SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S.Farm.)
Program Studi Ilmu Farmasi
Oleh:
Andreas Suseno Wimbo Hapsoro NIM : 078114057
FAKULTAS FARMASI
UNIVERSITAS SANATA DHARMA YOGYAKARTA
i SKRIPSI
Diajukan untuk Memenuhi Salah Satu Syarat Memperoleh Gelar Sarjana Farmasi (S.Farm.)
Program Studi Ilmu Farmasi
Oleh:
Andreas Suseno Wimbo Hapsoro NIM : 078114057
FAKULTAS FARMASI
UNIVERSITAS SANATA DHARMA YOGYAKARTA
iv
vii
KROMATOGRAFI LAPIS TIPIS (KLT)-DENSITOMETRI PADA PENETAPAN KADAR KURKUMIN DALAM SEDIAAN KAPSUL LUNAK OBAT HERBAL TERSTANDAR (OHT) RHEUMAKUR®” ini dapat diselesaikan oleh penulis. Skripsi ini disusun untuk memenuhi salah satu syarat memperoleh gelar Sarjana Farmasi (S. Farm) di Fakultas Farmasi Universitas Sanata Dharma, Yogyakarta.
Selama penyusunan skripsi ini, banyak pihak yang telah membantu penulis dalam menyelesaikannya, maka pada kesempatan ini penulis mengucapkan terima kasih kepada:
1. Ipang Djunarko, S.Si, Apt, M.Sc selaku Dekan Fakultas Farmasi Universitas Sanata Dharma Yogyakarta.
2. Christine Patramurti, M.Si., Apt. selaku Dosen Pembimbing yang telah banyak meluangkan waktunya dalam memberikan masukan, kritik, solusi, semangat baik selama penelitian, penyusunan skripsi maupun saat perkuliahan.
3. Yohanes Dwiatmaka, M.Si. selaku Dosen Pembimbing Akademik dan Dosen Penguji yang telah banyak memberikan masukan dan semangat baik selama penyusunan skripsi maupun dalam perkuliahan.
viii
skripsi, serta telah memberikan senyawa baku kurkumin kepada penulis sehingga sangat berguna dalam penelitian
6. Dr.C.J. Soegihardjo, Apt. atas saran dan diskusi yang telah diberikan dalam penyusunan skripsi.
7. Segenap dosen dan karyawan atas ilmu dan pengalaman yang berharga sehingga berguna dalam proses penyusunan skripsi.
8. Theresia Weliana Kosasih dan Lilis Dumaria Pasaribu selaku teman seperjuangan selama penelitian dan penyusunan skripsi.
9. Eliz, Yunita, Venny, Katiti, Lala, Toro, Katarina, Benny, dan Dian selaku teman yang melakukan penelitian dalam satu tema yang sama, terima kasih atas dukungannya.
10. Seluruh staf laboratorium kimia: Bimo, Kunto, Parlan, dan Wagiran yang telah membantu penulis selama penelitian di laboratorium.
11. Cinthya, Yosafat, Edhi, Siska, Dika, Bella, Vivi, Robby, Tika, dan Puput selaku teman yang sering bekerja kelompok bersama penulis, terima kasih atas kerjasama, pengalaman, dan semangat yang luar biasa selama ini.
12. Kak Dewi, Grace, Zi, dan Bayu, atas laporan dan saran yang sangat berguna bagi perkuliahan maupun penyusunan skripsi.
ix
memiliki kekurangan. Oleh karena itu, penulis mengharapkan kritik dan saran untuk perbaikan dan perkembangan selanjutnya. Semoga skripsi ini dapat bermanfaat demi perkembangan ilmu pengetahuan.
x
HALAMAN JUDUL... i
HALAMAN PERSETUJUAN PEMBIMBING ... ii
HALAMAN PENGESAHAN... iii
HALAMAN PERSEMBAHAN ... iv
PERNYATAAN KEASLIAN KARYA ...v
LEMBAR PERNYATAAN PERSETUJUAN PUBLIKASI KARYA ILMIAH.. vi
PRAKATA... vii
DAFTAR ISI...x
DAFTAR TABEL... xiv
DAFTAR GAMBAR ...xv
DAFTAR LAMPIRAN... xvii
INTISARI... xviii
ABSTRACT... xix
BAB I PENGANTAR ...1
A. Latar Belakang ...1
1. Permumusan masalah...3
2. Keaslian penelitian ...3
3. Manfaat penelitian...4
B. Tujuan Penelitian ...5
BAB II PENELAAHAN PUSTAKA...6
xi
E. Kromatografi Lapis Tipis...16
1. Kromatografi lapis tipis (KLT) secara umum...16
2. Jenis fase diam dan fase gerak KLT ...16
3. Migrasi dan retensi solut ...17
4. Pemisahan kromatografi lapis tipis ...17
5. Proses sorpsi-adsorpsi ...18
6. Profil puncak dan pelebaran puncak ...21
7. Puncak asimetri ...23
8. Resolusi kromatogram ...24
9. Analisis kualitatif ...25
10. Analisis kuantitatif ...26
11. Aplikasi penotolan sampel ...27
F. Densitometri...28
G. Validasi Metode Analisis ...32
1. Parameter validasi metode ...32
2. Kategori metode analisis ...38
H. Landasan Teori...39
I. Hipotesis...40
BAB III METODE PENELITIAN...41
xii
2. Definisi operasional ...42
C. Bahan...42
D. Alat...43
E. Cara Penelitian ...43
1. Pembuatan metanol pH 4 ...43
2. Pengaktifan silika gel G 60 ...43
3. Pembuatan dan penjenuhan fase gerak ...43
4. Penetapan panjang gelombang serapan maksimum...44
5. Penentuan kurva baku kurkumin...44
6. Penetapan perolehan kembali (recovery) dan koefisien variasi (KV) baku kurkumin ...45
7. Penentuan selektivitas kurkumin dalam sampel serta akurasi dan presisi baku yang ditambahkan ke dalam sampel ...46
F. Analisis Hasil ...47
1. Selektivitas ...47
2. Linearitas dan rentang ...48
3. Akurasi baku kurkumin...48
4. Presisi baku kurkumin...49
5. Akurasi baku kurkumin yang ditambahkan ke dalam sampel ...49
6. Presisi baku kurkumin yang ditambahkan ke dalam sampel ...49
xiii
Sampel...59
D. Pembuatan Kurva Baku...63
E. Validasi Metode Kromatografi Lapis Tipis-Densitometri ...65
1. Selektivitas ...65
2. Linearitas dan rentang ...67
3. Akurasi ...68
4. Presisi ...69
F. Penentuan Akurasi dan Presisi Baku Kurkumin dalam Sampel ...69
BAB V KESIMPULAN DAN SARAN...72
A. Kesimpulan ...72
B. Saran...72
DAFTAR PUSTAKA ...73
LAMPIRAN...77
xiv
Tabel I. Daftar fase gerak ...16
Tabel II. Parameter-parameter aplikasi penotolan yang direkomendasikan....27
Tabel III. Rentang kesalahan perolehan kembali yang diperbolehkan ...35
Tabel IV. Kriteria yang diperbolehkan untuk presisi pada kadar analit yang berbeda ...36
Tabel V. Kategori Analisis Kimia ...39
Tabel VI. Data pengukuran panjang gelombang serapan maksimum kurkumin...54
Tabel VII. Data nilai Rfbaku dan sampel ...60
Tabel VIII. Data kurva baku kurkumin ...63
Tabel IX. Resolusi puncak kurkumin dengan puncak senyawa 2...67
Tabel X. Hasil penentuan perolehan kembali (recovery) kurkumin baku ...68
Tabel XI. Hasil penentuan KV kurkumin baku ...69
Tabel XII. Perolehan kembali dan KV baku kurkumin dalam sampel...70
Tabel XIII. PerbandinganAUCkurkumin dalam sampel dan kurkumin dalam sampel yang ditambah baku ...70
xv
Gambar 2. Seyawa Kurkumin, Demetoksi Kurkumin, dan Bis-demetoksi
kurkumin...10
Gambar 3. Isomer geometrikcis-transdari kurkumin ...10
Gambar 4. Tautomerisasi bentuk keto-enol senyawa kurkuminoid ...12
Gambar 5. Strukutur kimia kurkumin dalam berbagai pH ...13
Gambar 6. Produk degradasi kurkumin pada pH alkali...14
Gambar 7. Struktur trans-6-(4-hydroxy-3- methoxyphenyl )-2,4-dioxo -5-hexenal...15
Gambar 8. Produk fotodegradasi kurkumin...15
Gambar 9. Cara penentuan harga Rf...18
Gambar 10. Interaksi antara analit dengan fase diam silika gel ...19
Gambar 11. Keadaan simetris dan pelebaran puncak kromatogram ...22
Gambar 12. Ilustrasi 3 prinsip utama yang menggambarkan puncak; (a). Pengaruh lintasan ganda (multiple-path effect); (b). Pengaruh difusi longitudinal; (c). Pengaruh transfer massa ...22
Gambar 13. Isoterm sorpsi serta profil-profil puncak yang dihasilkan. (a). Isoterm linier (b). Puncaktailingdan (c). Puncakfronting...23
Gambar 14. Ilustrasi resolusi pada KLT: (a) kromatogram; (b) profil kromatografi masing-masing bercak ...24
xvi
Gambar 18. Kromatogram panjang gelombang maksimum kurkumin ...54
Gambar 19. Kromatogram baku kurkumin 260 ppm...56
Gambar 20. Gugus polar dan gugus non polar pada kurkumin ...56
Gambar 21. Interaksi hidrogen antara kurkumin dengan fase diam ...57
Gambar 22. Interaksi kurkumin dengan fase gerak ...57
Gambar 23. Interaksi antara asam asetat glasial dengan silika gel...58
Gambar 24. Perbandingan kromatogram sampel (A) dan kromatogram baku kurkumin (B)...60
Gambar 25. Ilustrasi keadaan kesetimbangan pada pemisahan senyawa A (kurkumin) dengan senyawa lainnya ...61
Gambar 26. Hubungan antara konsentrasi kurkumin dengan AUC/50 ( replikasi I)...65
xvii
Lampiran 2. Hasil uji pH stabilitas kurkumin...79
Lampiran 3. Hasil kromatogram pengukuran panjang gelombang serapan maksimum ...81
Lampiran 4. Sistem KLT-Densitometri yang digunakan...84
Lampiran 5. Kromatogram kurva baku kurkumin ...86
Lampiran 6. Penimbangan baku dan contoh perhitungan kadar baku ...88
Lampiran 7. Kromatogram hasil validasi metode...90
Lampiran 8. Data penimbangan dan contoh perhitunganrecovery...93
Lampiran 9. Contoh perhitungan SD danCV...95
Lampiran 10. Kromatogram penentuan akurasi dan presisi baku dalam sampel..96
Lampiran 11. Data perhitungan nilai resolusi antara puncak kurkumin dengan puncak senyawa 2 dan contoh perhitungannya ...100
xviii
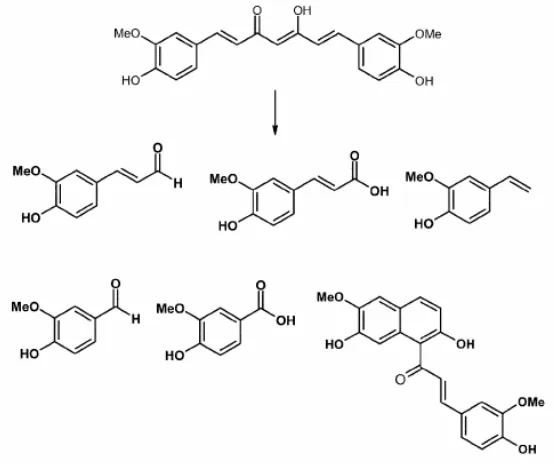
Kandungan kurkumin dalam ekstrak kunyit dimanfaatkan dalam produksi sediaan kapsul lunak Obat Herbal Terstandar (OHT). Oleh karena itu, stabilitas kadar kurkumin pada saat distribusi dan penyimpanan sediaan OHT penting untuk diketahui.
Metode penetapan kadar kurkumin dalam sediaan kapsul lunak OHT dilakukan secara KLT-Densitometri. Sistem KLT-Densitometri yang digunakan adalah fase normal dengan fase gerak kloroform : asam asetat glasial : heksana (85 : 10 :5) v/v dan fase diam silika gel G 60 yang diukur pada 425 nm.
Parameter validasi metode yang ditentukan adalah selektivitas, linearitas, akurasi, presisi, dan rentang. Hasil penelitian validasi baku menunjukkan metode KLT-Densitometri memiliki selektivitas yang baik dengan nilai resolusi pada pemisahan sampel adalah 2,2399. Metode ini memiliki linearitas yang baik pada konsentrasi kurkumin 100 – 500 ppm (r = 0,9996) dan rentang 260 - 500 ppm. Perolehan kembali dan KV untuk kadar rendah, sedang dan tinggi berturut-turut adalah 104,07%, 2,72%; 99,28%, 0,34%; dan 100,11%, 0,79%. Hasil validasi baku dalam matriks sampel memiliki perolehan kembali dan KV berturut-turut yaitu 102,07% dan 3,64%.
xix
Medicine (SBHM) soft capsule. Therefore, the stability of curcumin content during distribution and storage of SBHM dosage form important to know.
Curcumin content determination method performed by TLC-Densitometry. TLC-Densitometry system used is a normal phase with mobile phase chloroform: glacial acetic acid: hexane (85: 10: 5) v/v and stationary phase silica gel G 60 that was measured at 425 nm.
Specified validation parameters are selectivity, linearity, accuracy, precision, and range. The results show the validation of raw-Densitometry TLC method has good selectivity with the resolution of the separation of the sample is 2.2399. This method has good linearity of concentrations of curcumin from 100 to 500 ppm (r = 0.9996) and range about 260 – 500 ppm. The recoveries and CV for low, medium and high concentration respectively is 104.07%, 2.72%, 99.28%, 0.34%, and 100.11%, 0.79%. The validation raw material in the sample matrix had the recovery and CV, respectively, are 102.07% and 3.63%.
1
A. Latar Belakang
Kurkumin merupakan senyawa polifenol berwarna kuning oranye yang berkhasiat sebagai analgesik-antiinflamasi. Kurkumin yang terkandung dalam ekstrak simplisia rimpang kunyit (Curcumae domesticae Rhizoma) ditemukan bersama kedua turunannya yaitu demetoksi kurkumin dan bis-demetoksi kurkumin. Beberapa perusahaan telah mempergunakan kunyit sebagai bahan dasar pembuatan Obat Herbal Terstandar (OHT) (Usia, 2010).
Dalam dua dasawarsa terakhir penggunaan obat tradisional mengalami perkembangan yang sangat pesat (Usia, 2010). Minat masyarakat terhadap sediaan obat tradisional meningkat karena sediaan obat tradisonal diyakini sebagai obat alam yang memiliki akses hingga seluruh lapisan masyarakat, selain itu harganya terjangkau bagi seluruh lapisan masyarakat. Oleh karena itu, Badan Pengawas Obat dan Makanan melakukan peningkatan mutu obat tradisional melalui standarisasi bahan baku dan uji praklinik menjadi produk OHT (Kementerian Kesehatan Republik Indonesia, 1994).
Senyawa aktif yang diteliti pada penelitian ini adalah kurkumin. Kurkumin memiliki stabilitas yang rendah terhadap pH dan paparan cahaya (Stankovic, 2004). Hal ini mempengaruhi mutu sediaan karena kurkumin menjadi tidak stabil dalam proses distribusi dan penyimpanan sehingga dimungkinkan kadar kurkumin menjadi berkurang. Kadar kurkumin yang berkurang akan berpengaruh terhadap besarnya efek antiinflamasi yang ditimbulkan. Mutu sediaan OHT akan terjamin apabila jumlah dan kadar senyawa aktif pada setiap sediaan yang didistribusikan seragam dan sesuai dengan yang tertera pada kemasan. Untuk itu diperlukan penetapan kadar kurkumin dalam kapsul lunak OHT merk Rheumakur® pada nomor batch yang sama namun berbeda apotek untuk mengetahui stabilitas kurkumin dalam penyimpanan.
Penelitian ini merupakan serangkaian proses yaitu optimasi, validasi metode dan penetapan kadar kurkumin. Metode yang dilakukan pada penelitian analisis kurkumin ini adalah KLT-densitometri. Sistem KLT yang digunakan merupakan fase normal dengan fase diam silika gel G 60 dan fase gerak kloroform : asam asetat glasial : heksana (85 : 10 : 5) yang merupakan hasil optimasi. Metode yang digunakan adalah KLT-densitometri karena memiliki kelebihan yaitu dapat digunakan untuk analisis kualitatif dan kuantitatif secara simultan.
ditentukan melalui perhitungan perolehan kembali (98-102%), presisi baku kurkumin ditentukan melalui perhitungan koefisien variasi (<2%), selektivitas ditentukan melalui nilai resolusi (>1,5) dan kesamaan faktor retardasi sampel dan baku, serta linearitas dengan nilai r >0,999. Sedangkan akurasi dan presisi baku kurkumin dengan kadar 0,01% dalam matriks sampel yang baik secara berturut-turut ditunjukkan melalui nilai perolehan kembali 90-107% dan KV < 5,3% (Harmita, 2004; United States Pharmacopeial Convention, 1995; Yuwono dan Indrayatno, 2005).
1. Perumusan masalah
Berdasarkan latar belakang, maka diperoleh permasalahan sebagai berikut: Apakah metode KLT-Densitometri yang digunakan pada penetapan kadar kurkumin dalam sediaan kapsul lunak OHT “Rheumakur®” memenuhi parameter validasi meliputi selektivitas, linearitas, rentang , akurasi, dan presisi?
2. Keaslian penelitian
Sejauh pengetahuan penulis, penelitian validasi penetapan kadar kurkumin secara KLT-densitometri yang sudah pernah dilakukan, antara lain: Penentuan kadar kurkumin secara kromatografi lapis tipis-densitometri (Martono, 1996) dalam serbuk rhizoma Curcuma longa L. dengan fase gerak kloroform : etanol : air suling (25 : 0,96 : 0,04), Simultaneous determination of curcuminoids in curcuma samples using high performance thin layer chromatography (Gupta, Gupta, Kumar, 1999) dengan fase gerak kloroform : metanol (95 : 5),
Quantitative Analysis of Curcumin, Demethoxycurcumin and
Thailand by TLC-Densitometry(Pothitirat, Gritsanapan, 2005) danOccurrence of curcuminoids in curcuma longa: a quality standardization by HPTLC
(Paramasivam, Aktar, Poi, Banerjee, Bandyopadhyay, 2008) dengan fase gerak kloroform : metanol (48 : 2).
Penelitian yang dilakukan penulis memiliki perbedaan terhadap penelitian yang tercantum di atas. Penulis lebih menekankan pada penjaminan mutu obat tradisional khususnya stabilitas kadar kurkumin dalam sediaan kapsul Obat Herbal Terstandar, bukan dalam simplisia maupun ekstrak. Parameter lain yang membedakan dengan penelitian sebelumnya adalam sistem fase gerak yang digunakan penulis yaitu kloroform : asam asetat glasial : heksana (85 : 10 : 5) v/v. 3. Manfaat penelitian
a. Manfaat metodologis. Penelitian ini diharapkan mampu memberikan metode baru karena digunakan sistem fase gerak yang belum dilakukan pada penelitian sebelumnya. Selain itu, penelitian ini juga diharapkan dapat memberikan pemahaman mengenai parameter validasi antara lain selektivitas, linearitas, rentang, akurasi, dan presisi pada validasi metode penetapan kadar kurkumin secara Kromatografi Lapis Tipis-Densitometri
B. Tujuan Penelitian
6
A. Kapsul Lunak Obat Herbal Terstandar
Kapsul adalah sediaan padat yang terdiri dari obat dalam cangkang keras atau lunak yang dapat larut. Kapsul cangkang lunak umumnya terbuat dari gelatin. Cangkang gelatin lunak umumnya mengandung 6% hingga 13% air. Umumnya kapsul cangkang lunak diisi dengan cairan. Khususnya bahan aktif dilarutkan atau disuspensikan dalam bahan pembawa cair. Digunakan pula bahan pembawa minyak seperti minyak nabati. Keunggulan kapsul cangkang lunak adalah keseragaman kandungan dan disolusi obatnya lebih baik daripada kapsul berisi serbuk kering. Namun, kontak antara cangkang lunak atau keras dengan isi zat cair lebih besar dibandingkan dengan kapsul berisi serbuk kering (Direktorat Jenderal Pengawasan Obat dan Makanan RI, 1995).
ekstrak dan bahan lain bergantung pada dosis ekstrak yang hendak diadministrasikan (Naguib, 2000).
Gambar 1. Logo Obat Herbal Terstandar (OHT)
Berdasarkan PERMENKES No.246/MENKES/PER/V/1990 obat tradisional adalah bahan atau ramuan bahan yang berupa bahan tumbuhan, bahan hewan, bahan mineral, sediaan galenik atau campuran dan bahan-bahan tersebut, yang secara tradisional telah digunakan untuk pengobatan berdasarkan pengalaman. Sedangkan kapsul lunak Obat Herbal Terstandar adalah sediaan kapsul lunak obat bahan alam yang telah dibuktikan keamanan dan khasiatnya secara ilmiah dengan uji praklinik dan bahan bakunya telah distandarisasi (Kementerian Kesehatan Republik Indonesia, 1990).
B. Standarisasi Ekstrak
fraksi-fraksi, isolat senyawa tunggal ataupun tetap sebagai campuran dengan ekstrak lain (Badan Pengawas Obat dan Makanan Republik Indonesia, 2005).
Adapun jika sebagai produk jadi berarti ekstrak yang berada dalam sediaan obat jadi siap digunakan. Ekstrak tersebut bisa dalam bentuk ekstrak kering, ekstrak kental dan ekstrak cair yang proses pembuatannya disesuaikan dengan bahan aktif yang dikandung serta maksud penggunaannya, apakah akan dibuat menjadi sediaan dalam bentuk kapsul, tablet, cairan obat dalam, pil, dan lainnya (Badan Pengawas Obat dan Makanan Republik Indonesia, 2005).
Terpenuhinya standar mutu produk atau bahan ekstrak tidak terlepas dari pengendalian proses, artinya bahwa proses yang terstandar dapat menjamin produk yang terstandar tanpa penerapan pengujian atau pemeriksaan. Untuk menjaga kualitas bahan baku obat alam perlu dilakukan usaha budidaya dan standarisasi terhadap bahan baku tersebut, baik yang berupa simplisia maupun yang berbentuk ekstrak atau sediaan galenik (Badan Pengawas Obat dan Makanan Republik Indonesia, 2005).
Persyaratan mutu ekstrak terdiri dari berbagai parameter standar umum dan parameter standar spesifik. Pengertian standardisasi juga berarti proses menjamin bahwa produk akhir mempunyai nilai parameter tertentu yang konstan dan ditetapkan terlebih dahulu (Badan Pengawas Obat dan Makanan Republik Indonesia, 2005).
senyawa-senyawa tersebut terhadap pemanasan , udara, cahaya, logam berat dan derajat keasaman (Badan Pengawas Obat dan Makanan Republik Indonesia, 2005).
C. Ekstrak Rimpang Kunyit
Curcumae domesticae Rhizoma (rimpang kunyit) adalah rimpang
Curcuma domesticaVal. Pemerian yang dimiliki yaitu berbau khas aromatik, rasa agak pahit, agak pedas, dan lama kelamaan menimbulkan rasa tebal. Dosis lazim rimpang Kunyit yang diperbolehkan adalah 8 mg/kg (Direktorat Jenderal Pengawasan Obat dan Makanan RI, 1977).
Ekstrak kental rimpang kunyit adalah ekstrak yang dibuat dari rimpang tumbuhan Curcuma domestica Val., mengandung minyak atsiri ≥ 3,2% dan kurkuminoid ≥ 33,9% (Badan Pengawas Obat dan Makanan, 2004). Kandungan dari ekstrak rimpang Kunyit antara lain kurkuminoid, minyak atsiri, lemak 1 -3 %, karbohidrat 3%, protein 30%, pati 8%, vitamin C 45-55%, dan garam-garam mineral (Rukmana, 1994). Dalam kurkuminoid, kandungan kurkumin sebesar 77 %, demetoksi kurkumin 17 % dan bis-demetoksi kurkumin 3% (Anggarwal, 1995).
D. Kurkumin
Kurkumin adalah senyawa aktif polifenol dengan rumus kimia C21H20O6. Kurkumin memiliki nilai log P = 2,56 dan merupakan zat warna kuning utama (0,5 –6%) yang terdapat dalam rhizoma Curcuma longa L atau
demetoksi kurkumin (log P = 2,69) dan bis-demetoksi kurkumin (log P = 2,81) dikenal dengan nama kurkuminoid (Martono, 1996). Kurkumin memiliki panjang gelombang serapan maksimum pada 430 nm dalam pelarut metanol dan 425 nm dalam pelarut etanol (Anggarwal, 1995). Kurkumin larut dalam alkohol dan asam asetat glasial, tetapi tidak dapat larut dalam air dan eter (Windholz, 1981). Struktur kurkumin ditampilkan sebagai berikut :
OH
R1 R2
OH
O O
R1= -OCH3dan R2= -OCH3(Kurkumin) R1= -H dan R2= -OCH3(Demetoksi kurkumin) R1= -H dan R2= -H (Bis-demetoksi kurkumin)
Gambar 2. Seyawa Kurkumin, Demetoksi Kurkumin, danbis-demetoksi kurkumin (Anggarwal, 1995)
Selain konstituen mayor (kurkumin, demetoksi kurkumin, dan bis
demetoksi kurkumin), konstituen minor juga dapat diisolasi yang memiliki bentuk isomer geometrikal dari senyawa 1-3 (Gambar 2). Satu dari antaranya berbentuk isomer geometrik cis-trans (Gambar 3) memiliki titik lebur, stabilitas dalam larutan dan cahaya yang lebih rendah (Stankovic, 2004).
Kurkumin memiliki aktivitas sebagai antiinflamasi pada penyakit osteoartritis. Osteoartritis adalah penyakit reumatik dengan prevalensi yang terus meningkat sesuai dengan pertambahan usia. Secara klinis osteoartritis ditandai oleh nyeri, deformitas, pembesaran sendi, hambatan gerak; disamping itu juga terjadi inflamasi tingkat ringan sampai berat (Kalim, 1996; Rahardjo, 1994).
Ekstrak rimpang kunyit dengan kadar kurkuminoid 3,66 ± 0,65 % b/b dan 25 mL minyak atsiri rimpang temulawak yang mengandung kamfora, kamfen, kurkumen, bergamoten, germakren B, kurserenon, germakron dan xanthorrhizol mampu menurunkan angka leukosit di dalam cairan sinovial penderita osteoartritis. Sehingga produksi sitokin yang menyebabkan inflamasi menjadi berkurang (Kertia, Sudarsono, Imono, Mufrod, Catur, Rahardjo, dkk, 2005).
Kurkuminoid mempunyai aktivitas anti inflamasi yang menarik meliputi penghambatan lipid peroksidase (anti oksidan) serta penghambatan aktivitas enzim siklooksigenase dan lipooksigenase (Timmerman, 1995; Kohli, Ali, Ansari, dan Raheman, 2005). Kurkumin mempunyai aktivitas yang paling kuat (IC 50 =
0,05 цM) dibandingkan senyawa turunannya (Agnam dkk, 1995). Selain itu,
jumlah kurkumin yang aman dikonsumsi oleh manusia adalah 100 mg/hari (Commandeur dan Vermeulen, 1996). Oleh sebab itu, ekstrak rimpang kunyit dimanfaatkan sebagai bahan dalam pembuatan obat tradisional (Anggarwal, 1995).
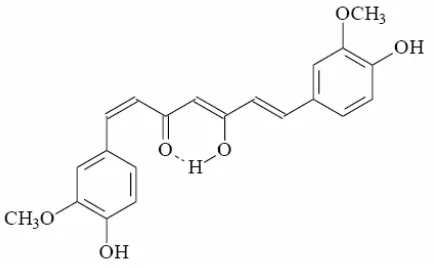
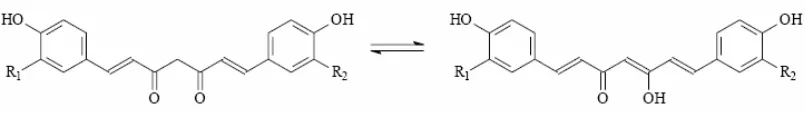
mengalami reaksi hidrolisis dan degradasi yang disebabkan oleh adanya gugus metilen aktif pada senyawa tersebut. Reaksi tersebut sangat dipengaruhi oleh pH lingkungannya (Tonnesen dan Karlsen, 1985a).
Kurkumin di dalam larutan mengalami tautomerisasi menjadi bentuk keto-enol bergantung pada pelarut yang digunakan. Sembilan puluh lima persen berada dalam bentuk enol (Stankovic, 2004).
Gambar 4. Tautomerisasi bentuk keto-enol senyawa kurkuminoid (Stankovic, 2004)
OH
Gambar 5. Strukutur kimia kurkumin dalam berbagai pH (Stankovic, 2004)
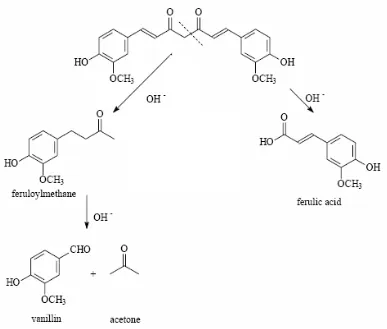
adalah vanilin dan aseton serta jumlahnya meningkat seiring bertambahnya waktu (Stankovic, 2004).
Gambar 6. Produk degradasi kurkumin pada pH alkali (Stankovic, 2004)
O
O O
O HO
Gambar 7. Strukturtrans-6-(4-hydroxy-3-methoxyphenyl)-2,4-dioxo-5-hexenal(Stankovic, 2004)
Instabilitas kurkumin juga dipengaruhi oleh adanya cahaya yang menyebabkan terjadinya degradasi fotokimia senyawa tersebut (Van der Goot, 1997). Prinsipnya, kurkumin tidak stabil terhadap cahaya, terutama dalam bentuk larutan. Setelah mengalami foto-iradiasi, akan terdeteksi produk siklisasi, sama halnya dengan produk dekomposisi asam vanilat, vanilin, dan asam ferulat (Sasaki, Sat, Abe, Sugimoto, dan Maitani, 1998).
E. Kromatografi Lapis Tipis 1. Kromatografi lapis tipis (KLT) secara umum
Kromatografi lapis tipis (KLT) merupakan metode pemisahan komponen-komponen berdasarkan perbedaan adsorpsi atau partisi oleh fase diam di bawah gerakan pelarut pengembang atau pelarut pengembang campur (Mulja dan Suharman, 1995). KLT dapat dipakai untuk dua tujuan, yaitu sebagai metode untuk mencapai hasil kualitatif, kuantitatif, dan preparatif. Tujuan kedua yaitu dipakai untuk menjajaki sistem pelarut dan sistem penyangga (Gritter, Bobit, dan Schwarting, 1991).
2. Jenis fase diam dan fase gerak KLT
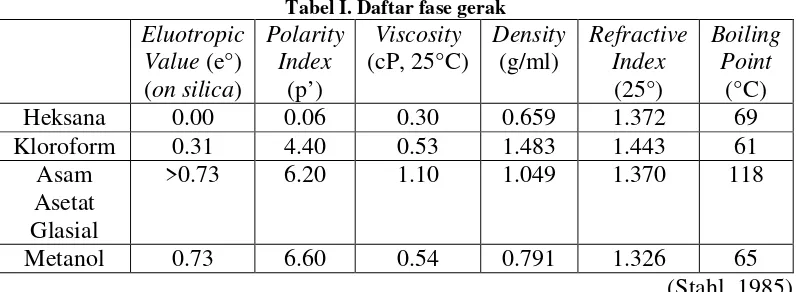
Fase diam yang banyak terpilih adalah silika gel. Silika gel G adalah silika gel yang dicampur perekat CaSO4 lebih kurang 13% (Jork, Funk, Fischer, dan Wimmer, 1990). Fase gerak dalam mediun KLT merupakan medium angkut dan terdiri atas satu atau beberapa bahan pelarut yang bergerak di dalam fase diam, karena ada gaya kapiler (Stahl, 1985). Berikut ini merupakan daftar fase gerak beserta indeks polaritasnya:
Tabel I. Daftar fase gerak Eluotropic
Heksana 0.00 0.06 0.30 0.659 1.372 69
Kloroform 0.31 4.40 0.53 1.483 1.443 61
Asam Asetat Glasial
>0.73 6.20 1.10 1.049 1.370 118
Metanol 0.73 6.60 0.54 0.791 1.326 65
3. Migrasi dan retensi solut
Kecepatan migrasi solut melalui fase diam ditentukan oleh perbandingan distribusinya (D), dan besarnya D ditentukan oleh afinitas relatif solut pada pada kedua fase (fase diam dan fase gerak). Dalam konteks kromatografi, nilai D didefinisikan sebagai perbandingan konsentrasi solut dalam fase diam (Cs) dan dalam fase gerak (Cm) (Gandjar dan Rohman, 2007).
(1) Jadi semakin besar nilai D maka migrasi solut semakin lambat; dan semakin kecil nilai D maka migrasi solut semakin cepat. Solut akan terelusi menurut perbandingan distribusinya. Jika perbedaan perbandingan distribusi solut cukup besar maka campuran-campuran solut akan mudah dan cepat dipisahkan (Gandjar dan Rohman, 2007).
4. Pemisahan kromatografi lapis tipis
Pemisahan kromatografi planar ini pada umumnya dihentikan sebelum semua fase gerak melewati seluruh permukaan fase diam. Solut pada kedua kromatografi ini dikarakterisasi dengan jarak migrasi solut terhadap jarak ujung fase geraknya. Faktor retardasi solut (Rf) didefinisikan sebagai:
Gambaran untuk menghitung Rfterdapat dalam gambar berikut ini:
Gambar 9. Cara penentuan harga Rf(Gandjar dan Rohman, 2007)
Nilai Rfdihitung dengan menggunakan perbandingan sebagaimana dalam persamaan berikut:
(3) (Gandjar dan Rohman, 2007) Nilai maksimum Rf adalah 1 dan ini dicapai ketika solut mempunyai perbandingan distribusi (D) dan faktor retensi (k’) sama dengan 0 yang berarti solut bermigrasi dengan kecepatan yang sama dengan fase gerak. Nilai minimum Rf adalah 0 dan ini teramati jika solut tertahan pada posisi titik awal di permukaan fase diam (Gandjar dan Rohman, 2007).
5. Proses sorpsi – adsorpsi
terus menerus selama pemisahan kromatografi karenanya sistem kromatografi berada dalam keadaan kesetimbangan dinamis. Solut akan terdistribusi diantara dua fase yang bersesuaian dengan perbandingan distribusinya (D) untuk menjaga keadaan kesetimbangan ini (Gandjar dan Rohman, 2007).
Adsorpsi merupakan penyerapan pada permukaannya saja. Adsorpsi pada permukaan melibatkan interaksi-interaksi elektrostatik seperti ikatan hidrogen, penarikan dipol-dipol, dan penarikan yang diinduksi oleh dipol. Solut akan bersaing dengan fase gerak untuk berikatan dengan sisi-sisi polar pada permukaan adsorben (Skoog, Holler, dan Nieman, 1998).
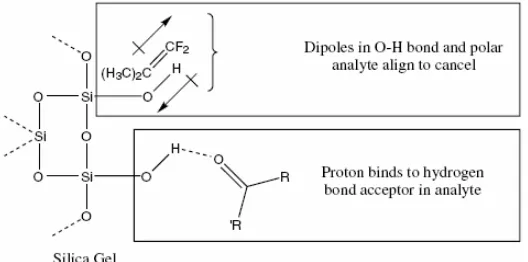
Silika gel memiliki permukaan yang terdiri atas gugus Si-O-Si dan gugus silanol (Si-OH). Gugus silanol bersifat sedikit asam dan polar karenanya gugus ini mampu membentuk ikatan hidrogen dengan solut-solut yang agak polar sampai sangat polar (Gandjar dan Rohman, 2007).
Gambar 10. Interaksi antara analit dengan fase diam silika gel (Skoog, dkk, 1998)
Semakin polar solut maka semakin tertahan kuat ke dalam adsorben silika gel ini. Solut-solut non polar tidak mempunyai afinitas atau mempunyai sedikit afinitas terhadap adsorben polar, sementara solut-solut yang terpolarisasi memiliki afinitas yang kecil terhadap adsorben polar disebabkan adanya interaksi dipol atau interaksi-interaksi yang diinduksi oleh dipol. Solut-solut polar, terutama yang mampu membentuk ikatan hidrogen, akan terikat kuat pada adsorben karenanya butuh fase gerak yang cukup polar untuk mengelusinya. Berikut adalah urutan polaritas solut-solut organik: alkana < alkena < aromatis < eter < ester < keton dan aldehid < tiol < amin dan amida < alkohol < fenol < asam-asam organik (Gandjar dan Rohman, 2007).
Adsorbsi solut oleh fase diam atau oleh adsorben sangat tergantung pada: (a) struktur kimia solut atau adanya gugus aktif tertentu yang berinteraksi dengan adsorben; (b) ukuran partikel adsorben. Semakin kecil ukuran partikel adsorben, maka luas permukaannya semakin luas sehingga interaksinya dengan solut juga semakin luas; (c) kelarutan solut dalam fase gerak. Solut yang semakin mudah larut dalam fase gerak akan semakin mudah lepas dari fase diam (Gandjar dan Rohman, 2007).
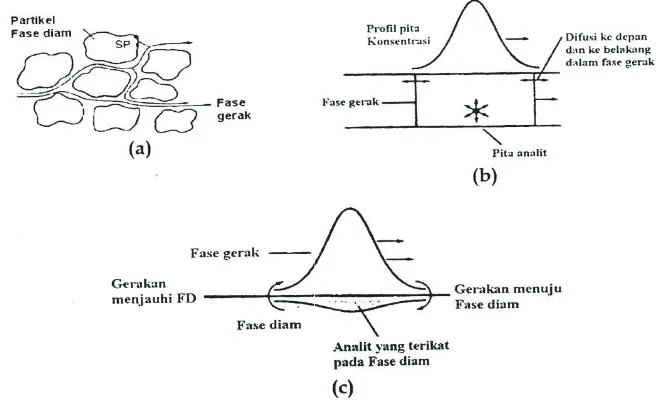
6. Profil puncak dan pelebaran puncak
Selama pemisahan kromatografi, solut individual akan membentuk profil konsentrasi yang simetri atau dikenal juga dengan profil Gaussian dalam arah aliran fase gerak. Profil, dikenal juga dengan puncak atau pita, secara perlahan-lahan akan melebar dan sering juga membentuk profil yang asimetrik karena solut-solut melanjutkan migrasinya ke fase diam. Prinsip yang mendasari alasan-alasan bentuk puncak dan pelebaran puncak dapat diringkas sebagai berikut:
a. Sorpsi dan desorpsi solut yang terus menerus antara fase diam dan fase gerak, secara inheren akan menghasilkan profil konsentrasi Gaussian yang akan melebar karena solut bermigrasi lebih lanjut.
b. Perjalanan solut melalui partikel fase diam sedikit berbeda, sehingga menyebabkan profil konsentrasinya melebar secara simetris. Keadaan seperti ini disebut dengan pengaruh lintasan ganda (multiple-path effect).
c. Spesies solut menyebar ke segala arah dengan difusi ketika berada di dalam fase gerak. Difusi terjadi dengan arah yang sama dan berlawanan dengan aliran fase gerak (longitudinal or axial diffusion) karenanya akan berkontribusi terjadinya pelebaran pita secara simetris.
gerak yang akan mengakibatkan adanya pelebaran puncak lebih lanjut. Desorpsi yang lambat dapat juga menghasilkan puncak yang asimetris atau condong.
e. Adanya variasi rasio distribusi solut dengan total konsentrasinya juga berperan terjadinya puncak yang asimetris atau condong (Gandjar dan Rohman, 2007).
Gambar 11. Keadaan simetris dan pelebaran puncak kromatogram (Gandjar dan Rohman, 2007)
Gambar 12. Ilustrasi 3 prinsip utama yang menggambarkan puncak; (a). Pengaruh lintasan ganda (multiple-path effect); (b). Pengaruh difusi longitudinal; (c). Pengaruh transfer massa
7. Puncak asimetri
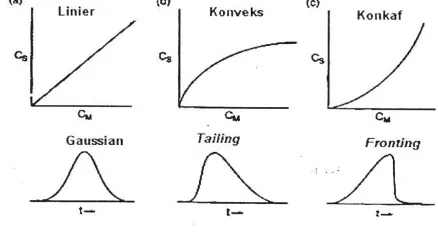
Profil konsentrasi solut yang bermigrasi akan simetris jika rasio distribusi solut (D) konstan selama kisaran konsentrasi keseluruhan puncak, sebagaimana ditunjukkan oleh isoterm sorpsi yang linier yang merupakan plot konsentrasi solut dalam fase diam (Cs) terhadap konsentrasi solut dalam fase gerak (Cm). Meskipun demikian, kurva isoterm akan berubah menjadi 2 jenis puncak asimetris yakni membentuk puncak yang berekor (tailing) dan adanya puncak pendahulu (fronting) jika ada perubahan rasio distribusi solut kearah yang lebih besar (Gandjar dan Rohman, 2007).
.
Gambar 13. Isoterm sorpsi serta profil-profil puncak yang dihasilkan. (a). Isoterm linier (b). Puncaktailingdan (c). Puncakfronting(Gandjar dan Rohman, 2007)
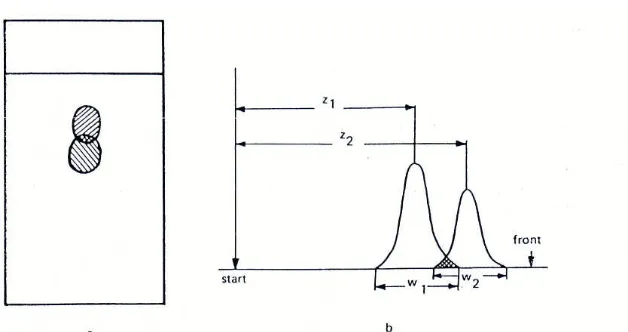
8. Resolusi kromatogram
Resolusi didefinisikan sebagai perbedaan antara waktu retensi 2 puncak yang saling berdekatan (Δz = z2– z1) dibagi dengan rata-rata lebar puncak (W1+ W2)/2 sebagaimana dalam ilustrasi dan persamaan berikut ini:
Gambar 14. Ilustrasi resolusi pada KLT: (a) kromatogram; (b) profil kromatografi masing-masing bercak (Sherma dan Fried, 1996)
(4) (Sherma dan Fried, 1996) Dari persamaan ini dapat diketahui bahwa yang sangat berpengaruh terhadap pemisahan suatu komponen adalah: jarak masing-masing bercak solut terhadap titik awal pengembanagn (z2 dan z1) serta lebar puncak masing-masing komponen yang dipisahkan ((W1dan W2) (Sherma dan Fried, 1996).
Gambar 15. Pengukuran resolusi 2 puncak yang berdekatan (Gandjar dan Rohman, 2007)
9. Analisis kualitatif
Ada 3 pendekatan untuk analisis kualitatif yaitu:
a. Perbandingan antara data retensi solut yang tidak diketahui dengan data retensi baku yang sesuai (senyawa yang diketahui) pada kondisi yang sama. Untuk kromatografi lapis tipis, faktor retardasi (Rf) baku dan senyawa yang tidak diketahui dibandingkan dengan cara kromatografi secara bersama-sama untuk menghilangkan adanya variasi kondisi bahan yang digunakan dan laboratorium.
c. Menggabungkan alat kromatografi dengan spektrometer massa. Cara ini memberikan informasi data spektra solut dengan waktu retensi tertentu. Spektra solut yang tidak diketahui dapat dibandingkan dengan spektra yang ada di base komputer atau diinterpretasi sendiri. Cara ini dapat dilakukan untuk solut yang belum ada baku murninya (Gandjar dan Rohman, 2007). 10. Analisis kuantitatif
Untuk menjamin kondisi yang digunakan dalam analisis kuantitatif stabil dan reprodusibel, baik pada penyiapan sampel atau proses kromatografi, berikut beberapa syarat yang harus dipenuhi dalam analisis kuantitatif: Analit (solut) harus telah diketahui dan terpisah sempurna dari komponen-komponen lain dalam kromatogram. Baku dengan kemurnian yang tinggi dan telah diketahui harus tersedia. Prosedur kalibrasi yang sudah diketahui harus digunakan. Penyerap atau adsorben yang digunakan murni, begitu pula pelarutnya (Skoog, dkk, 1998). Pengukuran respon dapat dilakukan dengan mengukur tinggi puncak atau dengan menghitung luas puncak (Gandjar dan Rohman, 2007).
disiapkan, selanjutnya diinjeksikan dan dianalisis dengan cara yang sama. Konsentrasi senyawa tersebut ditentukan dengan metode grafik dari plot kalibrasi atau secara numerik (Gandjar dan Rohman, 2007).
Larutan baku disiapkan dengan konsentrasi tertentu yang sudah diketahui. Sejumlah tertentu volume larutan ini diinjeksikan dan dianalisis, lalu respon detektor (luas puncak) diplotkan terhadap konsentrasi (Gandjar dan Rohman, 2007).
11. Aplikasi penotolan sampel
Pemisahan pada kromatografi lapis tipis yang optimal akan diperoleh hanya jika menotolkan sampel dengan ukuran bercak sekecil dan sesempit mungkin. Sebagaimana dalam prosedur kromatografi yang lain, jika sampel yang digunakan terlalu banyak maka akan menurunkan resolusi. Penotolan sampel yang tidak tepat akan menyebabkan bercak yang menyebar dan puncak ganda. Berdasarkan pada tujuan analisis, berbagai macam jumlah sampel telah disarankan untuk digunakan dan diringkas pada tabel II.
Tabel II. Parameter-parameter aplikasi penotolan yang direkomendasikan
Tujuan Diameter bercak Identifikasi 3 mm untuk
volume sampel 1μl 0,1-1 1-20 Uji
kemurnian
4 mm untuk
volume sampel 2μl 5 100
F. Densitometri
Densitometri merupakan salah satu dari metode analisa KLT kuantitatif. Penetapan kadar suatu senyawa dengan metode ini dilakukan dengan mengukur kerapatan bercak senyawa yang dipisahkan dengan cara KLT. Syarat-syarat untuk senyawa standar adalah murni, inert, dan stabil (Sastrohamidjojo, 1985).
Metode densitometri mempunyai cara kerja yang sederhana dan cepat. Pada metode densitometri diperlukan adsorben dan fase gerak yang murni. Untuk memperoleh hasil yang baik lazimnya digunakan adsorben siap pakai yang telah mengalami pencucian (Gritter dkk, 1991).
Alat densitometri mempunyai sumber sinar yang bergerak di atas bercak pemisahan pada lempeng kromatografi yang akan ditetapkan kadar komponennya. Lazimnya lempeng itu digerakkan menyusuri berkas sinar yang berasal dari sumber sinar tersebut(Sudjadi, 1988).
Gambar 16. Pemantulan Sinar
(Mintarsih, 1990) Sifat pemantulan ini akan menjadi sensitif dan selektif bila sinar yang datang adalah monokromatis. Disini biasanya dipilih sinar pada panjang gelombang yang diserap atau dipantulkan paling banyak oleh noda yang diteliti. Banyaknya sinar yang direfleksikan akan ditangkap oleh suatu alat yang disebut
reflection photomultiplier yang akan diteruskan ke pencatat atau rekorder untuk diubah menjadi suatu puncak atau kromatogram. Luas puncak atau tinggi puncak sesuai dengan konsentrasi senyawa pada noda yang diukur kerapatannya (Mintarsih, 1990).
Pada beberapa TLC scanner sudah dilengkapi alat pemproses data atau mikro komputer, sehingga integrasi luas puncak atau tinggi puncak tersebut dapat langsung direkam atau dicatat sebagai data sekaligus dengan kromatogramnya dan dapat pula direkam dan dicatat langsung sebagai kadarnya, melalui teknik pemprograman tertentu. Noda yang kecil dan intensif akan menghasilkan suatu puncak yang sempit dan tajam, sebaliknya noda yang lebar dan kurang intensif akan menghasilkan puncak yang lebar maupun tumpul (Mintarsih, 1990).
dapat dilakukan satu per satu, atau apabila satu pelat bercak yang diperoleh segaris semua maka dapat dilakukan penelusuran untuk semua bercak sekaligus. Sedangkan cara penelusuran vertikal, hanya dapat dilakukan satu per satu (Mintarsih, 1990).
Pada penelusuran bercak horisontal dengan penelusuran beberapa bercak sekaligus hanya dapat dilakukan apabila bercak-bercak tersebut benar-benar dalam satu baris. Cara ini akan mengalami kesulitan jika bercak yang sangat dekat dengan bercak yang ditetapkan, karena ada kemungkinan bercak yang tidak diinginkan ikut pula ditetapkan (Mintarsih, 1990).
Berdasarkan atas jalannya sinar, penelusuran dapat dilakukan dengan dua cara yaitu penelusuran lurus dan penelusuran zig-zag (naik turun). Pada penelusuran lurus, sinar yang mengenai bercak berjalan lurus dari kiri ke kanan, sedangkan pada penelusuranzig-zag, sinar yang mengenai bercak berjalanzig-zag
dari kiri ke kanan. Besarnya jarak, naik turunnya sinar dapat diatur menurut kebutuhan, yang diperhitungkan dengan besar kecilnya bercak, yang dalam operasi alat dikenal sebagai lebar penelusuran (scan widht) (Mintarsih, 1990).
Penelusuran bercak akan mendapatkan hasil yang baik apabila dilakukan pada panjang gelombang maksimum, karena perubahan konsentrasi pada bercak sedikit saja sudah terdeteksi. Pengukuran dilakukan dengan menelusuri bercak yang akan ditetapkan kadarnya pada kisaran panjang gelombang zat tersebut (Mintarsih, 1990).
kompak sehingga akan mempengaruhi hasil penelusuran dengan densitometri, yaitu berupa puncak yang lebar dan kasar. Puncak yang lebar disebabkan kurang kompaknya fase diam, sedangkan puncak yang kasar disebabkan permukaan pelat yang kurang rata (Mintarsih, 1990).
Pada umumnya tebal lapisan tipis pada lempeng yang digunakan adalah 0,20-0,25 mm maksimum 0,33 mm, untuk mengurangi efek hamburan sinar yang disebabkan oleh fase diam terhadap linearitas hubungan serapan dan konsentrasi dari senyawa yang diteliti. Hubungan antara serapan terhadap konsentrasi dilinearkan dengan dasar teori Kubelka-Munk, menggunakan kurva kerja linear yang telah diprogramkan oleh suatu mikro komputer. Kurva serapan konsentrasi tersebut ditentukan oleh harga parameter hamburan yang disebabkan oleh fase diam. Harga parameter hamburan tersebut tergantung ukuran dan distribusi partikel fase diam pada lempeng KLT ( Supardjan, 1987).
Ada dua cara penetapan kadar dengan alat densitometer. Pertama, setiap kali penetapan ditotolkan sediaan baku dari senyawa yang bersangkutan dan dielusi bersama dalam satu lempeng, kemudianAUC(luas daerah di bawah kurva) sampel dibandingkan dengan hargaAUCzat baku. Yang kedua, dengan membuat kurva baku hubungan antara jumlah zat baku denganAUC. Kurva baku diperoleh dengan membuat totolan zat baku pada pelat KLT dengan bermacam-macam konsentrasi (minimal tiga macam konsentrasi). Bercak yang diperoleh dicari
G. Validasi Metode Analisis
Validasi metode analisis merupakan serangkaian prosedur yang digunakan untuk membuktikan apakah suatu metode analisis yang digunakan tersebut sesuai yang diharapkan dengan akurasi dan presisi yang memadai(United States Pharmacopeial Convention, 1995).
1. Parameter validasi metode
a. Akurasi. Akurasi dapat diartikan sebagai kedekatan hasil analisis yang diperoleh menggunakan metode analisis tertentu dengan nilai yang sebenarnya. Hal tersebut diperoleh dengan cara membandingkan kadar terukur dari sejumah tertentu senyawa standar yang sengaja ditambahkan ke dalam sampel dengan jumlah tertentu pula. Harga perbandingan tersebut disebut perolehan kembali (United States Pharmacopeial Convention, 1995).
Nilai perolehan kembali untuk bahan obat dengan kadar kecil biasanya disepakati 90-110%, untuk kadar obat yang lebih besar 95-105%, akurasi untuk bahan baku disepakati 98-102%, sedang untuk bioanalisis rentang akurasi 80-120% masih bisa diterima (Mulja dan Suharman, 1995).
Kecermatan ditentukan dengan dua cara yaitu metode simulasi (
spiked-placebo recovery) atau metode penambahan baku (standard addition method).
kembali dapat ditentukan dengan cara membuat sampel plasebo (eksepien obat, cairan biologis) kemudian ditambah analit dengan konsentrasi tertentu (biasanya 80% sampai 120% dari kadar analit yang diperkirakan), kemudian dianalisis dengan metode yang akan divalidasi. Tetapi bila tidak memungkinkan membuat sampel plasebo karena matriksnya tidak diketahui seperti obat-obatan paten, atau karena analitnya berupa suatu senyawa endogen misalnya metabolit sekunder pada kultur kalus, maka dapat dipakai metode adisi (Harmita, 2004).
Metode adisi dapat dilakukan dengan menambahkan sejumlah analit dengan konsentrasi tertentu pada sampel yang diperiksa, lalu dianalisis dengan metode tersebut. Persen perolehan kembali ditentukan dengan menentukan berapa persen analit yang ditambahkan tadi dapat ditemukan. Kriteria kecermatan sangat tergantung kepada konsentrasi analit dalam matriks sampel dan pada keseksamaan metode (RSD). Selisih kadar pada berbagai penentuan (Xd) harus 5% atau kurang pada setiap konsentrasi analit pada mana prosedur dilakukan. Harga rata-rata selisih secara statistik harus 1,5% atau kurang. Kriteria tersebut dinyatakan secara matematik sebagai berikut:
Kadar analit dan perolehan kembali dalam metode penambahan baku dapat dihitung sebagai berikut:
(6) (Harmita, 2004)
Pada metode penambahan baku, pengukuran blanko tidak diperlukan lagi. Metode ini tidak dapat digunakan jika penambahan analit dapat mengganggu pengukuran, misalnya analit yang ditambahkan menyebabkan kekurangan pereaksi, mengubah pH atau kapasitas dapar, dan lain-lain. Kriteria kecermatan dilakukan sama seperti pada metode simulasi (Harmita, 2004).
Tabel III. Rentang kesalahan perolehan kembali yang diperbolehkan
Konsentrasi analit (%) Unit Rata-rata perolehan kembali
(%)
100 100 % 98-102
≥10 10 % 98-102
≥1 1 % 97-103
≥0.1 0.1% 95-105
0.01 100 ppm 90-107
0.001 10 ppm 90-107
0.0001 1 ppm 80-110
0.00001 100 ppb 80-110
0.000001 10 ppb 60-115
0.0000001 1 ppb 40-120
(Harmita, 2004)
b. Presisi. Keseksamaan adalah ukuran yang menunjukkan derajat kesesuaian antara hasil uji individual, diukur melalui penyebaran hasil individual dari rata-rata jika prosedur diterapkan secara berulang pada sampel-sampel yang diambil dari campuran yang homogen (Harmita, 2004).
Untuk menetapkan presisi bahan campuran dan bahan sisa pada artikel obat, formula berikut ini harus digunakan untuk menentukan metode ketertiruan yang tepat (interlaboratorium) (Harmita, 2004).
RSD < 2(1-0,5 log c) (7)
dan untuk keterulangan :
RSD < 2(1-0,5 log c) x 0,67 (8) c = konsentrasi analit sebagai fraksi desimal (contoh: 0,1% = 0,001) Keseksamaan dapat dihitung dengan cara sebagai berikut: Hasil analisis adalah x1, x2, x3, x4,...xn maka simpangan bakunya adalah
(9) Simpangan baku relatif atau koefisien variasi (KV) adalah:
(10) (Harmita, 2004)
Tabel IV. Kriteria yang diperbolehkan untuk presisi pada kadar analit yang berbeda
Konsentrasi analit (%) Unit Presisi (RSD) ( %)
100 100 % 1,3
0,00001 100 ppb 15
0,000001 10 ppb 21
0,0000001 1 ppb 30
kuantitatif. Penentuan LOD dengan cara membandingkan respon dari pengukuran analit terhadap respon blangko. Konsentrasi analit yang mampu memberikan respon 2-3 kali respon blangko disebut LOD(United States Pharmacopeial Convention, 1995). LOQ merupakan konsentrasi analit terkecil dalam sampel yang masih dapat dianalisis dengan hasil penentuan kualitatif yang menuju akurasi yang memadai. Penentuan LOQ pada metode instrumen dengan cara membandingkan respon dari pengukuran analit terhadap respon blanko konsentrasi analit yang mampu memberikan respon 10 kali respon blanko (United States Pharmacopeial Convention, 1995).
d. Selektivitas. Selektivitas dapat diartikan sebagai kemampuan dari suatu metode analisis untuk mengukur keberadaan analit dalam sampel secara tepat (United States Pharmacopeial Convention, 1995). Jadi metode yang digunakan hanya mengukur zat tertentu saja secara cermat dan seksama dengan adanya komponen lain yang mungkin ada dalam matriks sampel. Selektivitas seringkali dapat dinyatakan sebagai derajat penyimpangan (degree of bias) metode yang dilakukan terhadap sampel yang mengandung bahan yang ditambahkan berupa cemaran, hasil urai, senyawa sejenis, senyawa asing lainnya, dan dibandingkan terhadap hasil analisis sampel yang tidak mengandung bahan lain yang ditambahkan (Harmita, 2004).
keduanya. Jika cemaran dan hasil urai tidak dapat diidentifikasi atau tidak dapat diperoleh, maka selektivitas dapat ditunjukkan dengan cara menganalisis sampel yang mengandung cemaran atau hasil uji urai dengan metode yang hendak diuji lalu dibandingkan dengan metode lain untuk pengujian kemurnian seperti kromatografi, analisis kelarutan fase, dan Differential Scanning Calorimetry. Derajat kesesuaian kedua hasil analisis tersebut merupakan ukuran selektivitas. Pada metode analisis yang melibatkan kromatografi, selektivitas ditentukan melalui perhitungan daya resolusinya (Rs) (Harmita, 2004).
e. Linearitas dan rentang. Linearitas adalah rentang kadar terendah sampai kadar tertinggi yang ditentukan dengan metode analisis dan dihubungkan dengan tanggap detektor sehingga memberikan harga koefisien relasi pada tabel statistik (Mulja dan Suharman, 1995). Rentang adalah interval antara kadar terendah sampai kadar tertinggi dari analit yang dapat diukur secara kuantitif menggunakan metode analisis tertentu dan menghasilkan presisi dan keakuratan yang mencukupi (United States Pharmacopeial Convention, 1995). Persyaratan data linearitas yang bisa diterima jika memenuhi nilai koefisien korelasi (r) >0,999 (Snyder, Kirkland, dan Glajch, 1997).
f. Ketidakpastian. Ketidakpastian metode merupakan tingkat kebolehjadian dari hasil tes analisis pada sampel yang sama di bawah kondisi variasi tes normal (United States Pharmacopeial Convention, 1995).
2. Kategori metode analisis
mengukur secara kuantitatif sejumlah besar komponen dari serbuk obat atau senyawa aktif dalam sediaan obat jadi. Kategori II meliputi metode-metode analitik yang digunakan untuk penentuan kemurnian dalam serbuk obat atau penentuan senyawa degradasi dalam sediaan obat jadi. Kategori III meliputi metode-metode analitik yang digunakan untuk penentuan sifat-sifat khusus seperti kecepatan disolusi dan pelepasan obat. Parameter-parameter yang harus dipenuhi pada masing-masing kategori dapat dilihat pada tabel V.
Tabel V. Kategori Analisis Kimia
Parameter analitik
Kategori I Kategori II Kategori III Uji kuantitatif Uji kualitatif
Akurasi
*Mungkin diperlukan, tergantung sifat uji spesifik yang dilakukan (United States Pharmacopeial Convention, 1995).
H. Landasan Teori
dipengaruhi oleh pH dan paparan cahaya. Kurkumin memiliki panjang gelombang maksimum pada daerah panjang gelombang 425 nm dalam pelarut etanol.
KLT merupakan metode pemisahan komponen-komponen berdasarkan perbedaan adsorpsi atau partisi oleh fase diam di bawah gerakan pelarut pengembang atau pelarut pengembang campuran. Densitometri merupakan salah satu dari metode analisa KLT kuantitatif untuk penetapan kadar suatu senyawa dengan mengukur kerapatan bercak senyawa yang dipisahkan dengan cara KLT.
Validasi metode analisis adalah serangkaian prosedur yang digunakan untuk membuktikan apakah suatu metode analisis yang digunakan tersebut sesuai yang diharapkan dengan selektivitas, linearitas, rentang, akurasi, dan presisi yang memadai. Nilai selektivitas yang baik untuk metode KLT memiliki resolusi > 1,5. Linearitas dan rentang yang baik ditunjukkan melalui nilai faktor korelasi (r) >0,999. Nilai akurasi yang memadai untuk senyawa baku dengan kemurnian 100% adalah memiliki perolehan kembali 98-102%, sedangkan untuk analit dalam matriks sampel pada kadar 100 ppm adalah 90-107%. Nilai presisi yang baik untuk senyawa baku memiliki nilai KV < 2%, sedang untuk analit dalam matriks sampel pada kadar 100 ppm adalah 5,3%.
I. Hipotesis
41
A. Jenis dan Rancangan Penelitian
Penelitian ini termasuk jenis penelitian non eksperimental deskriptif karena tidak ada manipulasi dan perlakuan terhadap fenomena yang diamati.
B. Variabel dan Definisi Operasional 1. Klasifikasi variabel
a. Variabel bebas dalam penelitian ini adalah sistem kromatografi yang menghasilkan pemisahan paling optimum. Sistem kromatografi yang digunakan adalah fase normal dengan fase diam silika gel G 60 dan fase gerak kloroform:asam asetat glasial:heksana (85 : 10 : 5) v/v.
b. Variabel tergantung pada penelitian ini adalah parameter-parameter validasi yaitu selektivitas, linearitas, rentang, akurasi, dan presisi.
c. Variabel pengacau terkendali yang terdapat dalam penelitian ini adalah paparan cahaya, kemurnian pelarut dan pH. Paparan cahaya diminimalkan dengan menutup larutan dengan kertas aluminium dan pengerjaan dilakukan di ruang gelap. Pelarut dan fase gerak yang digunakan seluruhnya berkualitas
2. Definisi operasional
a. Kurkumin dalam sampel kapsul lunak OHT merupakan salah satu senyawa kurkuminoid, terdapat bersama minyak atsiri yang berasal dari Curcumae xanthorrizae Rhizoma yang disuspensikan oleh beeswax sehingga berbentuk semi padat dalam sediaan kapsul lunak.
b. Kromatografi lapis tipis (KLT) fase normal yang digunakan adalah seperangkat bejana kromatografi (CAMAG) dengan fase diam silika gel G 60 (E. Merck) dan fase gerak kloroform : asam asetat glasial : heksana (85 : 10 : 5) v/v.
c. Kadar kurkumin dalam larutan sampel ditetapkan dengan satuan ppm
d. Parameter validasi metode analisis yang ditetapkan adalah selektivitas, linearitas, rentang, akurasi, dan presisi.
C. Bahan
Bahan yang digunakan dalam penelitian ini adalah metanol p.a.
D. Alat
Alat yang digunakan dalam penelitian ini meliputi neraca; neraca analitik (OHAUS Carat Series PAJ 1003 dengan spesifikasimax60/120g; d = 0,01/0,1 mg ;dane= 1 mg); Densitometer (CAMAG TLC Scanner 3 CAT. No. 027.6485 SER. No.160602); labu takar 5,0 mL; labu takar 10,0 mL; labu takar 50,0 mL, labu takar 100,0 mL; cawan arloji; pipet mikro volume 0,1-2 μL; pipet mikro volume 0,5-5 mL; corong; flakon; pipet tetes; Bekker glass; sendok; pengaduk; ultrasonikator Retschtipe T460 (Schwing.1 PXE, FTZ-Nr. C-066/83 HF-Frequ. : 35 kHz); oven; dan bejana kromatografi (CAMAG).
E. Cara Penelitian 1. Pembuatan metanol pH 4
Metanol ditambahkan dengan asam asetat glasial dengan perbandingan 9:1 untuk setiap pembuatan larutan.
2. Pengaktifan silika Gel G 60
Lempeng silika gel G 60 dipanaskan di dalam oven pada suhu 105ºC selama 1 jam sebelum digunakan.
3. Pembuatan dan penjenuhan fase gerak
4. Penetapan panjang gelombang serapan maksimum
Lebih kurang 10,0 mg baku kurkumin ditimbang seksama, dilarutkan dalam metanol pH 4 , kemudian dimasukkan ke dalam labu takar dan diencerkan dengan metanol pH 4 hingga 10,0 mL (larutan induk 1000 ppm). Larutan induk diencerkan dengan metanol pH 4 hingga diperoleh seri larutan baku yang mengandung kurkumin 100; 300; dan 500 ppm. Pembuatan larutan baku kurkumin direplikasi sebanyak 3 kali. Semua larutan baku harus terlindung dari cahaya.
Seri larutan baku ditotolkan sebanyak 1,0 L pada lempeng silika gel G 60 kemudian segera dikembangkan dalam bejana kromatografi yang telah dijenuhkan dengan campuran kloroform : asam asetat glasial : heksana (85:10:5) v/v selama 15 menit. Pengembangan dilakukan setinggi 10 cm. Lempeng silika gel G 60 segera dikeluarkan dan dikeringkan setelah pengembangan selesai.
Bercak seri larutan baku yang mengandung kurkumin 100; 300; dan 500 ppm diukur dengan densitometer pada panjang gelombang 400-500 nm. Panjang gelombang maksimum ditentukan berdasarkan serapan maksimum yang dihasilkan oleh bercak tersebut.
5. Penentuan kurva baku kurkumin
mengandung kurkumin 100; 180; 260; 340; 420 dan 500 ppm. Semua larutan baku harus terlindung dari cahaya.
Seri larutan baku ditotolkan sebanyak 1,0 L pada lempeng silika gel G 60 kemudian segera dikembangkan dalam bejana kromatografi yang telah dijenuhkan dengan campuran kloroform : asam asetat glasial : heksana (85:10:5) v/v selama 15 menit. Pengembangan dilakukan setinggi 10 cm. Lempeng silika gel G 60 segera dikeluarkan dan dikeringkan setelah pengembangan selesai.
Bercak seri larutan baku kurkumin diukurAUC-nya dengan densitometer pada panjang gelombang maksimum yang telah diperoleh. Puncak kromatogram dan nilai AUC yang muncul diamati. Dengan metode regresi linear, nilai konsentrasi (ppm) diplotkan terhadap nilai AUC masing-masing seri larutan baku sehingga diperoleh persamaan y = bx + a dimana y merupakan nilai respon (AUC), x merupakan konsentrasi senyawa baku (ppm), a adalah intersept, dan b adalahslope. Pembuatan kurva baku direplikasi sebanyak 3 kali.
6. Penetapan perolehan kembali (recovery) dan koefisien variasi (KV) baku kurkumin
Seri larutan baku ditotolkan sebanyak 1,0 L pada lempeng silika gel G 60 kemudian segera dikembangkan dalam bejana kromatografi yang telah dijenuhkan dengan campuran kloroform : asam asetat glasial : heksana (85:10:5) v/v selama 15 menit. Pengembangan dilakukan setinggi 10 cm. Lempeng silika gel G 60 segera dikeluarkan dan dikeringkan setelah pengembangan selesai.
Tiga replikasi bercak seri larutan baku yang mengandung kurkumin 100; 260; dan 500 ppm diukur dengan densitometer pada panjang gelombang maksimum yang telah diperoleh. Puncak kromatogram dan nilai AUC yang muncul diamati. Kadar kurkumin dihitung dengan persamaan kurva baku yang telah diperoleh. Masing-masing seri kadar dihitung nilai perolehan kembali dan koefisien variasinya (KV).
7. Penentuan selektivitas kurkumin dalam sampel serta akurasi dan presisi baku yang ditambahkan ke dalam sampel
Lebih kurang 10,0 mg baku kurkumin dilarutkan dalam metanol pH 4 hingga 10,0 mL (larutan A). Sejumlah lebih kurang 1000,0 mg sampel ditambah metanol pH 4 sebanyak 40 mL (larutan B). Larutan B kemudian diultrasonikasi selama 15 menit. Sampel dipastikan sudah larut seluruhnya. Kemudian larutan B disentrifugasi selama 15 menit. Supernatan kemudian diambil seluruhnya dengan pipet. Supernatan ditambah metanol pH 4 hingga 50,0 mL (larutan C).
ditambahkan metanol pH 4 hingga 5,0 mL(larutan D). Semua larutan harus terlindung dari cahaya.
Sejumlah 3,2 mL larutan C dimasukkan dalam labu takar 5,0 mL (masing-masing 5 kali). Masing-masing labu takar ditambahkan metanol pH 4 hingga 5,0 mL (larutan E). Semua larutan sampel harus terlindung dari cahaya.
Masing-masing larutan D dan E ditotolkan sebanyak 1,0 L pada lempeng silika gel G 60 kemudian segera dikembangkan dalam bejana kromatografi yang telah dijenuhkan dengan campuran kloroform : asam asetat glasial : heksana (85:10:5) v/v selama 15 menit. Pengembangan dilakukan setinggi 10 cm. Lempeng silika gel G 60 segera dikeluarkan dan dikeringkan setelah pengembangan selesai. Bercak masing-masing larutan D dan E diukur dengan densitometer pada panjang gelombang maksimum yang telah diperoleh.
F. Analisis Hasil
Perhitungan kadar kurkumin dalam setiap larutan dilakukan dengan memasukkan nilai AUC sebagai y pada persamaan kurva baku y = bx + a sehingga diperoleh nilai x sebagai kadar dengan satuan ppm. Sedangkan kesahihan metode ditentukan dengan parameter selektivitas, linearitas, rentang, akurasi, dan presisi dengan rumus sebagai berikut:
1. Selektivitas
kurkumin sampel dengan Rf puncak kurkumin baku. Selain itu, pemisahan yang baik ditunjukkan dengan nilai resolusi (Rs) > 1,5. Berikut ini merupakan metode perhitungan Rfdan Rs:
(11)
(12) Z2 – z1 merupakan selisih antara nilai Rf maksimum kurkumin dengan nilai Rfmaksimum senyawa di dekatnya. W1adalah jarak yang ditempuh senyawa di dekat puncak kurkumin, sedangkan W2 adalah jarak yang ditempuh puncak kurkumin.
2. Linearitas dan rentang
Nilai linearitas ditunjukkan melalui nilai koefisien korelasi (r) yang diperoleh melalui penentuan kurva baku dengan metode regresi linear. Nilai r yang baik adalah > 0,999. Rentang yang baik ditunjukkan melalui seberapa besar rentang konsentrasi seri baku dari kadar paling rendah hingga paling tinggi yang masih memiliki linearitas, akurasi, dan presisi yang baik.
3. Akurasi baku kurkumin
Akurasi baku kurkumin ditentukan melalui nilai perolehan kembali (recovery). Metode memiliki akurasi bahan baku yang baik apabila memiliki nilai perolehan kembali 98-102% (Mulja dan Suharman, 1995).
4. Presisi baku kurkumin
Presisi baku kurkumin ditentukan melalui nilai koefisien variasi (KV) atau Coefficient Variation (CV). Metode dikatakan memenuhi presisi yang baik apabila nilai koefisien variasinya < 2% (Mulja dan Suharman, 1995).
(14) 5. Akurasi baku kurkumin yang ditambahkan ke dalam matriks sampel
Akurasi baku kurkumin dalam matriks sampel ditentukan melalui nilai perolehan kembali (recovery) kadar baku yang ditambahkan ke dalam sampel. Metode memiliki akurasi bahan baku yang baik apabila memiliki nilai perolehan kembali 90-107% pada kadar 100 ppm(Harmita, 2004).
(15) CF merupakan kadar kurkumin yang terukur dalam larutan sampel yang ditambah baku (larutan D). CAmerupakan kadar kurkumin dalam larutan sampel yang terukur (larutan E). C*Amerupakan konsentrasi teoritis baku kurkumin yang ditambahkan dalam volume pengenceran yang sama dengan larutan D dan E. 6. Presisi baku kurkumin yang ditambahkan ke dalam matriks sampel
Kadar kurkumin baku yang ditambahkan (x) = kadar kurkumin larutan D – kadar kurkumin larutan E
(16)
51
Penelitian ini bertujuan untuk mengetahui apakah metode KLT-Densitometri memenuhi parameter validasi yang ditetapkan. Metode yang digunakan adalah KLT-Densitometri karena metode ini mampu memisahkan senyawa dalam suatu campuran. Sampel yang dianalisis pada penelitian ini merupakan Obat Herbal Terstandar yang dipastikan mengandung lebih dari satu senyawa. Selain itu, metode KLT-Densitometri memiliki kelebihan yaitu dapat digunakan untuk analisis kualitatif dan kuantitatif secara simultan. Penelitian ini menggunakan KLT fase normal dengan fase diam silika gel G 60 dan fase gerak kloroform : asam asetat glasial : heksana (85 : 10 : 5).
Kurkumin lebih stabil pada pH 4 karena kurkumin merupakan senyawa yang bersifat asam lemah. Penambahan asam asetat glasial hingga pH 4 akan menjaga kurkumin tetap dalam bentuk molekulnya sehingga kurkumin tetap stabil dalam larutan. Pembuatan larutan baku maupun sampel dilakukan pada pH 4 dengan penambahan asam asetat glasial karena hasil menunjukkan kurkumin tetap stabil pada pH 4.
A. Penentuan Panjang Gelombang Serapan Maksimum
Penentuan panjang gelombang serapan maksimum bertujuan untuk memperoleh besar area di bawah kurva (AUC) kurkumin yang paling maksimum. Penentuan panjang gelombang serapan maksimum dilakukan dengan mengukur kerapatan bercak dari larutan baku kurkumin kadar rendah (100 ppm), tengah (300 ppm), dan tinggi (500 ppm) pada rentang panjang gelombang 400-500 nm.
Densitometer yang dipergunakan memiliki detektor visibel yang dapat mengukur sinar yang dipantulkan oleh bercak suatu senyawa pada panjang gelombang 380-800 nm. Untuk dapat diukur pada panjang gelombang tersebut, senyawa yang dianalisis harus memiliki gugus kromofor dan auksokrom sehingga energi eksitasi yang dibutuhkan kecil (Gandjar dan Rohman, 2007). Kedua gugus ini bertanggungjawab terhadap penyerapan sinar radiasi elektromagnetik pada kurkumin.
gugus auksokrom merupakan gugus yang memiliki atom dengan elektron bebas. Gugus auksokrom terikat secara langsung pada gugus kromofor. Pasangan elektron bebas pada elektron O akan berinteraksi dengan elektron π pada gugus kromofor. Pengikatan ini akan menyebabkan pergeseran panjang gelombang maksimum dan intensitas serapan maksimum dari kurkumin ke panjang gelombang yang lebih besar. Berikut ini merupakan gugus kromofor dan auksokrom yang terdapat pada struktur kurkumin (Gambar 17).
Gambar 17. Gugus kromofor dan auksokrom pada struktur kurkumin
Gambar 18. Kromatogram panjang gelombang maksimum kurkumin
Chan, Lam, Lee, dan Zhang (2004) menyatakan bahwa pergeseran panjang gelombang maksimum yang diperbolehkan untuk rentang panjang gelombang visibel adalah 3 nm. Kesesuaian panjang gelombang serapan maksimum kurkumin pada percobaan dengan panjang gelombang maksimum kurkumin teoritis menunjukkan bahwa senyawa yang dianalisis tetap stabil (tabel VI).
Tabel VI. Data pengukuran panjang gelombang serapan maksimum kurkumin
Panjang gelombang serapan maksimum Seri Kadar
Pengukuran panjang gelombang maksimum dilakukan terhadap bercak kurkumin, sehingga yang terukur merupakan senyawa murni karena fase gerak maupun pelarut telah menguap. Anggarwal (1995) menyatakan bahwa panjang gelombang maksimum senyawa murni kurkumin adalah 425 nm. Hasil pengukuran menunjukkan bahwa seluruh bercak larutan baku kurkumin memiliki panjang gelombang maksimum 425 nm dan tidak terjadi pergeseran. Hal ini menunjukkan bahwa kualitas baku dan stabilitas kurkumin dalam larutan masih baik.
Hasil pengukuran panjang gelombang maksimum pada rentang panjang gelombang ultraviolet hingga visibel, hanya terdapat satu puncak saja. Profil tersebut menunjukkan bahwa senyawa baku kurkumin memiliki kemurnian yang tinggi dan tidak mengandung senyawa lain maupun produk degradasinya.
B. Pengamatan Profil Kromatogram Baku Kurkumin
Pengamatan profil kromatogram kurkumin baku dilakukan untuk memastikan senyawa kurkumin dapat terelusi dengan baik. Parameter yang digunakan dalam pengamatan profil kromatografi adalah nilai faktor retardasi (Rf) dan warna bercak. Rfbiasa digunakan dalam analisis kualitatif. Hal ini disebabkan karena nilai Rf bersifat selektif untuk senyawa tertentu dalam kondisi tertentu pula.
nilai Rf ini ditentukan pula oleh polaritas senyawa analit, fase gerak, dan fase diam.
Gambar 19. Kromatogram baku kurkumin 260 ppm
Dari kromatogram (Gambar 19) dapat diketahui bahwa sistem fase gerak pada KLT mampu mengelusi kurkumin dengan baik. Hal ini ditunjukkan dengan nilai Rf baku kurkumin yaitu 0,85. Pada kromatogram baku kurkumin ini hanya terdapat 1 buah peak saja. Hal ini menunjukkan bahwa baku kurkumin yang digunakan memiliki kemurnian yang tinggi yaitu 100%.
O O
H3CO
HO
OCH3
OH
Gugus non polar
Gugus polar
Gambar 20. Gugus polar dan gugus non polar pada kurkumin
berinteraksi dengan silika gel G 60 dan asam asetat glasial. Sedang gugus nonpolar kurkumin berinteraksi dengan heksana. Gambar 21 dan 22 menunjukkan interaksi kurkumin dengan fase diam dan fase gerak.
O
Gambar 21. Interaksi hidrogen antara kurkumin dengan fase diam
Interaksi gugus polar kurkumin dengan fase diam merupakan interaksi hidrogen. Atom O pada silika gel, gugus metoksi dan fenol pada kurkumin memiliki pasangan elektron bebas yang mampu berinteraksi dengan atom H yang berada di dekatnya.
interaksi van Der Waals Heksana
interaksi Van der Waals kloroform
CH3 interaksi van Der Waals Heksana
interaksi hidrogen
Interaksi yang terjadi antara kurkumin dengan fase gerak adalah interaksi
Van der Waals antara atom C karbonil pada struktur kurkumin yang bersifat parsial positif dan atom Cl pada kloroform yang bersifat parsial negatif. Interaksi
Van der Waals juga terjadi pada gugus non polar kurkumin dengan heksana. Interaksi hidrogen terjadi antara atom O pada gugus karbonil, gugus metoksi, dan gugus fenol kurkumin dengan asam asetat glasial.
Si
Gambar 23. Interaksi antara asam asetat glasial dengan silika gel
Interaksi hidrogen yang terjadi antara kurkumin dengan silika, kurkumin dengan asam asetat glasial serta asam asetat glasial dengan silika gel bersifat kompetitif. Besarnya kekuatan masing-masing ikatan dipengaruhi oleh perbedaan elektronegatifitas antar atom pada senyawa masing-masing.
Kurkumin tidak larut dalam air sehingga dibutuhkan fase gerak yang dapat melarutkan kurkumin. Kloroform dan heksana berfungsi untuk membantu melarutkan kurkumin. Asam asetat glasial berfungsi untuk menjaga kurkumin tetap stabil dan secara kompetitif berinteraksi dengan silika. Kekuatan adsorpsi silika terhadap kurkumin lebih lemah dibandingkan dengan asam asetat glasial. Asam asetat glasial bersifat lebih polar daripada kurkumin sehingga pada saat bercak kurkumin dilewati oleh fase gerak yang mengandung asam asetat glasial, interaksi hidrogen kurkumin dengan silika akan tergantikan oleh ikatan hidrogen asam asetat dengan silika sehingga kurkumin berada dalam bentuk bebas. Kurkumin dalam bentuk bebas akan lebih mudah berinteraksi dengan fase gerak sehingga kurkumin lebih terbawa fase gerak. Hasil pengembangan menunjukkan bahwa harga Rf kurkumin 0,85. Harga Rf yang tinggi menunjukkan bahwa fase gerak dengan indeks polaritas 4,11 mampu mengelusi kurkumin dengan baik.
C. Preparasi Sampel dan Pengamatan Profil Kromatogram Kurkumin dalam Sampel
molekul lebih besar akan mengendap sehingga akan terpisah dengan kurkumin yang telah larut dalam metanol.
Pengamatan profil kromatogram kurkumin dalam sampel bertujuan untuk mengetahui dan memastikan bahwa kurkumin dalam sampel memiliki bercak dengan nilai Rf dan warna yang sama dengan baku. Nilai Rf dan warna bercak yang sama digunakan sebagai analisis kualitatif untuk mengetahui keberadaan senyawa analit dalam sampel. Gambar 24 A merupakan data kromatogram sampel dan 24 B merupakan kromatogram baku.
A
B
Gambar 24. Perbandingan kromatogram sampel (A) dan kromatogram baku kurkumin (B)
Tabel VII. Data nilai Rfbaku dan sampel Rf
Replikasi
Baku kurkumin Kurkumin dalam sampel
I 0,85 0,84
II 0,83 0,84
III 0,85 0,84
IV 0,84 0,83
V 0,84 0,84
Hasil kromatogram menunjukkan bahwa puncak kurkumin pada sampel (puncak 3) memiliki nilai Rf yang sama dengan baku. Nilai Rf baku yaitu 0,84 sedangkan Rf sampel yaitu 0,84. Bercak kurkumin sampel memiliki warna yang sama dengan bercak kurkumin baku yaitu kuning-oranye. Oleh karena itu, puncak 3 pada kromatogram sampel benar-benar merupakan puncak kurkumin.
Pada kromatogram sampel (Gambar 24 A) terlihat bahwa terjadi pemisahan antara puncak kurkumin dengan puncak senyawa lain. Pemisahan dapat terjadi karena setiap molekul memiliki keadaan kesetimbangan (equilibrium state) yang berbeda dengan fase gerak serta fase diam. Keadaan setimbang merupakan keadaan dimana analit berpindah dari keadaan bebas ke keadaan teradsorpsi atau sebaliknya. Keadaan bebas yaitu seluruh analit terlarut dalam fase gerak, sedangkan keadaan teradsorpsi yaitu analit tertahan pada permukaan fase diam.