Title:
Ethylene Coordination, Insertion and Chain Transfer at a Cationic Aluminum Center: a Comparative Study
with
Ab Initio
Correlated Level and Density Functional Methods
Author:
Giovanni Talarico*, Peter H. M. Budzelaar and Anton W. Gal
Affiliation:
Department of Inorganic Chemistry, University of Nijmegen, Toernooiveld 1, 6525 ED Nijmegen, The
Netherlands.
Correspondence
:
G. Talarico (current address) Dipartimento di Chimica, Università di Napoli, Via Mezzocannone 4, 80134,
Naples, Italy. e-mail: [email protected]
Description:
[image:1.595.57.515.328.786.2]Tables of cartesian coordinates optimized at MP2 level and the total energies computed at CCSD(T) level of all
species mentioned in the text and influence of basis sets (Tables S-I, S-II, S-III, and S-IV).
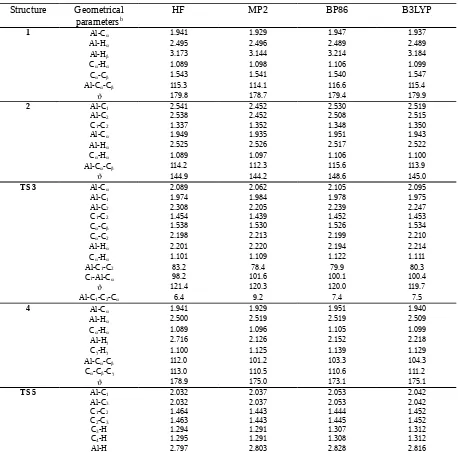
Table S-I.
Selected geometrical parameters
afor structures
1-10
calculated with different theoretical methods
Structure
Geometrical
parameters
bHF
MP2
BP86
B3LYP
1 Al-C 1.941 1.929 1.947 1.937
Al-H 2.495 2.496 2.489 2.489
Al-H 3.173 3.144 3.214 3.184
C-H 1.089 1.098 1.106 1.099
C-C 1.543 1.541 1.540 1.547
Al-C-C 115.3 114.1 116.6 115.4
179.8 178.7 179.4 179.9
2 Al-C1 2.541 2.452 2.530 2.519
Al-C2 2.538 2.452 2.508 2.515
C1-C2 1.337 1.352 1.348 1.350
Al-C 1.949 1.935 1.951 1.943
Al-H 2.525 2.526 2.517 2.522
C-H 1.089 1.097 1.106 1.100
Al-C-C 114.2 112.3 115.6 113.9
144.9 144.2 148.6 145.0
TS 3 Al-C 2.089 2.062 2.105 2.095
Al-C1 1.974 1.984 1.978 1.975
Al-C2 2.308 2.205 2.239 2.247
C1-C2 1.454 1.439 1.452 1.453
C-C 1.538 1.530 1.526 1.534
C-C2 2.198 2.213 2.199 2.210
Al-H 2.201 2.220 2.194 2.214
C-H 1.101 1.109 1.122 1.111
Al-C1-C2 83.2 78.4 79.9 80.3
C1-Al-C 98.2 101.6 100.1 100.4
121.4 120.3 120.0 119.7
Al-C1-C2-C 6.4 9.2 7.4 7.5
4 Al-C 1.941 1.929 1.951 1.940
C1-Al-C3 104.8 103.4 103.3 104.3
C2-H-C4 168.4 168.3 167.5 167.4
180.0 180.0 179.9 180.0
TS 6 Al-C 2.088 2.055 2.083 2.079
Al-C1 2.018 2.014 2.024 2.013
C1-C2 1.342 1.356 1.353 1.356
C-C 1.540 1.540 1.537 1.543
C-H 1.525 1.528 1.557 1.552
C1-H 1.458 1.452 1.461 1.453
Al-H 1.686 1.675 1.693 1.687
Al-H 2.373 2.419 2.400 2.380
C-H 1.096 1.104 1.112 1.106
C1AlC 91.4 92.1 92.5 92.4
174.9 176.0 177.6 176.0
C-Al-C1-H 2.2 3.9 1.0 1.6
TS 7 Al-H 1.662 1.654 1.663 1.661
Al-C 2.014 2.020 2.038 2.023
Al-C 2.318 2.233 2.242 2.261
C-C 1.435 1.421 1.423 1.431
C-H 1.737 1.727 1.745 1.742
Al-C-C 82.7 78.8 78.6 79.8
C-Al-H 86.2 88.7 88.9 88.4
Al-H-C 86.0 82.6 82.3 83.2
129.6 129.7 130.4 128.3
Al-C-C-H 0.0 0.0 0.0 0.0
8 Al-H 1.553 1.559 1.565 1.558
Al-C1 2.500 2.418 2.480 2.468
Al-C2 2.494 2.420 2.476 2.467
C1-C2 1.338 1.354 1.350 1.352
144.2 145.0 148.4 144.7
9 Al-H 1.548 1.561 1.566 1.557
Al-C1 2.848 2.638 2.721 2.731
Al-C2 2.847 2.639 2.724 2.731
C1-C2 1.329 1.348 1.342 1.344
C3-C4 1.329 1.349 1.343 1.345
179.9 180.0 180.0 180.0
10 Al-H 1.543 1.552 1.559 1.552
Al-N 1.864 1.880 1.901 1.880
N-C 1.319 1.335 1.334 1.335
N-Al-H 144.5 144.0 144.6 144.0
N-Al-N 71.1 71.7 70.7 71.6
N-C-N 110.6 111.1 111.1 111.0
179.9 179.8 179.9 179.9
a
In Å and deg.
bThe atom numbering scheme is H
2
C
1=CH
22for the olefin and Al-C-C-C for the growing chain.
Table S-II.
Basis set influence (at the MP2 level) of olefin coordination and insertion; energies relative to
-complex [HC(NH)
2Al CH
2CH
3(CH
2CH
2)]
+(kcal/mol).
Method
E
BSSEReactant
1
a(complexation)
(Insertion)
TS
3
Product
4
6-31G (d)
6.1
18.9
30.6
-9.0
6-31G (d,p)
6.0
18.6
30.2
-9.7
[image:2.595.60.508.630.754.2]Table S-III.
Basis set influence (at the MP2 level) on the energies of
-hydrogen transfer to the metal (BHE)
relative to precursor [HC(NH)
2AlCH
2CH
3]
+(kcal/mol).
Method
TS
7
(BHE)
Structure
8
Structure
9
a
E
BSSEb
Structure
10
aE
BSSEc6-31G (d)
39.2
14.1
9.9
5.2
36.9
5.8
6-31G (d,p)
39.8
15.5
10.9
5.1
38.1
5.7
6-311G (d,p)
38.7
16.2
11.2
4.1
38.1
4.1
6-311G (2d,p)
38.7
16.0
8.8
3.3
39.3
3.2
a
Corrected for BSSE;
brelative to
9
;
crelative to
8
.
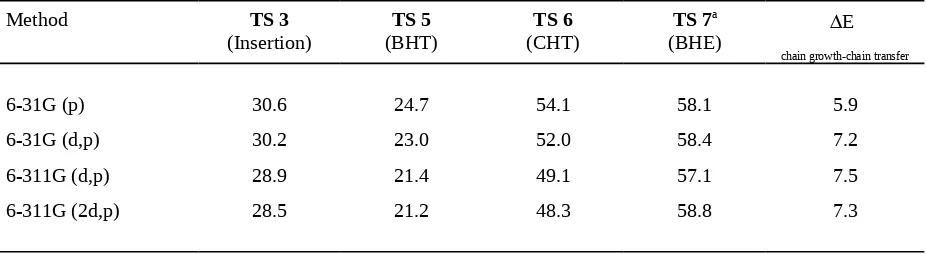
Table S-IV.
Basis set influence (at the MP2 level)on the energies of different reactions relative to
[HC(NH)
2AlCH
2CH
3(CH
2CH
2)]
+in kcal/mol.
Method
TS
3
(Insertion)
TS
5
(BHT)
TS
6
(CHT)
TS 7
a(BHE)
E
chain growth-chain transfer
6-31G (p)
30.6
24.7
54.1
58.1
5.9
6-31G (d,p)
30.2
23.0
52.0
58.4
7.2
6-311G (d,p)
28.9
21.4
49.1
57.1
7.5
6-311G (2d,p)
28.5
21.2
48.3
58.8
7.3
[image:3.595.54.517.474.601.2]Tables of cartesian coordinates optimized at MP2 level and the total energies computed at
CCSD(T) level of all species mentioned in the text.
Ethylene
Z-MATRIX (ANGSTROMS AND DEGREES)
CD Cent Atom N1 Length/X N2 Alpha/Y N3 Beta/Z J 1 1 C
2 2 C 1 1.334903( 1)
3 3 H 1 1.084765( 2) 2 121.704( 6)
4 4 H 1 1.084765( 3) 2 121.704( 7) 3 180.000( 10) 0 5 5 H 2 1.084765( 4) 1 121.704( 8) 3 180.000( 11) 0 6 6 H 2 1.084765( 5) 1 121.704( 9) 5 180.000( 12) 0 ---Standard orientation:
Center Atomic Coordinates (Angstroms) Number Number X Y Z 1 6 0.000000 0.000000 0.667451 2 6 0.000000 0.000000 -0.667451 3 1 0.000000 0.922890 1.237530 4 1 0.000000 -0.922890 1.237530 5 1 0.000000 -0.922890 -1.237530 6 1 0.000000 0.922890 -1.237530
DD1Dir will call FoFMem 1 times, MxPair= 42 NAB= 21 NAA= 0 NBB= 0.
The Euclidean norm of the A-vectors is 0.3131148D-05
DE(CORR)= -0.28156041D+00 E(CORR)= -0.78312630072D+02 NORM(A)= 0.10617637D+01
Dominant configurations: ***********************
Spin Case I J A B Value ABAB 8 8 9 9 -0.146796D+00 Largest amplitude= 1.47D-01
T4(AAA)= -0.16102092D-03 T4(AAB)= -0.47131497D-02 T5(AAA)= 0.12209276D-04 T5(AAB)= 0.23060927D-03
Time for triples= 16.96 seconds. T4(CCSD)= -0.97483413D-02
Reactant
1
Z-MATRIX (ANGSTROMS AND DEGREES)
CD Cent Atom N1 Length/X N2 Alpha/Y N3 Beta/Z J 1 1 Al
2 2 N 1 1.891674( 1)
3 3 H 2 1.013008( 2) 1 149.717( 14)
4 4 C 2 1.333918( 3) 3 121.463( 15) 1 179.358( 26) 0 5 5 H 4 1.090398( 4) 2 124.369( 16) 1 180.240( 27) 0 6 6 N 4 1.333545( 5) 2 111.243( 17) 3 -179.616( 28) 0 7 7 H 6 1.013044( 6) 4 121.543( 18) 2 180.161( 29) 0 8 8 C 1 1.928967( 7) 2 143.206( 19) 4 179.129( 30) 0 9 9 C 8 1.540758( 8) 1 114.093( 20) 2 -7.053( 31) 0 10 10 H 8 1.097322( 9) 9 110.571( 21) 1 121.658( 32) 0 11 11 H 8 1.097400( 10) 9 110.557( 22) 1 -121.889( 33) 0 12 12 H 9 1.092452( 11) 8 112.066( 23) 11 177.226( 34) 0 13 13 H 9 1.092423( 12) 8 112.058( 24) 10 -177.498( 35) 0 14 14 H 9 1.091995( 13) 8 109.686( 25) 13 119.138( 36) 0 ---Standard orientation:
Center Atomic Coordinates (Angstroms) Number Number X Y Z 1 13 0.002342 -0.259330 -0.030184 2 7 1.231135 1.178874 -0.038359 3 1 1.411494 2.175191 -0.070077 4 6 2.226355 0.292739 0.021973 5 1 3.284101 0.556042 0.050522 6 7 1.760208 -0.956435 0.046732 7 1 2.383295 -1.753758 0.094661 8 6 -1.879162 -0.683686 -0.058124 9 6 -2.802591 0.543194 0.068293 10 1 -2.073454 -1.397632 0.752214 11 1 -2.088957 -1.229839 -0.986558 12 1 -2.647926 1.078169 1.008149 13 1 -2.659933 1.250301 -0.752094 14 1 -3.846079 0.222258 0.044106
DD1Dir will call FoFDir 1 times, MxPair= 272 NAB= 136 NAA= 0 NBB= 0.
Spin AB IR= 101 I= 9 J= 9 Mu= 42 Nu= 42 IOp=1 MuP= 42 NuP= 42 FactIJ= 0.0 Fact= 0.0 A=-3.5498618990D-01 AP=-3.5498618990D-01
The Euclidean norm of the A-vectors is 0.5444416D-05
DE(CORR)= -0.79899121D+00 E(CORR)= -0.46970357664D+03 NORM(A)= 0.11463756D+01
Largest amplitude= 6.48D-02 T4(AAA)= -0.67798456D-03 T4(AAB)= -0.13157990D-01 T5(AAA)= 0.46312804D-04 T5(AAB)= 0.73750443D-03
Time for triples= 5294.96 seconds. T4(CCSD)= -0.27671948D-01
-complex
2
Z-MATRIX (ANGSTROMS AND DEGREES)
CD Cent Atom N1 Length/X N2 Alpha/Y N3 Beta/Z J 1 1 Al
2 2 C 1 1.934515( 1)
3 3 H 2 1.097569( 2) 1 109.748( 22)
4 4 H 2 1.097586( 3) 1 109.705( 23) 3 -115.453( 42) 0 5 5 C 2 1.541156( 4) 1 112.348( 24) 3 122.322( 43) 0 6 6 H 5 1.093321( 5) 2 111.786( 25) 4 177.321( 44) 0 7 7 H 5 1.093313( 6) 2 111.783( 26) 3 -177.068( 45) 0 8 8 H 5 1.092621( 7) 2 110.662( 27) 7 119.503( 46) 0 9 9 N 1 1.914328( 8) 2 132.140( 28) 4 72.535( 47) 0 10 10 C 9 1.331874( 9) 1 89.188( 29) 2 127.943( 48) 0 11 11 N 10 1.332112( 10) 1 55.697( 30) 9 -176.611( 49) 0 12 12 H 9 1.012748( 11) 1 149.778( 31) 10 -170.264( 50) 0 13 13 H 10 1.091092( 12) 9 124.356( 32) 1 -177.173( 51) 0 14 14 H 11 1.012731( 13) 10 120.592( 33) 9 -177.578( 52) 0 15 X 1 2.356851( 14) 2 109.037( 34) 5 -178.496( 53) 0 16 15 C 15 0.676532( 15) 1 90.005( 35) 2 -89.033( 54) 0 17 X 15 1.000000( 16) 16 90.000( 36) 1 0.000( 55) 0 18 16 C 15 0.676532( 17) 17 90.000( 37) 16 180.000( 56) 0 19 17 H 16 1.085671( 18) 18 121.224( 38) 1 -98.071( 57) 0 20 18 H 16 1.088460( 19) 18 121.272( 39) 19 -174.282( 58) 0 21 19 H 18 1.088516( 20) 16 121.266( 40) 19 174.351( 59) 0 22 20 H 18 1.085621( 21) 19 95.379( 41) 21 -171.010( 60) 0
Standard orientation:
Center Atomic Coordinates (Angstroms) Number Number X Y Z 1 13 0.095074 -0.077423 -0.007604 2 6 1.839091 -0.914495 -0.015470 3 1 1.938406 -1.556312 -0.900267 4 1 1.930321 -1.587784 0.846536 5 6 2.984146 0.116718 0.009228 6 1 2.954201 0.777255 -0.861487 7 1 2.947774 0.742007 0.905344 8 1 3.954844 -0.384803 0.002736 9 7 -0.670441 1.285601 1.097297 10 6 -1.012538 1.957135 -0.000839 11 7 -0.694162 1.279335 -1.102541 12 1 -0.812768 1.691462 2.014181 13 1 -1.494749 2.935884 0.001106 14 1 -0.859439 1.677958 -2.018733 15 6 -1.558473 -1.764619 -0.664759 16 6 -1.565430 -1.742026 0.688098 17 1 -2.300879 -1.220574 -1.240545 18 1 -0.867714 -2.400285 -1.215692 19 1 -0.879655 -2.358069 1.266951 20 1 -2.314859 -1.181070 1.237878
DD1Dir will call FoFDir 1 times, MxPair= 506 NAB= 253 NAA= 0 NBB= 0.
Spin AB IR= 199 I= 13 J= 13 Mu= 80 Nu= 80 IOp=1 MuP= 80 NuP= 80 FactIJ= 0.0 Fact= 0.0 A=-4.6532834924D-01 AP=-4.6532834924D-01
The Euclidean norm of the A-vectors is 0.7995369D-05
DE(CORR)= -0.10873041D+01 E(CORR)= -0.54805067865D+03 NORM(A)= 0.11990484D+01
Largest amplitude= 6.75D-02 T4(AAA)= -0.93831506D-03 T4(AAB)= -0.18510157D-01 T5(AAA)= 0.59723962D-04 T5(AAB)= 0.94510047D-03
Time for triples= 39574.64 seconds. T4(CCSD)= -0.38896945D-01
TS 3
(insertion)
Z-MATRIX (ANGSTROMS AND DEGREES)
CD Cent Atom N1 Length/X N2 Alpha/Y N3 Beta/Z J 1 1 Al
2 2 C 1 2.295057( 1)
3 3 N 2 1.333225( 2) 1 55.640( 20)
4 4 N 2 1.331391( 3) 3 111.618( 21) 1 -1.950( 38) 0 5 5 H 2 1.090439( 4) 3 124.105( 22) 4 180.149( 39) 0 6 6 H 3 1.012121( 5) 2 120.827( 23) 4 181.440( 40) 0 7 7 H 4 1.012346( 6) 2 120.743( 24) 3 -181.461( 41) 0 8 8 C 1 2.061399( 7) 3 111.386( 25) 2 112.566( 42) 0 9 9 C 8 1.530122( 8) 1 109.951( 26) 6 34.848( 43) 0 10 10 H 8 1.109545( 9) 9 111.160( 27) 1 90.189( 44) 0 11 11 H 8 1.088880( 10) 9 111.527( 28) 1 -155.107( 45) 0 12 12 H 9 1.093268( 11) 8 111.558( 29) 10 181.859( 46) 0 13 13 H 9 1.093052( 12) 8 112.710( 30) 12 -121.648( 47) 0 14 14 H 9 1.093612( 13) 8 109.982( 31) 12 118.790( 48) 0 15 15 C 1 2.204499( 14) 8 62.385( 32) 10 147.492( 49) 0 16 16 C 15 1.438190( 15) 1 61.865( 33) 8 169.981( 50) 0 17 17 H 15 1.087248( 16) 16 118.883( 34) 1 -101.005( 51) 0 18 18 H 15 1.085696( 17) 16 120.640( 35) 1 108.031( 52) 0 19 19 H 16 1.088578( 18) 15 116.316( 36) 18 -142.971( 53) 0 20 20 H 16 1.088778( 19) 15 116.475( 37) 17 148.779( 54) 0 ---Standard orientation:
Center Atomic Coordinates (Angstroms) Number Number X Y Z 1 13 -0.207037 0.344970 -0.020178 2 6 -2.394577 -0.349258 -0.022028 3 7 -1.497352 -0.686413 -0.948744 4 7 -1.854808 0.408108 0.930678 5 1 -3.440036 -0.658562 -0.042070 6 1 -1.763955 -1.244207 -1.750103 7 1 -2.419495 0.761232 1.693093 8 6 1.261648 -0.847931 0.797940 9 6 1.723308 -1.885233 -0.227802 10 1 0.419818 -1.235245 1.408186 11 1 2.034886 -0.645658 1.537433 12 1 2.574082 -1.522620 -0.810838 13 1 0.932148 -2.168721 -0.926704 14 1 2.046387 -2.797071 0.282255 15 6 1.827839 1.181163 0.120801 16 6 0.847391 1.907507 -0.640472 17 1 2.062833 1.514764 1.128569 18 1 2.637071 0.655942 -0.377217 19 1 0.491239 2.839227 -0.204518 20 1 0.987965 1.926943 -1.719962
DE(CORR)= -0.10956877D+01 E(CORR)= -0.54799820698D+03 NORM(A)= 0.12017481D+01
Largest amplitude= 5.13D-02 T4(AAA)= -0.10651453D-02 T4(AAB)= -0.19739589D-01 T5(AAA)= 0.68748974D-04 T5(AAB)= 0.11336342D-02
Time for triples= 31871.47 seconds. T4(CCSD)= -0.41609469D-01
Product
4
Z-MATRIX (ANGSTROMS AND DEGREES)
CD Cent Atom N1 Length/X N2 Alpha/Y N3 Beta/Z J 1 1 Al
2 2 N 1 1.895702( 1)
3 3 H 2 1.012714( 2) 1 149.667( 20)
4 4 C 2 1.333397( 3) 3 121.343( 21) 1 182.582( 38) 0 5 5 H 4 1.090527( 4) 2 124.314( 22) 1 181.091( 39) 0 6 6 N 4 1.332456( 5) 2 111.320( 23) 3 -180.221( 40) 0 7 7 H 6 1.012763( 6) 4 121.241( 24) 2 180.999( 41) 0 8 8 C 1 1.929450( 7) 2 145.499( 25) 4 -188.688( 42) 0 9 9 C 8 1.543353( 8) 1 101.225( 26) 2 115.795( 43) 0 10 10 H 8 1.096526( 9) 9 110.863( 27) 1 116.273( 44) 0 11 11 H 8 1.093646( 10) 9 112.944( 28) 1 -123.624( 45) 0 12 12 H 9 1.095784( 11) 8 110.214( 29) 11 -38.435( 46) 0 13 13 H 9 1.094680( 12) 8 110.953( 30) 10 -40.610( 47) 0 14 14 C 9 1.528807( 13) 8 110.504( 31) 13 121.357( 48) 0 15 15 H 14 1.124525( 14) 9 109.295( 32) 13 -178.534( 49) 0 16 16 H 14 1.098727( 15) 9 109.726( 33) 12 181.190( 50) 0 17 17 C 14 1.523621( 16) 9 114.956( 34) 15 118.929( 51) 0 18 18 H 17 1.092587( 17) 14 109.529( 35) 15 -177.520( 52) 0 19 19 H 17 1.091710( 18) 14 111.549( 36) 18 119.589( 53) 0 20 20 H 17 1.092283( 19) 14 110.849( 37) 18 -119.342( 54) 0 ---Standard orientation:
Center Atomic Coordinates (Angstroms) Number Number X Y Z 1 13 0.725852 0.577823 -0.033618 2 7 2.089184 -0.250857 -1.057483 3 1 2.519920 -0.431565 -1.956038 4 6 2.616884 -0.723385 0.072207 5 1 3.524015 -1.326631 0.121762 6 7 1.893878 -0.363845 1.132127 7 1 2.168293 -0.630085 2.069944 8 6 -0.766593 1.800247 -0.066208 9 6 -1.935186 0.857240 0.290260 10 1 -0.887831 2.207480 -1.077065 11 1 -0.683478 2.652277 0.614379 12 1 -2.024729 0.759736 1.378018 13 1 -2.887397 1.261033 -0.068306 14 6 -1.712145 -0.526433 -0.320439 15 1 -0.747131 -0.953090 0.068490 16 1 -1.599730 -0.437281 -1.409758 17 6 -2.786886 -1.555119 0.008436 18 1 -3.748442 -1.217391 -0.385386 19 1 -2.561498 -2.526538 -0.435835 20 1 -2.885944 -1.682028 1.088790
DE(CORR)= -0.10887892D+01 E(CORR)= -0.54806355421D+03 NORM(A)= 0.11940110D+01
Largest amplitude= 4.94D-02 T4(AAA)= -0.97112351D-03 T4(AAB)= -0.17978105D-01 T5(AAA)= 0.57810712D-04 T5(AAB)= 0.89721687D-03
Time for triples= 40030.42 seconds. T4(CCSD)= -0.37898457D-01
TS 5
(BHT)
Z-MATRIX (ANGSTROMS AND DEGREES)
CD Cent Atom N1 Length/X N2 Alpha/Y N3 Beta/Z J 1 1 Al
2 2 C 1 2.325295( 1)
3 3 N 2 1.330950( 2) 1 55.816( 20)
4 4 N 2 1.330971( 3) 3 111.628( 21) 1 0.003( 38) 0 5 5 H 2 1.091224( 4) 3 124.186( 22) 4 -179.999( 39) 0 6 6 H 3 1.011984( 5) 2 120.196( 23) 4 180.000( 40) 0 7 7 H 4 1.011985( 6) 2 120.197( 24) 3 -179.993( 41) 0 8 8 C 1 2.036598( 7) 2 128.333( 25) 3 89.977( 42) 0 9 9 C 8 1.442251( 8) 1 115.784( 26) 4 138.301( 43) 0 10 10 H 8 1.093762( 9) 9 113.286( 27) 1 -116.091( 44) 0 11 11 H 8 1.093798( 10) 9 113.281( 28) 10 -127.827( 45) 0 12 12 H 9 1.090867( 11) 8 118.000( 29) 11 -134.701( 46) 0 13 13 H 9 1.090902( 12) 8 117.993( 30) 10 134.790( 47) 0 14 14 H 9 1.291127( 13) 8 108.434( 31) 11 116.132( 48) 0 15 15 C 1 2.036608( 14) 4 120.565( 32) 7 65.631( 49) 0 16 16 C 15 1.442206( 15) 14 33.499( 33) 1 -180.095( 50) 0 17 17 H 15 1.093715( 16) 16 113.288( 34) 14 -116.004( 51) 0 18 18 H 15 1.093772( 17) 16 113.281( 35) 17 -127.830( 52) 0 19 19 H 16 1.090847( 18) 15 118.002( 36) 18 -134.669( 53) 0 20 20 H 16 1.090868( 19) 15 117.987( 37) 17 134.820( 54) 0 ---Standard orientation:
Center Atomic Coordinates (Angstroms) Number Number X Y Z 1 13 -0.336740 -0.000024 -0.000124 2 6 -2.662035 0.000011 0.000093 3 7 -1.914127 0.000033 1.101030 4 7 -1.914268 -0.000093 -1.100965 5 1 -3.753259 0.000089 0.000155 6 1 -2.351627 0.000114 2.013558 7 1 -2.351876 -0.000008 -2.013440 8 6 0.926403 -1.597585 0.000447 9 6 2.334204 -1.284241 -0.000530 10 1 0.600805 -2.122131 0.903305 11 1 0.599927 -2.123396 -0.901401 12 1 2.903459 -1.481723 0.908832 13 1 2.902078 -1.481656 -0.910813 14 1 2.466597 0.000080 -0.000521 15 6 0.926364 1.597581 -0.000553 16 6 2.334129 1.284289 0.000620 17 1 0.600925 2.122353 -0.903280 18 1 0.599677 2.123059 0.901382 19 1 2.903696 1.482581 -0.908348 20 1 2.901585 1.481035 0.911266
DE(CORR)= -0.10975322D+01 E(CORR)= -0.54800683470D+03 NORM(A)= 0.12063145D+01
Largest amplitude= 7.92D-02 T4(AAA)= -0.11447073D-02 T4(AAB)= -0.20291923D-01 T5(AAA)= 0.60916811D-04 T5(AAB)= 0.12846929D-02
Time for triples= 30544.22 seconds. T4(CCSD)= -0.42873260D-01
TS 6
(CHT)
Z-MATRIX (ANGSTROMS AND DEGREES)
CD Cent Atom N1 Length/X N2 Alpha/Y N3 Beta/Z J 1 1 Al
2 2 C 1 2.289492( 1)
3 3 N 2 1.333169( 2) 1 55.734( 20)
4 4 N 2 1.332019( 3) 3 111.557( 21) 1 1.403( 38) 0 5 5 H 2 1.090245( 4) 3 124.195( 22) 4 -180.029( 39) 0 6 6 H 3 1.012233( 5) 2 121.032( 23) 4 179.393( 40) 0 7 7 H 4 1.012343( 6) 2 121.062( 24) 3 -179.168( 41) 0 8 8 C 1 2.055119( 7) 2 131.609( 25) 3 86.285( 42) 0 9 9 C 8 1.540563( 8) 1 108.381( 26) 3 -29.017( 43) 0 10 10 H 8 1.099746( 9) 1 128.409( 27) 4 109.636( 44) 0 11 11 H 8 1.102800( 10) 9 112.024( 28) 10 112.611( 45) 0 12 12 H 9 1.093485( 11) 8 111.284( 29) 11 -180.668( 46) 0 13 13 H 9 1.092749( 12) 8 112.147( 30) 12 121.033( 47) 0 14 14 H 9 1.092367( 13) 8 110.539( 31) 13 119.925( 48) 0 15 15 C 1 2.014515( 14) 4 123.930( 32) 2 -123.754( 49) 0 16 16 C 15 1.356055( 15) 1 119.881( 33) 4 142.832( 50) 0 17 17 H 15 1.090591( 16) 16 117.859( 34) 1 163.030( 51) 0 18 18 H 16 1.089955( 17) 15 121.890( 35) 17 183.760( 52) 0 19 19 H 16 1.088000( 18) 15 122.907( 36) 18 179.055( 53) 0 20 20 H 1 1.675007( 19) 4 140.694( 37) 2 177.469( 54) 0 ---Standard orientation:
Center Atomic Coordinates (Angstroms) Number Number X Y Z 1 13 0.214580 -0.167061 -0.231476 2 6 2.432054 -0.058787 0.327860 3 7 1.431217 0.162063 1.180438 4 7 1.981824 -0.321037 -0.898024 5 1 3.487228 -0.027967 0.600428 6 1 1.611636 0.366379 2.155281 7 1 2.620601 -0.523150 -1.656940 8 6 -1.112951 1.281086 -0.834837 9 6 -1.438116 2.176772 0.375680 10 1 -2.019459 1.164841 -1.446534 11 1 -0.414878 1.769545 -1.535031 12 1 -2.139783 1.685803 1.055623 13 1 -0.543788 2.438757 0.946331 14 1 -1.903457 3.109312 0.048432 15 6 -1.176959 -1.615132 -0.389525 16 6 -2.050317 -1.826574 0.626066 17 1 -1.063454 -2.401896 -1.136189 18 1 -2.259935 -1.061817 1.373868 19 1 -2.601345 -2.756787 0.747788 20 1 -1.376453 -0.222599 -0.752223
DE(CORR)= -0.10999708D+01 E(CORR)= -0.54795930098D+03 NORM(A)= 0.12042885D+01
Largest amplitude= 7.31D-02 T4(AAA)= -0.10315790D-02 T4(AAB)= -0.19988196D-01 T5(AAA)= 0.63055603D-04 T5(AAB)= 0.10399748D-02
Time for triples= 40486.87 seconds. T4(CCSD)= -0.42039549D-01
TS 7
(BHE)
Z-MATRIX (ANGSTROMS AND DEGREES)
CD Cent Atom N1 Length/X N2 Alpha/Y N3 Beta/Z J 1 1 Al
2 2 C 1 2.280567( 1)
3 3 N 2 1.332497( 2) 1 55.846( 14)
4 4 N 2 1.332448( 3) 3 111.705( 15) 1 -0.739( 26) 0 5 5 H 2 1.090007( 4) 3 124.146( 16) 4 -179.961( 27) 0 6 6 H 3 1.012236( 5) 2 121.303( 17) 4 180.566( 28) 0 7 7 H 4 1.012172( 6) 2 121.357( 18) 3 -180.382( 29) 0 8 8 C 1 2.020224( 7) 2 141.541( 19) 4 89.840( 30) 0 9 9 C 8 1.421860( 8) 1 78.802( 20) 4 -131.187( 31) 0 10 10 H 8 1.087993( 9) 9 117.084( 21) 1 -108.026( 32) 0 11 11 H 8 1.088346( 10) 9 117.091( 22) 10 -144.102( 33) 0 12 12 H 9 1.087381( 11) 8 121.129( 23) 10 151.087( 34) 0 13 13 H 9 1.087419( 12) 8 121.105( 24) 11 -151.167( 35) 0 14 14 H 1 1.655026( 13) 3 121.442( 25) 6 62.068( 36) 0 ---Standard orientation:
Center Atomic Coordinates (Angstroms) Number Number X Y Z 1 13 -0.165616 -0.002285 -0.193289 2 6 2.098828 0.001909 0.077382 3 7 1.353090 1.103193 -0.003832 4 7 1.357427 -1.102296 -0.003081 5 1 3.182382 0.004091 0.195794 6 1 1.770273 2.024733 0.032813 7 1 1.777048 -2.022521 0.036858 8 6 -1.885507 0.000517 0.866554 9 6 -2.382172 0.000233 -0.465741 10 1 -1.994477 -0.920104 1.436038 11 1 -1.989868 0.923207 1.434225 12 1 -2.742139 0.914413 -0.931676 13 1 -2.745412 -0.913722 -0.929663 14 1 -1.065313 -0.002631 -1.582409
DD1Dir will call FoFDir 1 times, MxPair= 272 NAB= 136 NAA= 0 NBB= 0.
Spin AB IR= 116 I= 11 J= 11 Mu= 25 Nu= 25 IOp=1 MuP= 25 NuP= 25 FactIJ= 0.0 Fact= 0.0 A=-5.8784089079D-01 AP=-5.8784089079D-01
The Euclidean norm of the A-vectors is 0.7921390D-05
DE(CORR)= -0.80144047D+00 E(CORR)= -0.46964188144D+03 NORM(A)= 0.11528126D+01
Largest amplitude= 4.77D-02 T4(AAA)= -0.76629002D-03 T4(AAB)= -0.14404643D-01 T5(AAA)= 0.54027398D-04 T5(AAB)= 0.92636733D-03
Time for triples= 5133.25 seconds. T4(CCSD)= -0.30341866D-01
Structure
8
Z-MATRIX (ANGSTROMS AND DEGREES)
CD Cent Atom N1 Length/X N2 Alpha/Y N3 Beta/Z J 1 1 Al
2 2 C 1 2.306458( 1)
3 3 N 2 1.332523( 2) 1 55.656( 16)
4 4 N 2 1.332393( 3) 3 111.254( 17) 1 -2.973( 30) 0 5 5 H 2 1.090741( 4) 3 124.357( 18) 4 180.106( 31) 0 6 6 H 3 1.012754( 5) 2 120.861( 19) 4 182.620( 32) 0 7 7 H 4 1.012765( 6) 2 120.864( 20) 3 -182.674( 33) 0 8 8 H 1 1.559433( 7) 2 144.998( 21) 3 91.890( 34) 0 9 X 1 2.322742( 8) 8 105.607( 22) 3 128.321( 35) 0 10 9 C 9 0.677134( 9) 1 90.101( 23) 8 -89.294( 36) 0 11 X 9 1.000000( 10) 10 90.000( 24) 1 0.000( 37) 0 12 10 C 9 0.677134( 11) 11 90.000( 25) 10 180.000( 38) 0 13 11 H 10 1.085885( 12) 12 121.199( 26) 1 -99.082( 39) 0 14 12 H 10 1.089198( 13) 12 121.249( 27) 13 -174.186( 40) 0 15 13 H 12 1.089290( 14) 10 121.241( 28) 13 174.140( 41) 0 16 14 H 12 1.085835( 15) 10 121.211( 29) 15 -174.039( 42) 0 ---Standard orientation:
Center Atomic Coordinates (Angstroms) Number Number X Y Z 1 13 -0.054830 -0.005857 0.854284 2 6 1.799969 0.001565 -0.516634 3 7 1.175337 -1.100394 -0.102941 4 7 1.177494 1.099129 -0.088674 5 1 2.703135 0.004380 -1.128188 6 1 1.547741 -2.018494 -0.312890 7 1 1.552045 2.019176 -0.285949 8 1 -0.550385 -0.011841 2.332871 9 6 -1.984684 -0.672037 -0.446127 10 6 -1.970101 0.682140 -0.452036 11 1 -1.597479 -1.242410 -1.285111 12 1 -2.468448 -1.228395 0.355616 13 1 -2.442414 1.255687 0.344529 14 1 -1.572337 1.236885 -1.296477
DD1Dir will call FoFDir 1 times, MxPair= 272 NAB= 136 NAA= 0 NBB= 0.
Spin AB IR= 116 I= 11 J= 11 Mu= 25 Nu= 25 IOp=1 MuP= 25 NuP= 25 FactIJ= 0.0 Fact= 0.0 A=-4.4940089897D-01 AP=-4.4940089897D-01
The Euclidean norm of the A-vectors is 0.4715827D-05
DE(CORR)= -0.79894981D+00 E(CORR)= -0.46968210595D+03 NORM(A)= 0.11513837D+01
Largest amplitude= 5.49D-02 T4(AAA)= -0.71205693D-03 T4(AAB)= -0.14028945D-01 T5(AAA)= 0.49145168D-04 T5(AAB)= 0.77126737D-03
Time for triples= 5105.30 seconds. T4(CCSD)= -0.29482004D-01
Structure
9
Z-MATRIX (ANGSTROMS AND DEGREES)
CD Cent Atom N1 Length/X N2 Alpha/Y N3 Beta/Z J 1 1 Al
2 2 C 1 2.318399( 1)
3 3 N 2 1.331648( 2) 1 55.576( 24)
4 4 N 2 1.331678( 3) 3 111.155( 25) 1 -0.009( 46) 0 5 5 H 2 1.090815( 4) 3 124.413( 26) 4 180.001( 47) 0 6 6 H 3 1.012840( 5) 2 120.854( 27) 4 180.005( 48) 0 7 7 H 4 1.012850( 6) 2 120.837( 28) 3 -180.003( 49) 0 8 8 H 1 1.560808( 7) 2 179.952( 29) 3 -188.226( 50) 0 9 X 1 2.551389( 8) 8 88.104( 30) 3 89.957( 51) 0 10 9 C 9 0.674040( 9) 1 89.981( 31) 8 -89.992( 52) 0 11 X 9 1.000000( 10) 10 90.000( 32) 1 0.000( 53) 0 12 10 C 9 0.674040( 11) 11 90.000( 33) 10 180.000( 54) 0 13 11 H 10 1.084237( 12) 12 121.278( 34) 1 -104.131( 55) 0 14 12 H 10 1.087232( 13) 12 121.444( 35) 13 -174.058( 56) 0 15 13 H 12 1.087227( 14) 10 121.451( 36) 13 174.087( 57) 0 16 14 H 12 1.084240( 15) 10 121.279( 37) 15 -174.179( 58) 0 17 X 1 2.551031( 16) 8 88.093( 38) 3 -89.954( 59) 0 18 15 C 17 0.674043( 17) 1 89.973( 39) 8 89.984( 60) 0 19 X 17 1.000000( 18) 18 90.000( 40) 1 0.000( 61) 0 20 16 C 17 0.674043( 19) 19 90.000( 41) 18 180.000( 62) 0 21 17 H 18 1.084237( 20) 20 121.277( 42) 1 104.183( 63) 0 22 18 H 18 1.087236( 21) 20 121.443( 43) 21 174.031( 64) 0 23 19 H 20 1.084244( 22) 18 121.278( 44) 22 174.108( 65) 0 24 20 H 20 1.087228( 23) 18 121.452( 45) 23 -174.159( 66) 0 ---Standard orientation:
Center Atomic Coordinates (Angstroms) Number Number X Y Z 1 13 -0.000050 -0.563158 -0.002092 2 6 -0.000201 1.755236 0.002946 3 7 0.000032 1.000046 1.099747 4 7 -0.000138 1.004870 -1.097199 5 1 -0.000404 2.846048 0.005536 6 1 -0.000060 1.421631 2.020677 7 1 -0.000346 1.430778 -2.016148 8 1 -0.000136 -2.123960 -0.006780 9 6 2.549199 -0.649817 0.673632 10 6 2.550676 -0.645585 -0.674441 11 1 2.804459 0.239039 1.239624 12 1 2.386109 -1.564824 1.237757 13 1 2.388390 -1.556875 -1.244769 14 1 2.805738 0.247205 -1.234304 15 6 -2.548806 -0.650102 0.673727 16 6 -2.550539 -0.645701 -0.674351 17 1 -2.804843 0.238435 1.239869 18 1 -2.385130 -1.565104 1.237698 19 1 -2.806831 0.246847 -1.234044 20 1 -2.387531 -1.556765 -1.244839
DE(CORR)= -0.10892501D+01 E(CORR)= -0.54800471518D+03 NORM(A)= 0.12058560D+01
Largest amplitude= 6.09D-02 T4(AAA)= -0.96958325D-03 T4(AAB)= -0.19599966D-01 T5(AAA)= 0.62690319D-04 T5(AAB)= 0.10007197D-02
Structure
10
Z-MATRIX (ANGSTROMS AND DEGREES)
CD Cent Atom N1 Length/X N2 Alpha/Y N3 Beta/Z J 1 1 Al
2 2 C 1 2.279010( 1)
3 3 N 2 1.334747( 2) 1 55.565( 8)
4 4 N 2 1.335142( 3) 3 111.110( 9) 1 -0.010( 14) 0 5 5 H 2 1.090040( 4) 3 124.453( 10) 4 179.995( 15) 0 6 6 H 3 1.013450( 5) 2 121.872( 11) 4 179.956( 16) 0 7 7 H 4 1.013430( 6) 2 121.875( 12) 3 -180.062( 17) 0 8 8 H 1 1.552160( 7) 3 143.960( 13) 2 180.006( 18) 0 ---Standard orientation:
Center Atomic Coordinates (Angstroms) Number Number X Y Z 1 13 1.099682 -0.000794 0.000021 2 6 -1.179328 0.000605 0.000033 3 7 -0.423887 1.100995 0.000065 4 7 -0.424645 -1.100785 -0.000219 5 1 -2.269368 0.000926 0.000263 6 1 -0.830552 2.029275 -0.000416 7 1 -0.832089 -2.028702 0.000660 8 1 2.651835 0.003724 0.000106
DD1Dir will call FoFMem 1 times, MxPair= 110 NAB= 55 NAA= 0 NBB= 0.
The Euclidean norm of the A-vectors is 0.4974486D-05
DE(CORR)= -0.51164566D+00 E(CORR)= -0.39132909977D+03 NORM(A)= 0.10964382D+01
Largest amplitude= 6.60D-02 T4(AAA)= -0.46563710D-03 T4(AAB)= -0.87034305D-02 T5(AAA)= 0.35542099D-04 T5(AAB)= 0.55224452D-03
Time for triples= 301.27 seconds. T4(CCSD)= -0.18338135D-01