parsimony and distance analyses was concordant with the species phy-logeny estimated using morphological criteria. The phylogenetic inferences in this study are based on sequence data from a singular gene in the rDNA array and may be distorted by unknown selective constraints. To produce a more representative tree for these medi-cally important sandflies, it will be necessary to analyse additional Larroussius taxa (e.g., the African species P.
longipes and P. pedifer) and data from mitochondrial
and/or nuclear genes with morphological comparisons of all life stages.
Acknowledgements
The authors are greatly indebted to M. Gallego, versity of Barcellona, Spain; F. Morillas Marquez, Uni-versity of Granada, Spain; T. Naucke, UniUni-versity of Bonn, Germany; and M. Cadi-Soussi, University of Rabat, Morocco, for their kind collaboration in col-lecting sandfly specimens. This work received financial support from “Grant 1% Ministero della Sanita`” and from Regione Campania, Italy.
References
Artemiev, M.M., 1991. A classification of the subfamily Phlebotomi-nae. In: Maroli, M. (Ed.), Proceedings of the First International Symposium on Phlebotomine Sandflies. Parassitologia 33 (Suppl. 1), 69–77.
Ashford, R.W., 1991. A new morphological character to distinguish
Sergentomyia and Phlebotomus. Parassitologia 33 (Suppl. 1), 79–
83.
Benadbennbi, I., Pesson, B., Cadi-Soussi, M., Morillas Marquez, F., 1999. Morphological and isoenzymatic differentiation of sympatric populations of Phlebotomus perniciosus and P. longicuspis (Diptera: Psichodidae) in northern Morocco. J. Med. Entomol. 36, 116–120.
Collantes, F., Martı´nez-Ortega, E., 1997. Sobre la validez taxono´mica de Phlebotomus longicuspis (Nitzulescu, 1931) (Diptera, Psychodidae). Boletı´n de la Asociacio´n Espan˜ola de Entomo´logos 21, 141–146.
Collins, F.H., Paskewitz, S.M., 1996. A review of the use of ribosomal DNA (rDNA) to differentiate among cryptic Anopheles species. Insect Molec. Biol. 5, 1–9.
H., Kaltenbach, M., Le´ger, N., 1998. Syste´matique mole´culaire des Phlebotominae, e´tude pilote. Paraphylie du genre Plebotomus. C. R. Acad. Sci. III 321, 849–855.
Esseghir, S., Ready, P.D., Killick-Kendrick, R., Ben-Ismail, R., 1997. Mitochondrial haplotypes and phylogeography of Phlebotomus vectors of Leishmania major. Insect Molec. Biol. 6, 211–225. Felsenstein, J., 1985. Confidence limits on phylogenies, an approach
using the bootstrap. Evolution 39, 783–791.
Killick-Kendrick, R., 1990. Phlebotomine vectors of the leishmaniases: a review. Med. Vet. Entomol. 4, 1–24.
Kimura, M., 1980. A simple method for estimating evolutionary rates of base substitution through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120.
Kumar, S., Tamura, K., Nei, M., 1993. MEGA, Molecular Evolution-ary Genetics Analysis. Version 1.01. Pennsylvania State Univer-sity, University Park.
Lewis, D.J., 1982. A taxonomic review of the genus Phlebotomus (Diptera, Psychodidae). Bull. Br. Mus. (Nat. Hist.) (Entomol.) 37, 217–243.
Lewis, D.J., Young, D.G., Minter, D.M., 1977. Proposals for a stable classification of the phlebotomine sandflies (Diptera, Psychodidae). Syst. Entomol. 2, 319–332.
Marinucci, M., Romi, R., Mancini, P., Di Luca, M., Severini, C., 1999. Phylogenetic relationships of seven palearctic members of the
mac-ulipennis complex (Diptera, Culicidae) inferred from ITS2
sequence analysis. Insect Molec. Biol. 8, 469–480.
Miller, B.R., Crabtree, M.B., Savage, H.M., 1996. Phylogeny of four-teen Culex mosquito species, including the Culex pipiens complex, inferred from the internal transcribed spacers of ribosomal DNA. Insect Molec. Biol. 5, 93–107.
Miller, B.R., Crabtree, M.B., Savage, H.M., 1997. Phylogenetic relationships of the Culicomorpha inferred from 18S and 5.8S ribosomal DNA sequences (Diptera: Nematocera). Insect Molec. Biol. 6, 105–114.
Pesson, B., Vallon, M., Floer, M.T., Kristensen, A.R., 1991. Etude isoenzymatique de populations me´diterrane´ennes de Phle´botomes du sous-genre Larroussius. In: Maroli, M. (Ed.), Proceedings of the First International Symposium on Phlebotomine Sandflies. Par-assitologia 33 (Suppl. 1), 471–476.
Rispail, P., 1990. Approche phe´ne´tique et cladistique du genre
Phle-botomus Rondani and Berte´, 1840 (Diptera: Psychodidae). Apport
des caracte`res morphologiques imaginaux. The`se de Doctorat, Universite´ des Sciences et Techniques du Languedoc, Montpell-ier, France.
Rispail, P., Le´ger, N., 1991. Application of numerical taxonomic methods on Phlebotominae. In: Maroli, M. (Ed.), Proceedings of the First International Symposium on Phlebotomine Sandflies. Par-assitologia 33 (Suppl. 1), 485–492.
Rispail, P., Le´ger, N., 1998a. Numerical taxonomy of Old World Phle-botominae (Diptera, Psychodidae). 1. Considerations of morpho-logical characters in the genus Phlebotomus Rondani and Berte´ 1840. Mem. Inst. Oswaldo Cruz 93, 773–785.
Rispail, P., Le´ger, N., 1998b. Numerical taxonomy of Old World Phle-botomine (Diptera, Psychodidae). 2. Restatement of classification upon subgeneric morphological characters. Mem. Inst Oswaldo Cruz 93, 787–793.
Saitou, N., Nei, M., 1987. The neighbour-joining method, a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425.
Seccombe, A.K., Ready, P.D., Huddleston, L.M., 1993. A catalogue of the Old World Phlebotomine sandflies (Diptera, Psychodidae, Phlebotominae). Occasional Papers on Systematic Entomology 8, 1–57.
Severini, C., Silvestrini, F., Mancini, P., La Rosa, G., Marinucci, M., 1996. Sequence and secondary structure of the rDNA second internal transcribed spacer in the sibling species Culex pipiens L. and Cx. quinquefasciatus Say (Diptera: Culicidae). Insect Molec. Biol. 5, 181–186.
Shimada, T., 1992. Distribution of split 5.8S ribosomal DNA in Dip-tera. Insect Molec. Biol. 1, 45–49.
Swofford, D.L., 1993. PAUP, Phylogenetic Analysis Using Parsimony, version 3.1.1. Distributed by the Illinois Natural History Survey, Champaign, IL.
Thompson, J.D., Higgins, D.G., Gibson, T.J., 1994. CLUSTAL W, improving the sensitivity of progressive multiple sequence align-ment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680. Van de Peer, Y., De Wachter, R., 1994. TREECON for Windows: a