TESIS – SM 142501
APLIKASI METODE
UPGMA
UNTUK IDENTIFIKASI
KEKERABATAN JENIS VIRUS DAN PENYEBARAN EPIDEMI
EBOLA MELALUI PEMBENTUKAN POHON FILOGENETIK
TRI ANDRIANI NRP 1213 201 045
DOSEN PEMBIMBING
Prof. Dr. MOHAMMAD ISA IRAWAN, M.T.
PROGRAM MAGISTER JURUSAN MATEMATIKA
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM INSTITUT TEKNOLOGI SEPULUH NOPEMBER
TESIS – SM 142501
APPLICATION OF UPGMA METHOD FOR THE IDENTIFICATION
TYPE VIRUS TYPE AND EBOLA EPIDEMIC SPREADING THROUGH
ESTABLISHMENT PHYLOGENETIC TREES
TRI ANDRIANI NRP 1213 201 045
SUPERVISOR
Prof. Dr. MOHAMMAD ISA IRAWAN, M.T.
MASTER’S DEGREE
MATHEMATICS DEPARTMENT
FACULTY OF MATHEMATICS AND NATURAL SCIENCES SEPULUH NOPEMBER INSTITUTE OF TECHNOLOGY SURABAYA
ix
DAFTAR ISI
HALAMAN JUDUL
LEMBAR PENGESAHAN ... i
ABSTRAK ... iii
ABSTRACT ... v
KATA PENGANTAR . ... vii
DAFTAR ISI ... ix
DAFTAR TABEL ... xi
DAFTAR GAMBAR . ... xiii
BAB I PENDAHULUAN ... 1
1.1Latar Belakang ... 4
1.2Rumusan Masalah ... 4
1.3Batasan Masalah... 4
1.4Tujuan Penelitian ... 5
1.5Manfaat Penelitian ... 5
1.6Kontribusi Hasil Penelitian ... 5
BAB II KAJIAN PUSTAKA DAN DASAR TEORI ... 7
2.1Penelitian Terdahulu ... 7
2.2Penyakit Virus Ebola (EVD) ... 8
2.3Bioinformatika ... 10
2.4Sekuens ... 10
2.4.1. Sekuens Protein ... 12
2.4.2. Sekuens DNA ... 13
2.5Alignment ... 14
2.5.1 Matriks Penalti ... 15
2.5.2 Matriks Penskoran ... 16
2.6Algoritma Needleman Wunsch . ... 18
x
2.7.1 Metode Progressive ... 24
2.8Filogenetik Molekuler ... 25
2.8.1. Pohon ... 25
2.8.2. Pohon Filogenetik (Phylogenetic tree) ... 29
2.8.3. Metode-metode Pembentukan Pohon ... 31
2.9Metode Berbasis Jarak (Distance Based Method) ... 33
2.10 Metode UPGMA ... 35
BAB III METODOLOGI PENELITIAN ... 41
3.1Tahapan Penelitian ... 41
3.1.1 Studi Literatur ... 42
3.1.2 Pengambilan Data ... 42
3.1.3 Pembuatan Program Pensejajaran ... 42
3.1.4 Pembuatan Pohon Filogenetik Metode UPGMA ... 43
3.1.5 Hasil Pembentukan Pohon Filogenetik ... 45
3.1.6 Analisis dan Pembahasan ... 45
BAB IV HASIL DAN PEMBAHASAN ... 47
4.1Identifikasi Kekerabatan Jenis-jenis Virus Ebola ... 47
4.1.1 Pengumpulan Data ... 47
4.1.2 Pensejajaran Sekuens ... 50
4.1.3 Matriks Jarak ... 60
4.1.4 Pohon Filogenetik Metode UPGMA untuk Identifikasi Kekerabatan Jenis-jenis Virus Ebola ... 67
4.2 Penyebaran Epidemi Virus Ebola ... 73
4.1.1 Data Epidemi ... 73
4.1.2 Pohon Filogenetik untuk Penyebaran Epidemi Ebola ... 74
4.3 Pembahasan ... 76
4.4 Validasi Pohon Filogenetik ... 80
BAB IV KESIMPULAN DAN SARAN ... 81
xi
DAFTAR TABEL
Tabel 2.1. Kasus Ebola di Afrika ... 9
Tabel 2.2. Asam Amino dan Kode resmi ... 12
Tabel 2.3. Kode Standart Genetik ... 13
Tabel 2.4. Tabel Dua Dimensi Sekuens ... 18
Tabel 4.1. Hasil Jarak Evolusi Pasangan Sekuens Data Uji ... 61
Tabel 4.2.Hasil Jarak Evolusi Pasangan Sekuens Protein ... 62
Tabel 4.3. Matriks Jarak Pasangan Sekuens Data Uji ... 67
Tabel 4.4. Matriks Jarak Pasangan Sekuens Protein ... 67
xv
DAFTAR LAMPIRAN
LAMPIRAN A: Sekuens Beberapa Jenis Virus Ebola ... 85
LAMPIRAN B: Sekuens DNA Virus Ebola ... 87
xiii
DAFTAR GAMBAR
Gambar 2.1 (a) Pohon berakar dan (b) Pohon tidak berakar ... 27
Gambar 2.2 Pohon berakar dengan empat spesies mamalia ... 28
Gambar 2.3 Pohon tidak .berakar dengan empat spesies mamalia ... 28
Gambar 2.4 Pohon dengan panjang pohon ... 29
Gambar 2.5 (a) Pohon Filogenetik berakar dan (b) Pohon Filogenetik tidak Berakar ... 30
Gambar 2.6 Struktur dari Pohon Filogenetik berakar ... 31
Gambar 2.7 Pohon filogenetik dibangun oleh Metode UPGMA ... 39
Gambar 3.1. Diagram Alir Penelitian ... 41
Gambar 3.2. Multiple Alignment oleh Metode Progressive ... 43
Gambar 3.3. Diagram Alir Proses Pensejajaran hingga Pohon Filogenetik ... 44
Gambar 3.3. Diagram Alir Proses Pensejajaran hingga Pohon Filogenetik ... 44
Gambar 4.1Pohon untk Proses Pensejajaran ... 58
Gambar 4.2 Pensejajaran sekuens matriks protein ... 60
Gambar 4.3 Pohon Filogenetik Metode UPGMA data uji ... 71
Gambar 4.4 Pohon Filogenetik identifikasi kekerabatan jenis-jenis virus ebola simulasi MATLAB ... 72
Gambar 4.5 Hasil Pensejajaran sekuens DNA ... 74
Gambar 4.6 Pohon filogenetik Metode UPGMA untuk penyebaran epidemi ebola ... 75
vii
KATA PENGANTAR
Segala puji syukur dan kemuliaan hanya kepada Tuhan atas segala limpahan
kasih karunia, sehingga penulis dapat menyelesaikan tesis yang berjudul “Aplikasi Metode UPGMA untuk Identifikasi Kekerabatan Jenis Virus dan Penyebaran
Epidemi Ebola Melalui Pembentukan Pohon Filogenetik”
Tesis ini disusun sebagai salah satu prasyarat kelulusan Program Magister
Jurusan Matematika Fakultas Matematika dan Ilmu Pengetahuan Alam, Institut
Teknologi Sepuluh Nopember Surabaya. Penulis menyadari bahwa tulisan Tesis ini
masih ada kekurangan, sehingga kritik dan saran dari pembaca sangat penulis
harapkan untuk kedepannya.
Penyusunan Tesis ini tidak terlepas dari bantuan dan dukungan dari banyak
pihak. Oleh karena itu, penulis mengucapkan terima kasih kepada:
1. Prof. Ir. Joni Hermana, M.Sc.ES., Ph.D., selaku Rektor Institut Teknologi
Sepuluh Nopember (ITS) Surabaya yang telah memberikan fasilitas kepada
penulis selama menempuh pendidikan sehingga dapat menyelesaikan Tesis ini.
2. Direktorat Jenderal Pendidikan Tinggi (DIKTI) selaku penyandang dana yang
telah memberikan beasiswa BPPDN.
3. Prof. Dr. Ir. Adi Soeprijanto, M.T., selaku Direktur Program Pascasarjana ITS.
4. Dr. Imam Mukhlash, S.Si, M.T. selaku Ketua Jurusan Matematika ITS.
5. Dr. Subiono, MS., selaku Ketua Program Studi Pascasarjana Matematika ITS.
6. Prof. Dr. Mohammad Isa Irawan, M.T.,selaku dosen pembimbing yang telah
meluangkan waktu untuk memberikan arahan, nasehat, dan motivasi kepada
penulis sehingga dapat menyelesaikan Tesis ini dengan baik.
7. Dr. Drs. Haiyanto, M.Si., Dr. Budi Setiyono, S.Si., MT. dan Dr. Dwi Ratna
Sulistyaningrum, S.Si., MT. selaku dosen penguji yang telah memberikan
masukan kritik dan saran yang membantu penulis untuk memperbaiki tulisan
viii
8. Dr. Mahmud Yunus, M.Si selaku dosen wali yang selama ini sudah banyak
mendidik dan membantu selama penulis menempuh studi S2.
9. Seluruh dosen Jurusan Matematika, yang selama ini sudah banyak mendidik dan
membekali penulis dengan berbagai ilmu pengetahuan selama penulis mengikuti
proses perkuliahan dan seluruh staf dan karyawan Jurusan Matematika ITS yang
telah memberikan bantuan, kemudahan, dan kelancaran.
10.Ayah dan Ibu, kedua kakak dan adik tercinta serta seluruh keluarga atas
perhatian, doa dan segala dukungannya selama ini.
11.Teman-teman seperjuangan Program Magister Matematika ITS angkatan 2013
yang telah menemani, memotivasi, dan segala bantuannya.
12.Keluarga Besar Pascasarjana Matematika ITS dan semua pihak yang telah
membantu proses penulisan Tesis ini.
Penulis berharap semoga tulisan Tesis ini dapat bermanfaat untuk kemajuan dan
perkembangan ilmu pengetahuan, khususnya disiplin ilmu Komputasi dan dapat
memberikan kontribusi bagi kemajuan ITS.
Surabaya, Januari 2016
iii
Aplikasi Metode UPGMA untuk Identifikasi Kekerabatan Jenis
Virus dan Penyebaran Epidemi Ebola Melalui Pembentukan Pohon
Filogenetik
Nama : Tri Andriani
NRP : 1213201045
Dosen Pembimbing : Prof. Dr. M. Isa Irawan, MT.
ABSTRAK
Penyakit ebola atau dalam bahasa medis Ebola Virus Disease (EVD) adalah penyakit
yang disebabkan oleh sejenis virus dari genus Ebolavirus (EBOV), famili Filoviridae.
Virus ebola diklasifikasikan ke dalam 5 jenis, yaitu Zaire ebolavirus (ZEBOV),
Sudan ebolavirus (SEBOV), Bundibugyo ebolavirus (BEBOV), Tai Forest ebolavirus
yang juga dikenal sebagai Cote d’Ivoire ebolavirus (CIEBOV), dan Reston
ebolavirus (REBOV). Identifikasi kekerabatan jenis virus ebola dan penyebarannya
dapat dilakukan dengan menggunakan pohon filogenetik. Pada penelitian ini, pohon
filogenetik dibangun dengan Metode UPGMA yang didalamnya terdapat Multiple
Alignment. Multiple Alignment menggunakan Metode Progressive yang didalamnya
terdapat pensejajaran berpasangan menggunakan Algoritma Needleman Wunsch.
Hasil pembentukan pohon fillogenetik disimpulkan bahwa hubungan kekerabatan
jenis virus ebola tidak dapat disimpulkan secara umum, sebab tergantung pada type
protein yang dibandingkan.. Misal pada type minor nucleoprotein jenis Zaire
ebolavirus dekat dengan Sudan ebolavirus. Pada type membrane associated protein
VP 24 jenis Zaire ebolavirus dekat dengan Bundibugyo ebolavirus. Berdasarkan
pohon filogenetik data DNA, jenis Tai Forest ebolavirus dekat dengan Bundibugyo
ebolavirus tetapi letak negara penyebaran epidemi ebola berjauhan. Jarak genetik
untuk jenis Bundibugyo ebolavirus dengan Tai Forest ebolavirus adalah 0.3725.
Jenis Tai Forest ebolavirus mirip dengan Bundibugyo ebolavirus tidak dipengaruhi
oleh kedekatan daerah penyebaran epidemi ebola.
v
Aplication of UPGMA Method for the Kinship Identification Type
Virus Types
and Ebola Epidemic Spreading Through Establishment
of Phylogenetic Trees
Name : Tri Andriani
NRP : 1213201045
Supervisor : Prof. Dr. M. Isa Irawan, MT.
ABSTRACT
Ebola disease or in medical language Ebola Virus Disease (EVD) is a disease caused
by a virus of the genus Ebolavirus (EBOV), family Filoviridae. Ebola virus is
classified into five types, namely Zaire ebolavirus (ZEBOV) Sudan ebolavirus
(SEBOV), Bundibugyo ebolavirus (BEBOV), Tai Forest ebolavirus also known as
Cote d'Ivoire ebolavirus (CIEBOV), and Reston ebolavirus (REBOV). Identification
of kinship types of Ebola virus and its spread can be performed using phylogenetic
tree. In this study, the phylogenetic tree constructed by UPGMA method in which
there are Multiple Alignment. Progressive Multiple Alignment using a method in
which there are pairwise alignments using the Needleman Wunsch algorithm. Results
fillogenetik tree formation was concluded that kinship types of Ebola virus can not be
inferred in general, because depending on the type of protein compared .. Eg the
minor type nucleoprotein Zaire ebolavirus species close to Sudan ebolavirus. On the
type of membrane associated protein VP 24 types Zaire ebolavirus close to
Bundibugyo ebolavirus. Based on phylogenetic trees DNA data, the type of Tai
Forest ebolavirus close to Bundibugyo ebolavirus but the layout state ebola epidemic
spread far apart. Genetic distance for this type of Bundibugyo ebolavirus with Tai
Forest ebolavirus is 0.3725. Tai Forest ebolavirus type similar to Bundibugyo
ebolavirus not influenced by the proximity of ebola epidemic spreading area.
1
BAB 1
PENDAHULUAN
Pada bagian ini diberikan ulasan mengenai hal-hal yang melatarbelakangi
usulan penelitian, rumusan masalah yang akan diselesaikan dalam penelitian,
batasan masalah, tujuan penelitian, dan manfaat penelitian.
1.1 Latar Belakang
Penyakit ebola atau dalam bahasa medis Ebola Virus Disease (EVD)
adalah penyakit yang disebabkan oleh sejenis virus dari genus Ebolavirus
(EBOV), dari keluarga Filoviridae. Ebola yang dikenal juga sebagai demam
berdarah ebola atau Ebola Haemorrhagic Fever (EHF) telah ada sebagai epidemi
menular sejak tahun 1976 di Afrika Tengah. Epidemi ialah mewabahnya penyakit
dalam daerah tertentu dengan jumlah yang melebihi batas jumlah normal atau
yang biasa. Virus ebola dapat ditularkan melalui kontak langsung oleh cairan
tubuh seperti darah, keringat, air liur, lendir, sperma, dan air mata dari pasien
EVD. Selain ditularkan manusia, EVD dapat menular melalui binatang seperti
gorila, simpanse, monyet, dan kelelawar buah. Masa inkubasi biasanya dimulai
dua hari hingga tiga minggu. Pada tahap awal, pasien EVD biasanya menunjukkan
gejala seperti demam, sakit tenggorokan, nyeri otot, sakit kepala dan tubuh lemah.
Gejala lanjut dari EVD adalah pendarahan serta menurunnya fungsi hati dan
ginjal. Menurut analisa sejarah wabah ebola, tingkat kematian dari pasien EVD
adalah 40% sampai 90%. Meskipun EVD dianggap ancaman potensial bagi
kesehatan masyarakat, sampai saat ini belum tersedia obat atau vaksin berlisensi
untuk penyakit ini (Li dkk, 2014).
Penyakit ebola (EVD) pertama kali ditemukan di Afrika, daerah selatan
Sudan dan Zaire pada tahun 1976 pada tubuh seekor monyet. Pada tanggal 23
Maret 2014, Organisasi Kesehatan Dunia (WHO) melaporkan wabah baru infeksi
virus Ebola (EBOV) yang dimulai pada bulan Desember 2013 di Republik Guinea
dan menyebar ke negara-negara Afrika Barat lainnya, yaitu Sierra Leone dan
2
terdapat sebanyak 3.354 kasus dan 2.120 diantaranya meninggal. Jumlah kasus
yang dilaporkan di Guinea, Liberia dan Sierra Leone untuk periode Januari
sampai September 2014 adalah 1009 kasus dan 574 diantaranya meninggal
(Clercq, 2014).
Virus ebola diklasifikasikan ke dalam 5 jenis, yaitu Zaire ebolavirus
(ZEBOV), Sudan ebolavirus (SEBOV), Bundibugyo ebolavirus (BEBOV), Tai
Forest ebolavirus yang juga dikenal sebagai Cote d’Ivoire ebolavirus (CIEBOV),
dan Reston ebolavirus (REBOV). Reston ebolavirus (REBOV) adalah
satu-satunya virus yang tidak menyerang manusia, namun menyerang monyet
(Bovendo dkk, 2012). Untuk mengetahui seberapa mirip lima jenis virus ebola
yang ada, sangat perlu melakukan identifikasi kekerabatan kelima jenis virus
ebola tersebut. Selama ini belum ada penelitian mengenai kekerabatan jenis virus
ebola. Salah satu cara identifikasi kekerabatan adalah dengan membangun pohon
filogenetik.
Konstruksi pohon filogenetik baru-baru ini menjadi perhatian banyak
peneliti karena ketersediaan data biologis yang luas. Untuk mengkonstruksi pohon
filogenetik, terdapat beberapa metode yang dapat digunakan, yaitu Metode
Berbasis Jarak (misalnya, neighbor-joining dan unwight pair group method with
arithmetic average), Metode Berbasis Fitur (misalnya, maximum parsimony), dan
Metode Berbasis Probabilitas (misalnya, maximum likelihood) (Shen dkk, 2008).
Irawan dan Amiroch (2014) melakukan konstruksi pohon filogenetik
menggunakan Metode Berbasis Jarak untuk identifikasi host dan penyebaran
epidemi SARS. Dalam penelitiannya, Algoritma Neighbor Joining digunakan
untuk mengkonstruksi pohon filogenetik yang disimulasikan dalam Matlab. Input
untuk mengkonstruksi sebuah pohon filogenetik dengan Metode Berbasis Jarak
berupa matriks jarak. Matriks jarak diperoleh dari penyejajaran antar sequence
dengan menggunakan Metode Super Pairwise Alignment (SPA). Output dari
pensejajaran ini berupa jumlah perbedaan antar sequence yang menentukan jarak
genetiknya. Dari matriks jarak tersebut, jarak genetik diubah menjadi jarak
evolutioner menggunakan Model Jukes Cantor yang selanjutnya dibentuk pohon
filogenetik menggunakan Algoritma Neighbor Joining. Akan tetapi, algoritma
3
filogenetik dari data sekuens yang similaritasnya sangat tinggi. Apabila Algoritma
Neighbor Joining tetap digunakan untuk membentuk pohon filogenetik dari data
sekuens yang similaritasnya sangat tinggi berakibat akan diperoleh beberapa
pohon yang berbeda. Dengan kata lain, pohon yang dihasilkan tidak stabil.
Dalam membangun pohon filogenetik menggunakan Metode UPGMA
langkah awal adalah mendapatkan multiple alignment (MA) dari multiple sekuens
yang diberikan. Hasil dari MA berupa suatu himpunan sekuens yang panjangnya
sama. MA dapat menunjukkan multiple sequence berada pada keluarga yang sama
atau tidak. Selain itu, MA dapat menunjukkan semua hubungan atau relasi antar
famili dari multiple sequence yang ada. Berdasarkan pembagian keluarga, dapat
ditentukan keadaan evolusi masing-masing sekuens dalam keluarga. Secara umum
digunakan pohon topologi untuk menggambarkan hubungan di antara multiple
sequence, pohon topologi tersebut selanjutnya dikenal dengan pohon filogenetik
(Shen dkk, 2008).
UPGMA (Unwight Pair Group Method with Arithmetic Average) atau
metode kelompok pasangan unweight dengan rataan aritmatika adalah metode
paling sederhana dari semua metode clustering yang digunakan untuk membangun
pohon filogenetik. Metode clustering yang paling intuitif digunakan untuk
membangun pohon filogenetik adalah metode UPGMA. Metode ini merakit dua
kelas terdekat untuk menjadi kelas yang baru, ke dalam sebuah cluster setiap
waktu sampai semua kelas dirakit menjadi satu kelas. UPGMA digunakan untuk
membangun pohon filogenetik dengan cara yang mirip dengan Metode sistem
clustering, perbedaan utamanya adalah formula yang digunakan untuk
menghitung jarak kelas (Shen dkk, 2008).
Dengan memanfaatkan clustering, Metode UPGMA digunakan untuk
membangun pohon filogenetik. Kelebihan Metode UPGMA adalah metode ini
paling sederhana dari semua metode clustering yang digunakan untuk membangun
pohon filogenetik. Metode ini membutuhkan kecepatan substitusi dari nukleotida
atau asam amino menjadi seragam dan tidak berubah melalui proses evolusi
secara keseluruhan. Dengan kata lain, hipotesis mengukur waktu molekuler
dipenuhi. Pada setiap node induk, panjang cabang dari node induk ke dua simpul
4
mengenai konstruksi filogenetik menggunakan Metode UPGMA untuk
identifikasi kekerabatan beberapa jenis virus ebola dan asal penyebaran epidemi
ebola menggunakan pohon filogenetik.
1.2 Rumusan Masalah
Berdasarkan uraian latar belakang yang ada, permasalahan yang akan
dibahas dalam penelitian ini adalah
1. Bagaimana membentuk pohon filogenetik epidemi ebola berdasarkan jenis
virus menggunakan Metode UPGMA?
2. Bagaimana identifikasi kekerabatan beberapa jenis virus ebola dan asal
penyebaran epidemi ebola menggunakan pohon filogenetik?
1.3 Batasan Masalah
Permasalahan yang akan dibahas dalam penelitian ini dibatasi sebagai
berikut:
1. Sekuens yang disejajarkan adalah sekuens protein lima jenis virus ebola
baik yang menyerang manusia maupun binatang, sekuens DNA host dan
individu lain yang terinfeksi berdasarkan data lokasi dan tanggal
pengambilan sample.
2. Data sekuens protein yang digunakan diambil dari database Uniprot
(www.uniprot.org).
3. Data sekuens DNA yang digunakan diambil dari database National Center
for Biotechnologi Information (www.ncbi.nlm.nih.gov).
4. Data sekuens DNA yang digunakan untuk penyebaran epidemi ebola di
negara-negara Afrika.
5. Pensejajaran sekuens menggunakan Metode Progressive dengan bantuan
MATLAB.
6. Pohon filogenetik disimulasikan menggunakan Metode UPGMA dengan
5
1.4 Tujuan Penelitian
Dari perumusan masalah yang ada, maka tujuan dari penelitian ini adalah
1. Mendapatkan pohon filogenetik dengan menggunakan Metode UPGMA
dengan obyek virus ebola.
2. Mengetahui kekerabatan jenis virus dan asal penyebaran epidemi ebola
dengan menggunakan pohon filogenetik.
1.5 Manfaat Penelitian
Hasil penelitian ini diharapkan dapat memberikan manfaat sebagai
berikut:
1. Sebagai tambahan referensi untuk penelitian berikutnya mengenai proses
kontruksi pohon filogenetik dengan menggunakan Metode Berbasis Jarak,
yaitu Metode UPGMA.
2. Mengetahui penerapan pohon filogenetik untuk menyelesaikan masalah
dalam bidang kesehatan, terutama untuk mengetahui kekerabatan dan asal
penyebaran epidemi ebola.
3. Mengetahui tingkat kemiripan jenis-jenis virus ebola sehingga dapat
membantu peneliti bidang kesehatan dalam pembuatan vaksin.
1.6 Kontribusi Hasil Penelitian
Kontribusi hasil penelitian ini terhadap pengembangan ilmu adalah dapat
membantu peneliti dalam bidang kesehatan untuk mengambil tindakan lebih lanjut
7
BAB 2
KAJIAN PUSTAKA DAN DASAR TEORI
Pada bagian ini diberikan ulasan mengenai penelitian terdahulu dan
teori-teori yang diperlukan dalam proses penelitian. Penelitian terdahulu yang diulas
dalam bab ini adalah penelitian mengenai konstruksi pohon filogenetik yang
dilakukan Irawan dan Amiroch (2014). Adapun beberapa teori yang diberikan
meliputi penyakit virus ebola (EVD), sekuens, protein, DNA, pensejajaran
sekuens, multiple alignment, Metode Progressive, Algoritma Needleman Wunsch,
matriks penalti dan matriks penskoran, filogenetik molekuler, pohon, pohon
filogenetik, metode berbasis jarak dan Metode UPGMA.
2.1 Penelitian Terdahulu
Penelitian pertama berkaitan dengan identifikasi host dan penyebaran
epidemi SARS oleh Irawan dan Amiroch (2014). Irawan dan Amiroch melakukan
konstruksi pohon filogenetik menggunakan Metode Berbasis Jarak untuk
identifikasi host dan penyebaran SARS. Dalam penelitiannya, Algoritma
Neighbor Joining digunakan untuk mengkonstruksi pohon filogenetik yang
disimulasikan dalam Matlab. Input untuk mengkonstruksi sebuah pohon
filogenetik Metode Berbasis Jarak berupa matriks jarak. Matriks jarak diperoleh
dari pensejajaran antar sekuen dengan menggunakan Metode Super Pairwise
Alignment (SPA). Output dari penyejajaran ini berupa jumlah perbedaan antar
sekuens yang menentukan jarak genetiknya. Dari matriks jarak tersebut, jarak
genetik diubah menjadi jarak evolutioner menggunakan model Jukes Cantor yang
selanjutnya dibentuk pohon filogenetik menggunakan Algoritma Neighbor
Joining. Hasil penelitian menunjukkan, dengan menggunakan pohon filogenetik
dapat dibuktikan data sekuens protein berbagai binatang yang dicurigai sebagai
host dari SARS Coronavirus dan data sekuens DNA pasien yang terinfeksi SARS.
Dari hasil pembentukan pohon filogenetik diketahui epidemi berawal pada tanggal
16 Desember 2002 di Guangzhou China Selatan yang kemudian menyebar ke
8
selanjutnya menyebar ke Hanoi, Toronto, Singapura, Taiwan dan HongKong
sehingga kasus SARS menjadi wabah internasional. Penerapan pensejajaran super
pairwase alignment (SPA) berhasil diterapakan untuk mensejajarkan sequence
human SARS Coronavirus dengan coronavirus lain yang dibawa oleh binatang
(Irawan dkk, 2014).
2.2 Penyakit Virus Ebola (EVD)
Penyakit ebola atau dalam bahasa medis Ebola Virus Disease (EVD)
adalah penyakit yang disebabkan oleh sejenis virus dari genus Ebolavirus
(EBOV), famili Filoviridae. Ebola yang dikenal juga sebagai demam berdarah
ebola atau Ebola Haemorrhagic Fever (EHF) telah ada sebagai epidemi menular
sejak tahun 1976 di Afrika Tengah. Virus ebola diklasifikasikan ke dalam 5 jenis,
yaitu Zaire ebolavirus (ZEBOV), Sudan ebolavirus (SEBOV), Bundibugyo
ebolavirus (BEBOV), Tai Forest ebolavirus juga dikenal sebagai Cote d’Ivoire
ebolavirus (CIEBOV), dan Reston ebolavirus (REBOV). Reston ebolavirus
(REBOV) adalah satu-satunya virus yang tidak menyerang manusia, namun
menyerang monyet (Bovendo dkk, 2012). Selama ini belum ada penelitian
mengenai kekerabatan jenis-jenis virus ebola tersebut.
Penyakit ebola (EVD) pertama kali ditemukan di Afrika, daerah selatan
Sudan dan Zaire pada tubuh seekor monyet. Pada tanggal 23 Maret 2014,
Organisasi Kesehatan Dunia (WHO) melaporkan jumlah kasus EVD yang terjadi
di Afrika pada tahun 1976 hingga tahun 2014. Sejak ditemukannya EVD pada
tahun 1976 hingga tahun 2014, dilaporkan terdapat sebanyak 3.354 kasus dan
2.120 diantaranya meninggal. Jumlah kasus yang dilaporkan di Guinea, Liberia
dan Sierra Leone untuk periode Januari sampai September 2014 adalah 1009
kasus dan 574 diantaranya meninggal (Clercq, 2014). Adapun data kasus ebola
9
Tabel 2.1: Kasus ebola di Afrika
Tahun Negara Kota Kasus Meninggal Spesies
1976
hewan primata (misalnya, monyet, gorila dan simpanse). Masa inkubasi biasanya
dimulai dua hari hingga tiga minggu setelah terjangkit virus. Pada tahap awal,
pasien EVD biasanya menunjukkan gejala seperti demam, sakit tenggorokan,
nyeri otot, sakit kepala dan tubuh lemah. Gejala lanjut dari EVD adalah muntah,
diare, pendarahan serta menurunnya fungsi hati dan ginjal. Menurut analisa
sejarah wabah ebola, tingkat kematian dari pasien EVD adalah 40% sampai 90%
10
Virus ebola mudah menyebar dengan cepat. Pertama kali infeksi dimulai
dari penularan hewan yang terinfeksi ke manusia. Dari situ nantinya manusia
meneruskan rantai penyakit ini ke manusia yang lain. Penyebaran virus ebola
antar manusia bisa melalui berbagai macam cara antara lain melalui makanan,
jarum suntik, berpegangan tangan, dan kontak langsung oleh cairan tubuh
penderita, seperti darah, keringat, air liur, lendir, sperma, dan air mata dari pasien
EVD atau melalui binatang yang rawan terinfeksi.
2.3 Bioinformatika
Bioinformatika (bioinformatics) telah dikembangkan dalam ruang, yang
telah diduduki oleh sejumlah disiplin ilmu terkait. Bioinformatika adalah ilmu
yang mempelajari penerapan teknik komputasi untuk mengelola dan menganalisis
informasi biologis. Bidang ini mencakup penerapan metode-metode matematika,
statistika dan informatika untuk memecahkan masalah-masalah biologis, terutama
dengan menggunakan sekuens DNA dan asam amino serta informasi yang
berkaitan dengannya. Contoh topik utama bidang ini meliputi basis data untuk
mengelola informasi biologis, pensejajaran sekuens (sequence alignment),
prediksi struktur untuk meramalkan bentuk struktur protein maupun struktur
sekunder RNA, analisis filogenetik, dan analisis ekspresi gen. Ini termasuk ilmu
kuantitatif seperti matematika dan biologi komputasi, biometri dan biostatistik,
ilmu komputer, sibernetika. Serta ilmu biologi seperti evolusi molekuler,
genomics dan proteomik, genetika dan biologi sel. Bioinformatika merupakan
perluasan langsung dari biologi, matematika dan komputasi ke dalam bidang baru
dalam data set yang besar (Polanski dkk, 2007).
2.4 Sekuens
Istilah sekuens biologis pada umumnya digunakan untuk menyatakan
sekuens DNA, sekuens RNA dan sekuens protein. Dalam pengertian biologi
molekuler, sekuens biologi terdiri dari banyak makromolekul, dimana semua
makromolekul memiliki fungsi-fungsi yang spesifik dalam kondisi tertentu.
Makromolekul tersebut dapat dibagi ke dalam sejumlah besa mikromolekul
11
didasarkan pada empat nukleotida, sedangkan sekuens pada protein didasarkan
pada 20 asam amino. Jika diperhatikan nukleotida sekuens DNA atau asam amino
dalam protein adalah unit-unit dasar, maka sekuens biologi hanyalah kombinasi
dari unit-unit dasar (Shen dkk, 2008).
Banyak cara yang dapat dilakukan untuk merepresentasikan struktur dari
sekuens biologis. Cara yang paling sering digunakan adalah dengan mendeskripsikan
sekuens tersebut ke dalam bentuk struktur primer, sekunder dan tersier (struktur tiga
dimensi). Untuk sekuens protein, struktur primernya mendeskripsikan kombinasi
asam amino penyusun protein. Sedangkan untuk sekuens DNA/RNA, struktur
primernya mendeskripsikan komponen-komponen nukleotida. Struktur primer
sekuens biologi menentukan komponen nukleotida atau asam aminonya. Struktur
tersier atau 3D dari sekuens biologi menggambarkan susunan 3D (posisi koordinat)
dari atom konstituen dalam molekul. Struktur sekunder dari sekuens protein
menunjukkan struktur khusus dari masing-masing segmen protein, bisa berupa
struktur helix, untai atau struktur lainnya. Super struktur sekunder juga sering
digunakan untuk mendeskripsikan suatu keadaan antara struktur sekunder dan tesier,
yang terdiri dari sebagian besar kelompok molekul kompak (domain).
Menurut Shen (Shen dkk, 2008), digunakan deskripsi untuk sekuens
biologi sebagai berikut.
A = ( ) B = ( ) C = ( ) (2.3)
dengan huruf capital A, B dan C merepresentasikan sekuens,
merepresentasikan unit-unit dasar sekuens pada posisi ke- , yang
elemen-elemennya diperoleh dari himpunan * +. Pada persamaan (2.3),
adalah panjang sekuens A, B dan C. Jika A, B dan C merupakan sekuens DNA/RNA maka dan * + atau * +. jika A, B
dan C merupakan sekuens protein maka dan
* +.
Multiple sequence (group sekuens) adalah kumpulan dari sekuens yang
dinotasikan sebagai
* + (2.4) Untuk setiap merupakan sekuens terpisah yang didefinisikan pada dan
12
analisis sekuens, dapat diketahui bahwa sekuens merupakan mutasi dari
sekuens , namun tidak dapat diketahui apakah keduanya memiliki makna yang
sama secara biologi, sehingga kedua sekuens tersebut belum bisa dikatakan
homolog, namun hanya bisa dikatakan mirip (Shen dkk, 2008).
2.4.1 Sekuens Protein
Protein adalah salah satu bio-molekuler yang penting peranannya dalam
makluk hidup. Untuk sekuens protein, struktur primernya mendeskripsikan
kombinasi asam amino penyusun protein. Adapun kode huruf dan nama asam
amino dapat dilihat pada tabel 2.2.
Tabel 2.2. Asam Amino dan Kode resmi
No 1 – Kode Huruf 3 – Kode Huruf Nama
13
2.4.2 Sekuens DNA
Asam deoksiribonukleat (deoxyribonucleid acid) atau disebut DNA
merupakan sebuah polimer yang terbentuk dari molekul kecil yang disebut
nukleotida dan berperan sebagai dasar hereditas. Informasi mengenai hereditas ini
digambarkan dalam sebuah sekuens DNA. Sekuens DNA adalah polimer linear
berdimensi satu yang disusun oleh empat nukleotida berbeda, yaitu Adenin (A),
Cytosin (C), Guanin (G) dan Tymin (T). Dengan kata lain sekuens DNA adalah
sekuens yang disusun oleh empat huruf A, C, T dan G selanjutnya empat sekuens
basa. Identifikasi sekuens DNA tersebut dapat memberikan informasi mengenai
perbedaan spesies yang satu dengan yang lainnya, walaupun mereka mempunyai
nukleotida yang sama (Hochreiter, 2008).
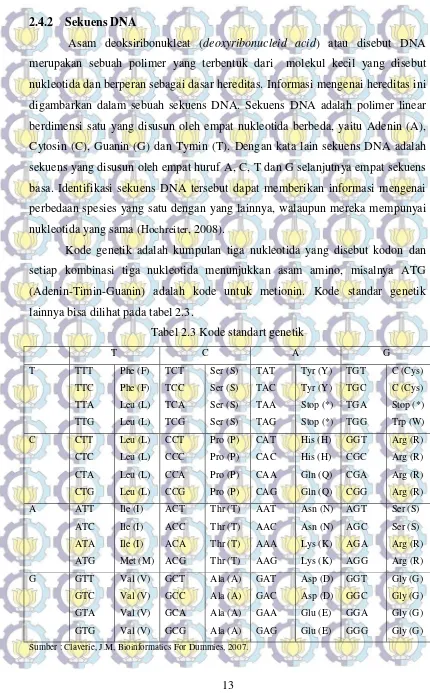
Kode genetik adalah kumpulan tiga nukleotida yang disebut kodon dan
setiap kombinasi tiga nukleotida menunjukkan asam amino, misalnya ATG
(Adenin-Timin-Guanin) adalah kode untuk metionin. Kode standar genetik
lainnya bisa dilihat pada tabel 2.3.
Tabel 2.3 Kode standart genetik
T C A G
14
2.5 Alignment
Untuk mengkonfirmasi hubungan antar mutasi, pendekatan umum adalah
untuk membandingkan perbedaan dalam keluarga sekuens (family of sequences),
yang dapat dilihat sebagai operasi dalam aritmatika. Hal ini disebut sebagai
sequences alignment atau alignment. Pensejajaran sekuens atau sequence
alignment adalah proses penyusunan atau pengaturan dua atau lebih sekuens
sehingga persamaan sekuens-sekuens tersebut tampak nyata. Kunci pensejajaran
sekuens adalah menentukan perpindahan mutasi. Jika dan adalah dua sekuens
yang didefinisikan pada persamaan 2.3. Penyisipan simbol ”–“ ke dalam dan
bertujuan untuk membentuk dua sekuens baru, yaitu dan . Selanjutnya
elemen-elemen dari dan menjadi range dari * +
* +, dengan adalah himpunan quaternary (himpunan yang terdiri dari 4 elemen) dan adalah himpunan yang terdiri dari 5 elemen.
Definisi 2. Sekuens adalah perluasan sekuens , dimana adalah sekuens
dengan penambahan gap yang diberi simbol “ –“.
Pensejajaran sekuens adalah sebuah alat penting dalam analisis posisi dan
tipe mutasi tersembunyi dalam sekuens biologi serta mengizinkan sebuah
komparasi yang tepat. Pensejajaran sekuens juga penting karena dapat
digunakan untuk penelitian penyakit genetik dan epidemi. Sebagai contoh,
adalah mungkin untuk menentukan asal, variasi, varians, difusi, dan
pengembangan epidemi dan kemudian menemukan virus dan bakteri yang
bertanggung jawab dan obat yang sesuai. Jadi pensejajaran sekuens sangat
penting dalam bidang bioinformatika dan biomedis karena berfungsi sebagai
prediktif kuat yang sangat baik. Dalam rangka untuk mendapatkan algoritma
level tinggi yang lebih baik, maka dibutuhkan teori-teori matematika (Shen dkk,
15
2.5.1 Matriks Penalti
Tujuan pensejajaran sekuens adalah untuk menemukan perluasan yang
diberikan oleh grup sehingga semua sekuens dalam memiliki tingkat
perbedaan yang lebih rendah atau tingkat kemiripan yang lebih tinggi. Dalam
bioinformatika, tingkat perbedaan biasanya diukur menggunakan matriks penalti
atau matriks penskoran. Matriks penalti dan matriks penskoran digunakan untuk
mengoptimalkan hasil pensejajaan (Shen dkk, 2008).
Matriks penalti menunjukkan tingkat perbedaan untuk tiap-tiap unit
molekul, seperti nukleotida atau asam amino, dalam sekuens biologi. Matriks
penalti dapat dinotasikan sebagai berikut :
( ))
Dalam bioinformatika, matriks penalti pada pensejajaran sekuens DNA
ditetapkan oleh matriks Hamming. Didefinisikan matriks Hamming untuk
adalah
( ) { 2.1
Contoh 2.1. Misalkan diketahui sekuens-sekuens berikut:
( )
( )
( ) ( )
( )
( )
Tentukan skor penalti minimum dari sekuens berpasangan tersebut !
Jawab :
Dapat disimpulkan bahwa B merupakan sekuens mutasi dari , dan
masing-masing dan atau dan adalah perluasan sekuens dan . Dengan
menggunakan matriks Hamming yaitu:
16 maka diperoleh
( ) ( )
Oleh karena itu, skor penalti ( ) lebih kecil dari pada skor penalti ( ).
2.5.2 Matriks Penskoran
Matriks penskoran menggunakan matriks Blosum, yang disebut “BLOSUM p” (BLOck Substitution Matrix). Matriks penskoran BLOSUM adalah langsung berasal dari blok dengan kesamaan tertentu, yaitu kesamaan sekuens
yang berbeda tidak dihitung berdasarkan model asumsi yang mungkin salah. Data
ini didasarkan pada data base blok dimana sub sekuens yang sama dikelompokkan
ke dalam blok. Disini p mengacu pada identitas % dari blok misalnya blosum 62
berasal dari blok dengan identitas 62%. Matriks skor yang paling populer untuk
pensejajaran berpasangan adalah blosum 62 matriks (Hochreiter, 2008).
Adapun perhitungan matriks blosum dengan langkah-langkah sebagai
berikut:
1. Sekuens dengan paling tidak identitas berkumpul satu sama lain.
Setiap cluster menghasilkan sekuens frekuensi (frekuensi asam amino
relatif pada setiap posisi). Sekuens frekuensi mewakili semua sekuens satu
cluster dan sekuens yang sama, yaitu tidak ada frekuensi. Frekuensi akan
ditentukan kemudian.
2. Sekuens frekuensi sekarang dibandingkan dengan satu sama lain. Pasang
asam amino dan dihitung oleh yang mana asam amino dihitung
sesuai dengan frekuensi mereka. Jika dalam kolom ada asam amino
dan asam amino maka hitungan untuk kolom memberikan
{( )
(2.2)
dengan, ( ) ( ), dimana faktor menyumbang symetri dan
17
3. Hitung ∑ dan ∑ ( ), dimana adalah
panjang sekuens dan adalah nomor pada sekuens. Sekarang adalah
dinormalisasi untuk mendapatkan probabilitas
(2.3)
Akhirnya mulai dari untuk .
4. Yang probabilitas dari kejadian asam amino adalah
∑ (2.4)
Probabilitas tidak sedang bermutasi ditambah jumlah dari pobabilitas
mutasi. adalah dibagi dengan 2 karena mutasi dari ke dan ke
dihitung menggunakan langkah 2.
5. Rasio kemungkinan dan serta rasio log-odds.
{
(2.5)
Dayhoffm memperkenalkan Percent or Point Accepted Mutation (PAM)
matrices. PAM sesuai dengan unit evolusi misalnya 1 PAM = 1 poin mutasi atau
100 asam amino dan 250 PAM = 250 poin mutasi atau 100 asam amino. Oleh
karena itu unit evolusi adalah waktu bahwa rata-rata n% mutasi terjadi pada posisi
tertentu dan bertahan. Untuk PAM 250 1/5 asam amino tetap tidak berubah. PAM
n adalah diperoleh dari PAM 1 sampai n kali perkalian matrik (Durbin dkk, 2002).
Matriks PAM adalah matriks Markov dan memiliki bentuk
[
] (2.6)
18
2.6 Algoritma Needleman Wunsch
Algoritma Needleman Wunsch merupakan algoritma global alignment
untuk sekuens yang berpasangan. Langkah-langkah dalam menjalankan Algoritma
Needleman Wunsch sebagai berikut:
1. Menyusun dua sekuens dalam tabel dua dimensi.
Jika diberikan sekuens ( ) dan
( ) maka tabel dua dimensi dari sekuens tersebut terdapat pada tabel 2.4, dengan ( ) diperoleh dari langkah selanjutnya.
Tabel 2.4 Tabel Dua Dimensi Sekuens
...
( ) ( ) ( ) ... ( )
( ) ( ) ( ) ... ( )
( ) ( ) ( ) ... ( )
... ... ... ... ... ...
( ) ( ) ( ) ... ( )
2. Menghitung elemen ( ) dari tabel dua dimensi
Masing-masing elemen ( ) yang terdapat pada Tabel Dua
Dimensi yaitu, ( ) yang ada di sisi kiri atas, ( ) yang
ada di sisi kiri dan ( ) yang ada di atas. Langkah awal yakni
menentukan skor ( ) dan skor ( ). Skor penalti pada virtual symbol
dengan elemen ( ) dapat dihitung menggunakan rumus sebagai
berikut:
( ) * ( ) ( ) ( ) ( ) +
3. Algoritma Traceback
Traceback berguna untuk menentukan backward pathway yang
selanjutnya akan digunakan untuk menentukan letak penambahan simbol virtual “–“. Metode Backward untuk mencari lintasan dan untuk mencari DNA yang optimum. Nilai akhir ( ) adalah skor maksimum dari
pensejajaran sekuens ( ) dan ( ) menjadi titik awal dan ( )
19 Kemungkinan lintasan :
1. Jika ( ) ( ) s( )
maka diagonal : ( ) ( )
2. Jika ( ) ( ) s( )
Cek nilai di samping dan di atas, pilih nilai terbesar.
(i) Atas : ( ) ( )
Maka ( ) ( )
(ii) Samping ( ) ( )
Maka ( ) ( )
Adapun penulisan hasil dari pensejajaran dengan cara sebagai berikut: 1. Jika alur mundurnya dimulai dari ke sudut kiri atas maka notasikan
pasangan dari asam nukleat .
2. Jika alur mundunya horizontal, maka sisipkan virtual symbol pada sekuens
vertikal dan notasikan sebagai ( ).
3. Jika alur mundurnya vertikal, maka sisipkan virtual symbol pada sekuens
horizontal dan notasikan ( ).
Contoh 2.2 Misalkan diketahui sekuens-sekuens berikut:
{
Diketahui: ( ) {
dan
Jawab:
t t g a
0 -8 -16 -24 -32
t -8 5 -3 -11 -19
g -16 -3 2 2 -6
a -24 -11 -6 -1 7
a -32 -19 -14 -9 4
g -40 -27 -22 -9 -4
Hasil pensejajaran:
20
Berikut perhitungan untuk mengisi baris dan kolom pada tabel di atas
( )
( )
( )
( )
( )
( )
( )
( ) { ( ) ( ) ( ) ( ) }
( ) * ( ) ( ) ( ) ( )
* +
* +
( ) * ( ) ( ) ( ) ( )
* ( ) +
* +
( ) * ( ) ( ) ( ) ( )
* ( ) +
* +
( ) * ( ) ( ) ( ) ( )
* +
* +
21
( ) * ( ) ( ) ( ) ( )
* ( ) +
* +
( ) * ( ) ( ) ( ) ( )
* ( ) +
* +
( ) * ( ) ( ) ( ) ( )
* ( ) +
* +
( ) * ( ) ( ) ( ) ( )
* +
* +
( ) * ( ) ( ) ( ) ( )
* +
* +
( ) * ( ) ( ) ( ) ( )
* ( ) +
* +
( ) * ( ) ( ) ( ) ( )
* ( ) +
* +
22
( ) * ( ) ( ) ( ) ( )
* ( ) +
* +
Berikut perhitungan untuk mencari lintasan menggunakan Metode Backward.
( ) ( ) ( )
( )
Cek nilai di samping dan di atas. Pilih nilai paling besar, terdapat
maka ( ) ( )
( ) ( ) ( )
maka ( ) ( )
( ) ( ) ( )
( ) maka ( ) ( )
( ) ( ) ( )
( ) maka ( ) ( )
( ) ( ) ( )
maka ( ) ( )
Algoritma Nedleman Wunsch merupakan metode yang digunakan untuk
mendapatkan sekuens berpasangan menjadi sejajar atau diperoleh panjang sama.
Yang mana didalam algoritma itu terdapat perhitungan dalam entri baris dan
kolom yang terdapat pada tabel dua dimensi. Jika terdapat nilai yang sama dalam
baris dan kolom, misal pada contoh 2.2 pada ( ) dan ( ) diperoleh nilai
yaitu 2, maka lihat aturan algoritma traceback. Pada ( ) dalam menentukan
23
( ) ( ) ( ) ( )
Maka arah lintasannya diagonal, sehingga dipilih ( ).
Jika dalam perhitungan untuk entri baris dan kolom terdapat skor yang
sama, pilih salah satu nilai yang maksimal. Sebagai gambaran perhitungan pada
contoh 2.2 dapat dilihat untuk entri ( ) dihitung sebagai berikut
( ) * ( ) ( ) ( ) +
* +
* +
2.7 Multiple Alignment
Multiple alignment (MA), yaitu pensejajaran beberapa sekuens sekaligus.
MA adalah kunci utama dalam bidang bioinformatika. Contohnya, untuk
mempelajari evolusi biologis, para peneliti menganalisa perubahan stuktur
berdasarkan MA khusus sekuens DNA atau protein. Untuk mempelajari genome
virus, MA juga digunakan untuk mendapatkan proses evolusi dari virus spesifik.
Biasanya untuk sebuah MA melibatkan ratusan sekuens yang mana tedapat
ratusan juta panjang pasangan basa. Diberikan multiple sekuens untuk
mendapatkan MA, pertama kali harus dikonstruksi sebuah algoritma dengan
terlebih dahulu memformulasi prinsip-prinsip komputasi (Shen dkk, 2008).
Hasil dari MA berupa suatu himpunan sekuens yang panjangnya sama.
MA dapat menunjukkan multiple sequence berada pada keluarga yang sama atau
tidak. Selain itu, MA dapat menunjukkan semua hubungan atau relasi antar famili
dari multiple sequence yang ada. Berdasarkan pembagian keluarga, dapat
ditentukan keadaan evolusi masing-masing sekuens dalam keluarga. Secara umum
digunakan pohon topologi untuk menggambarkan hubungan di antara multiple
sequence, pohon topologi tersebut selanjutnya dikenal dengan pohon filogenetik
24
Studi tentang MA berkembang kedua arah. Yang pertama membahas
kompleksitas komputasi untuk solusi dengan pinalti minimum, yang mana banyak
publikasi mempertimbangkan pada masalah yang sangat sulit. Karena itu, adalah
sulit untuk mencapai MA dengan penalti minimum secara teori. Masalah MA
menjadi masalah dari kompleksitas komputasi (Shen dkk, 2008).
Sebagai contoh MA, dengan menggunakan tiga sekuens yang disejajarkan
yaitu = VIVALASVEGAS, = VIVADAVIS dan = VIVADALLAS. MA
dari tiga sekuens tersebut ditunjukkan oleh yang terlihat seperti berikut.
Ketika dilakukan untuk sekuens terkait, MA dapat membantu para peneliti
mengidentifikasi domain dan daerah lainnya yang menarik. Selain itu bisa dengan
mudah beradaptasi definisi pensejajaran berpasangan (pairwise alignment) untuk
menutupi kasus ini (Cristianini dkk, 2006). Masalah MA dipecahkan dengan
menggunakan beberapa metode yang berbeda , seperti classical, progressive, dan
iterative algorithms.
2.7.1 Metode Progressive
Adanya hubungan timbal balik antara pensejajaran dan hubungan
filogenetik antar sekuens di dalamnya memunculkan ide bahwa suatu pensejajaran
yang baik dapat dibuat berdasarkan hubungan filogenetiknya dalam bentuk sebuah
pohon. Namun demikian, hasil pensejajaran sekaligus juga pohon filogenetik dari
suatu sekuens yang belum disejajarkan merupakan hal yang rumit. Pendekatannya
adalah dengan menghasilkan suatu alignment sementara lalu membuat pohon dari
pensejajaran sementara tersebut, kemudian mengoptimasi pensejajaran tersebut
berdasarkan informasi kekerabatan antar sekuens yang terdapat dalam pohon
(Naznin dkk, 2012).
Metode progressive menghasilkan Multiple Alignment dari sejumlah
pensejajaran secara berpasangan (Pairwase Alignment). Metode Progressive
25
menurut skor untuk berpasangan mereka, sehingga biasanya sekuens yang
mensejajarkan terbaik ditambahkan ke pelurusan pertama (Mojbak dkk, 2010).
Langkah-langkah pensejajaran sekuens menggunakan Metode Progressive
sebagai berikut.
1. Melakukan pensejajaran berpasangan untuk setiap pasang sekuens.
2. Membentuk matriks jarak dari hasil pensejajaran dari setiap pasang
sekuens. Entri dalam matriks jarak adalah beda hasil pensejajaran
pasangan sekuens.
3. Membangun pohon filogentik dari pensejajaran dengan jarak evolusi.
4. Hasil pensejajaran diperoleh melalui pohon filogenetik yang telah
dikontruksi.
Metode Progressive menggunakan Metode Dinamic Programming untuk
membentuk pensejajaran sekuens secara keseluruhan dimulai dengan sekuens
paling terkait atau kelompok sekuens ke pensejajaran awal (Ulum dkk, 2013).
2.8 Filogenetik Molekuler
Filogenetik molekuler adalah ilmu yang mempelajari hubungan
evolusioner antara organisme, gen, atau protein, menggunakan kombinasi biologi
molekuler dan teknik statistik. Hubungan filogenetik biasanya digambarkan dalam
bentuk pohon biner. Struktur pohon menggambarkan kemungkinan hubungan
keturunan leluhur antara varian diketahui yang ada di masa lalu, dimana
leluhurnya masih mempunyai hubungan kekerabatan dengan varian sekarang atau
node eksternal (Polanski dkk, 2007).
2.8.1 Pohon
Sebagaimana dinyatakan pada sub bab 2.5, hubungan filogenetik dapat
direpresentasikan dalam bentuk pohon, biasanya diposisikan terbalik.
Pengamatan, biasanya dalam bentuk sekuens, hanya tersedia di bawah pohon.
Tugas molekuler filogenetik adalah untuk menemukan struktur (topologi) dari
pohon, dan panjang cabang, yang mewakili struktur keterkaitan dari sekuens yang
26
Sebuah pohon adalah sebuah grafik yang terdiri dari node dan cabang, di
mana dua node yang terhubung oleh jalan yang unik. Sebuah pohon biner adalah
pohon dengan cabang diarahkan, sehingga masing-masing node memiliki lebih
dari dua keturunan. Sebuah pohon filogenetik adalah pohon yang node dan cabang
memiliki interpretasi sebagai spesies atau sekuens molekul dan hubungan di
antara mereka (Polanski dkk, 2007).
Dalam pohon terdapat istilah-istilah yang digunakan, adapun penjelasan
mengenai istilah-istilah yang ada hubungannya dengan pohon sebagai berikut:
a. Node
Node dalam pohon filogenetik disebut unit taksonomi. biasanya unit
taksonomi diwakili oleh sekuens (DNA atau RNA, nukleotida atau asam amino).
Sekuens tersebut sesuai dengan spesies atau individu dalam populasi yang
baisanya diwakili oleh parameter yang menggambarkan individu, seperti panjang,
sudut, atau warna.
b. Cabang
Cabang di pohon filogenetik menunjukkan keturunan atau hubungan
keturunan antar node.
c. Node terminal.
Node terminal juga disebut node eksternal, daun, atau ujung pohon. Untuk
pohon filogenetik, nama-nama node terminal unit taksonomi adalah unit
taksonomi yang masih ada atau unit taksonomi operasional.
d. Akar
Akar adalah node asal atau nenek moyang dari semua node.
e. Pohon berakar atau pohon tidak berakar.
Pada Gambar. 2.1, disajikan contoh pohon berakar dibandingkan pohon
tidak berakar untuk set yang sama node yang masih ada A, B, C, D, E. Dalam
pohon berakar, arah jalur evolusi (waktu) selalu ditentukan. Dalam pohon tidak
berakar, node yang masih ada secara unik ditentukan tetapi ada banyak jalur
27
(a) (b)
Gambar 2.1 (a) Pohon berakar dan (b) Pohon tidak berakar
f. Topologi
Topologi adalah pola percabangan pohon. Jumlah kemungkinan
topologi pada umumnya sangat besar. Jika jumlah Tus yang masih ada
adalah n, jumlah pohon tidak berakar berlabel berbeda adalah
( )
( ) (2.2)
dan jumlah pohon berakar berlabel berbeda adalah
( )
( ) (2.3)
Ekspresi di atas dapat diturunkan menggunakan prosedur iterasi dengan
menambahkan cabang pohon yang ada.
g. Panjang cabang
Panjang dari cabang menentukan matriks dari pohon. Pada pohon
filogenetik, panjang cabang diukur dalam satuan evolusi
waktu. Berlalunya waktu evolusi menghasilkan akumulasi perubahan
evolusioner. Karena itu, ketika menyimpulkan matriks dari pohon filogenetik,
jumlah dari evolusi berubah diantara spesies estimator dari panjang cabang.
Sebuah pohon berakar merupakan evolusi diarahkan dari nenek
moyang ke semua node terminal. Node internal lainnya dari
pohon merupakan nenek moyang kelompok tertentu dari Otus. Dengan
menghapus akar dari pohon berakar dan bergabung dengan dua cabang turun
dari akar menjadi cabang tunggal, satu memperoleh pohon tidak berakar.
Pohon tersebut dilakukan tidak mengandung informasi tentang arah evolusi akar
A B C D E
A B
C
D
28
dan menentukan hanya hubungan evolusi antara Otus. Gambar 2.2
menunjukkan pohon berakar dengan empat spesies mamalia dan gambar 2.3
menunjukkan pohon tidak berakar dengan empat spesies mamalia.
Gambar 2.2 Pohon berakar dengan empat spesies mamalia
Gambar 2.3 Pohon tidak berakar dengan empat spesies mamalia
Dua representasi yang diberikan untuk setiap pohon. Perhatikan bahwa
dua intern node dari pohon berakar mewakili masing-masing nenek moyang dari
kelompok {lumba-lumba, paus, babi} dan nenek moyang dari kelompok yang
lebih kecil {Lumba-lumba dan paus}. Pohon yang dijelaskan dalam gambar 2.2
dan gambar 2.3 disebut sebagai pohon filogenetik pohon. Setiap pohon filogenetik
yang daunnya diberi label oleh Otu tertentu, dikatakan untuk menghubungkan
Otu.
Panjang masing-masing cabang pohon adalah angka positif yang
merupakan tingkat keterkaitan antara spesies atau urutan yang sesuai ke kelenjar
di titik akhir dari cabang dan sering dihitung sebagai produk dari panjang interval
waktu yang secara historis memisahkan spesies atau urutan dan nilai tertentu dari
tingkat evolusi, yang mencoba untuk memperhitungkan fakta bahwa beberapa Horse
Ping
Dolphin Whale
Whale Horse
Ping
Dolphin
𝑥 Pin g Root
Horse Dolphin Whale Ping
Root
Horse
Dolphin
29
spesies atau gen berevolusi lebih cepat dari pada yang lain. Pada gambar. 2.2
hanya memberikan pola percabangan yang benar, tidak ada panjang cabang.
Panjang cabang sering ditampilkan sebagai label di sebelah cabang yang sesuai.
Pohon yang terdapat panjang pohon dapat dilihat pada gambar 2.4.
Gambar 2.4. Pohon dengan panjang pohon
2.8.2 Pohon Filogenetik (Phylogenetic tree)
Pohon filogenetik atau pohon evolusi adalah grafik tanpa siklus atau
pohon yang menunjukkan hubungan evolusi di antara berbagai spesies biologi
berdasarkan kedekatan genetik berbagai spesies (Ruzgar dkk, 2011). Tujuan dari
filogeni adalah untuk merekonstruksi sejarah kehidupan dan menjelaskan
keanekaragaman makhluk hidup saat ini. Hal ini dapat direpresentasikan sebagai
pohon genealogis besar (pohon kehidupan). Prinsip yang mendasari filogeni
adalah mencoba untuk mengelompokkan makhluk hidup sesuai dengan tingkat
kemiripan. Dalam konteks ini, asumsikan bahwa dua spesies yang lebih serupa
(seperti manusia dan kera), semakin dekat kekerabatan mereka dengan nenek
moyang mereka. Filogenetik merupakan jenis khusus dari filogeni yang
bergantung pada perbandingan gen yang berasal dari beberapa spesies untuk
merekonstruksi pohon genealogis pada spesies ini dan mencari tahu siapa kerabat
terdekat misalnya dalam keluarga (Claverie dkk, 2007).
Untuk menggambarkan hubungan evolusi antara gen dan organisme
dalam suatu hubungan kekerabatan yang erat dengan menggunakan pohon
𝑥
𝑥6
𝑥
𝑥
𝑥
𝑥 2 2
2 1
3 5
5
30
filogenetik. Disebut pohon filogenetik karena bentuknya menyerupai struktur
pohon. Istilah yang digunakan pada pohon filogenetik merujuk ke berbagai bagian
dari pohon (misalnya akar, cabang, node dan daun). Node eksternal atau daun
merepresentasikan taxa atau disebut OTUs (Operational Taxonomic Units), istilah
tersebut juga mewakili berbagai jenis taxa yang sebanding. Sebagai contoh,
sebuah keluarga organisme, individu atau strain virus dari satu spesies atau dari
spesies yang berbeda. Node internal atau disebut HTU (Hipothetical Taxonomic
Units) menekankan bahwa mereka adalah leluhur hipotesis OTUs. Sebuah cluster
merupakan sekelompok taxa yang berbagi cabang yang sama memiliki asal
monofiletik (Lemey dkk, 2009).
akar
(a) (b)
Gambar 2.5 (a) Pohon filogenetik berakar dan (b) Pohon filogenetik tidak berakar
Pada gambar 2.5 kedua pohon memiliki topologi yang sama. Pada gambar
di atas, Taxa A, B dan C membentuk cluster, memiliki leluhur bersama H, karena
asalnya monofiletik. Sedangkan C, D dan E tidak membentuk cluster tanpa
memasukkan strain tambahan dan tidak berasal dari monofiletik disebut
paraphyletic. Percabangan pola disebut sebagai topologi pohon. Pada pohon
berakar yang ditunjukkan pada gambar 2.5 (a), sebagai node internal atau OTU
yaitu A, B, C, D, dan F. Sedangkan node internal atau HTU yaitu G, H, I, J dan K,
31
K ke node eksternal D). Pada pohon tidak berakar yang ditunjukkan pada gambar
2.5 (b) tidak memiliki simpul akar, hanya garis antara node cabang. Sebuah pohon
tidak berakar hanya memposisikan sekelompok individu tanpa menunjukkan arah
proses evolusi. Dalam sebuah pohon tidak berakar, tidak ada indikasi yang
mewakili nenek moyang dari semua OTU (Lemey dkk, 2009).
(a) (b)
Gambar 2.6 Struktur dari pohon Filogenetik berakar
Pada gambar 2.6 menunjukkan pohon yang sama seperti pada gambar 2.5
tetapi dalam bentuk yang berbeda. Pada gambar 2.6, kedua gambar memiliki
topologi yang identik. Cabang diinternal node dapat diputar tanpa merubah
topologi pohon (Lemey dkk, 2009).
2.8.3 Metode-metode Pembentukan Pohon
Pohon filogenetik dapat dibentuk atas dasar pendekatan yang sangat
berbeda, yang mungkin dibagi menjadi metode berorientasi data dan metode
berorientasi model. Contoh metode berorientasi data adalah metode berbasis jarak.
Metode jarak, pohon yang dibangun dengan menggabungkan sekuens dengan
jarak kecil di antara mereka. Contoh lain adalah Metode Maximum Parsimony,
dengan Metode Maximum Parsimony pohon yang dibentuk menjelaskan data
yang diamati menggunakan nilai terkecil. Tidak ada diasumsikan model evolusi
dalam Metode Jarak dan Metode Maximium Parsimony. Mungkin ini menjadi
alasan mengapa pendekatan berorientasi data lebih menarik bagi ahli biologi dan
32
Pendekatan model antara lain, Metode Maximum Likelihood dan metode
berdasarkan Coalescent tersebut. Dalam Metode Maximum Likelihood, model
probabilistik evolusi diasumsikan dan cocok untuk sekuens data untuk
memaksimalkan kemungkinan semua pohon. Menghitung likelihood adalah
komputasi secara intensif, tetapi metode ini dapat dilakukan dengan beberapa
cara, termasuk evolusi di bawah tekanan selektif, yang mungkin membantu dalam
identifikasi protein aktif (Polanski dkk, 2007).
Menerapkan metode filogenetik untuk berbagai gen dari gen keluarga
untuk merekonstruksi sejarah keluarga dengan cara yang sama. Menurut Shen
(Shen dkk, 2008) metode-metode untuk membangun pohon filogenetik adalah
sebagai berikut:
1. Metode Berbasis Jarak
Setiap hasil alignment dapat digunakan untuk menghitung matriks
jarak antar sekuens. Bedasarkan pada matriks jarak, akan dapat dihasilkan
pohon filogenetik yang sesuai. Metode yang paling populer disebut
UPGMA (Unweighted Pair Group Method with Aritmatic) dan
Neighbor-Joining.
2. Metode Berbasis Fitur
Metode jenis ini menggunakan fitur (karakteristik) dari output
alignment untuk membangun pohon filogenetik. Metode berbasis fitur
yang digunakan dalam filogenetik adalah Metode Maximum Parsimony.
Penentuan pohon dengan tree length terkecil tidak dilakukan berdasarkan
matriks distance seperti pada ME. Perhitungan branch length dan tree
length pada metode MP didapatkan dari jumlah substitusi minimum antar
character state setiap situs pada sequence alignment.
3. Metode Berbasis Probabilitas
Penggunaan metode berbasis probabilitas ini untuk membangun
pohon filogenetik dimulai dengan membangun suatu model probabilitas
untuk mutasi sekuens, kemudian membangun pohon flogeetik didasarkan
33
2.9 Metode Berbasis Jarak (Distance Based Methods)
Metode jarak adalah salah satu metode pembentukan pohon filogenetik
dari sekumpulan jarak antar setiap pasangan sekuens yang telah disejajarkan.
Sekumpulan jarak tersebut dituliskan dalam bentuk matriks yang disebut matriks
jarak (Isaev, 2007). Adapun bentuk matriks jarak dapat dilihat pada contoh 3.
Contoh 2.3 : Diberikan N = 5 dan diberikan matriks jarak sebagai beikut.
0 11 8 9 8
11 0 13 14 13
8 13 0 9 8
9 14 9 0 9
8 13 8 9 0
Pada contoh 2.3 menunjukkan matriks jarak dari lima sekuens (OTU)
dengan himpunan sekuens * +. Setiap elemen matriks tersebut
merepresentasikan jarak genetik antar sekuens yang terlibat. Misalnya, jarak
antara OTU dan adalah 8. Angka tersebut menyatakan perbedaan genetik
sekuens dan sebesar 8 satuan. Perbedaan tersebut terjadi karena proses
evolusi yang terjadi didalam struktur genetiknya. Angka-angka tersebut bisa
dikatakan sebagai waktu evolusi atau perbedaan banyaknya gen akibat evolusi.
Terdapat asumsi bahwa diberi matriks jarak berpasangan antara sekuens.
Misal adalah sebuah himpunan dan adalah sebuah fungsi,
dikatakan sebagai distance function atau fungsi jarak pada jika
( ) untuk setiap 2.5
( ) untuk 2.6
( ) ( ) untuk setiap 2.7
Memenuhi ketidaksamaan segitiga ( ) ( ) ( ) untuk setiap
2.8
Jika adalah distance function atau fungsi jarak pada , maka untuk
34
dengan menetapkan ( ) untuk semua , dan ( )
untuk semua , tetapi fungsi jarak ini sangat tidak informatif.
Himpunan yang dipakai disini adalah himpunan berhingga
* + yang merupakan himpunan sekuens (OTU) yang akan dibentuk pohon filogenetik-nya. Diasumsikan bahwa fungsi jarak terdefinisi di dan relevan
secara biologi, maksudnya adalah sesuai dengan informasi genetik yang ada pada
sekuens di . Sebagai contoh ( ) ( ) berarti OTU dan lebih
jauh hubungan evolusi atau kekerabatannya dibanding OTU dan . Untuk
menyederhanakan penulisan, ( ) ditulis sebagai dengan * +.
Berdasarkan fungsi jarak tersebut dapat diperoleh matriks jarak (distance matrix)
( ) dengan definisi formal sebagai berikut.
Definisi 3. Misalkan adalah suatu fungsi jarak, disebut sebagai matriks
jarak yang didefinisikan oleh
[
]
dengan dan n adalah jumlah OTU yang terlibat (Isaev, 2007).
Pengelompokan program menghasilkan sebuah pensejajaran dan pohon
dari set sekuens protein. Metode jarak bekerja pada jumlah perubahan diantara
masing-masing pasangan dalam kelompok untuk mengkonstruksi pohon
filogenetik dalam kelompok. Pasangan sekuens yang mempunyai jumlah
perubahan terkecil diantara mereka disebut neighbors. Dalam pohon filogenetik,
sekuens-sekuens ini menggunakan secara bersama-sama satu titik dan
masing-masing dihubungkan titik oleh sebuah cabang. Tujuan dari metode jarak adalah
untuk mengidentifikasi pohon pada posisi neighbors dengan benar, dan juga
mempunyai cabang yang menghasilkan data dengan jarak sedekat mungkin.
35
penemuan neighbors terdekat diantara kelompok sekuens dengan metode jarak
(Feng dkk, 1996).
2.10Metode UPGMA
Metode UPGMA (Unwight Pair Group Method with Arithmetic Average)
adalah metode untuk konstruksi pohon yang mengasumsikan rata-rata perubahan
sepanjang pohon adalah konstan dan jaraknya kira-kira ultrameric (ultrameric
biasanya diekspresikan sebagai molecular clock tree). Metode UPGMA dimulai
dengan kalkulasi panjang cabang diantara sekuen paling dekat yang saling
berhubungan, kemudian rata-rata jarak antara sekuens ini atau kelompok sekuens
dan sekuens berikutnya atau kelompok sekuens dan berlanjut sampai semua
sekuens yang termasuk dalam pohon. Akhirnya metode ini memprediksi posisi
root dari pohon (Shen dkk, 2008).
Metode UPGMA adalah metode paling sederhana dari semua metode
clustering yang digunakan untuk membentuk pohon filogenetik. Metode ini
membutuhkan kecepatan substitusi dari nukleotida atau asam amino menjadi
seragam dan tidak berubah melalui seluruh proses evolusi. Dengan kata lain,
memenuhi hipotesis mengukur waktu molekuler. Pada setiap node induk, panjang
cabang dari node induk ke dua simpul anak adalah sama (Isaev, 2007).
Metode UPGMA mengasumsikan sebuah molecular clock dan rooted tree.
Metode ini secara normal menghitung skor similaritas yang didefinisikan sebagai
jumlah total dari jumlah sekuens yang identik dan jumlah substitusi konservatif
dalam pensejajaran dua sekuens dengan gap yang diabaikan. Skor identitas antara
sekuens menunjukkan hanya identitas yang mungkin ditemukan dalam
pensejajaran. Untuk analisis filogenetik digunakan skor jarak antara dua sekuens.
Skor diantara dua sekuens adalah jumlah posisi yang tidak cocok (mismatch)
dalam pensejajaran atau jumlah posisi sekuen yang harus diubah untuk
menghasilkan sekuens yang lain. Gap mungkin diabaikan dalam kalkulasi atau
diberi perlakuan seperti substitusi. Ketika sebuah skoring atau matriks substitusi
digunakan, kalkulasi menjadi lebih komplek tetapi secara prinsip tetap sama (Shen