PENGEMBANGAN ANALISIS LISTERIA MONOCYTOGENES
UNTUK JAJANAN PEMPEK DENGAN REAL-TIME
POLYMERASE CHAIN REACTION (rt-PCR)
ARIF MURTAQI AKHMAD MS
SEKOLAH PASCASARJANA INSTITUT PERTANIAN BOGOR
BOGOR 2016
iii
PERNYATAAN MENGENAI TESIS DAN
SUMBER INFORMASI SERTA PELIMPAHAN HAK CIPTA*
Dengan ini saya menyatakan bahwa tesis berjudul Pengembangan AnalisisListeria monocytogenes untuk Jajanan Pempek dengan Real-time Polymerase
Chain Reaction (rt-PCR) adalah karya saya dengan arahan dari komisi
pembimbing dan belum diajukan dalam bentuk apa pun kepada perguruan tinggi mana pun. Sumber informasi yang berasal atau dikutip dari karya yang diterbitkan maupun tidak diterbitkan dari penulis lain telah disebutkan dalam teks dan dicantumkan dalam Daftar Pustaka di bagian akhir tesis ini.
Dengan ini saya melimpahkan hak cipta dari karya tulis saya kepada Institut Pertanian Bogor.
Bogor, Januari 2016
Arif Murtaqi Akhmad MS NRP. F251120181
iv
RINGKASAN
ARIF MURTAQI AKHMAD MS. Pengembangan Analisis Listeria
monocytogenes untuk Jajanan Pempek dengan Real-time Polymerase Chain
Reaction (rt-PCR). Dibimbing oleh WINIATI P. RAHAYU dan SITI
NURJANAH.
Listeria (L.) monocytogenes merupakan bakteri patogen yang dapat
menyebabkan bahaya serius bagi manusia. L. monocytogenes dapat menyebabkan komplikasi meningitis. Pangan jajanan anak sekolah sebagai salah satu pangan siap saji memiliki risiko bahaya L. monocytogenes yang tinggi terutama bila dikonsumsi oleh anak sekolah dasar. Tujuan dari penelitian ini adalah untuk mengembangkan metode deteksi L. monocytogenes pada jajanan pempek menggunakan rt-PCR. Isolasi DNA L. monocytogenes dilakukan dengan membandingkan tiga metode ekstraksi yaitu: fenol:kloroform, pemanasan, dan kit komersial (QIAamp DNA Blood Mini Kit). Isolat DNA L. monocytogenes diamplifikasi menggunakan PCR dengan dua variabel primer yaitu LIM 2 dan LIMRE untuk deteksi gen iap, dan primer DG69 dan DG74 untuk deteksi gen
hlyA. Pengujian dengan rt-PCR diawali dengan denaturasi awal pada 94 °C
selama 5 menit diikuti dengan 30 siklus yang terdiri dari 45 detik denaturasi pada 94 °C, 45 detik annealing pada 55 °C, 45 detik ekstensi pada 72 °C dan ekstensi akhir pada 72 °C selama 7 menit. Kemudian dilanjutkan dengan program melting dengantransisi suhu 72-94 °C dan laju 0.5 °C/detik.
Metode fenol:kloroform menunjukkan ekstraksi DNA terbaik yang lebih
murni. Primer DG69/DG74 lebih spesifik dibandingkan dengan primer LIM 2/LIMRE. Kondisi running yang digunakan menghasilkan kurva amplifikasi
spesifik sampai siklus ke-27. Deteksi DNA dari kultur murni L. monocytogenes menghasilkan persamaan kurva standar CT = 37.9-3.11 C dengan nilai r sebesar
-0.990 dan limit deteksi (LOD) 3.2 x 103 CFU/mL. Deteksi DNA pada sampel pempek menghasilkan persamaan kurva standar CT = 41.03-3.69 C dengan nilai r
sebesar -0.997 dan limit deteksi (LOD) 6.3 x 103 CFU/g. Primer DG69 dan DG74 dapat dipertimbangkan sebagai metode spesifik dan sensitif yang dapat diandalkan untuk mendeteksi L. monocytogenes pada jajanan pempek.
Kata kunci: Listeria monocytogenes, metode deteksi, pempek, primer, real-time PCR
v
SUMMARY
ARIF MURTAQI AKHMAD MS. Development of Real-time Polymerase Chain Reaction Analysis for DNA Detection of Listeria monocytogenes in Pempek Snack. Supervised by WINIATI P. RAHAYU and SITI NURJANAH
Listeria (L.) monocytogenes is an important foodborne pathogen which can
cause serious human listeriosis. L. monocytogenes can cause meningitis
complications. School children snacks have high potential of risk of
L. monocytogenes because these foods are consumed by children. The aim of this
study was to develop rt-PCR method of L. monocytogenes detection in fish based snack. DNA extraction of L. monocytogenes was conducted by comparing three extraction methods: phenol:chloroform, heating, and commercial kit (QIAamp DNA Blood Mini Kit). DNA isolate was amplified by rt-PCR using two variables which were primer LIM 2 and LIMRE for iap gene detection, and primer DG69 and DG74 for hlyA gene detection. The cycling conditions included an initial denaturation at 94 °C for 5 min followed by 30 cycles of 45 s denaturation at 94 °C, 45 s annealing at 55 °C, 45 s extension at 72 °C and 7 min final extension at 72 °C. The thermocycling program was followed by a melting program of transition from 72 to 94 °C with a rate of 0.5 °C/s.
Phenol:chloroform method gave the best extraction result, while primer DG69 and DG74 gave more specific result on standard PCR compared to primer LIM 2 and LIMRE. The running condition showed the fluorescence amplification curve up to 27th cycle. Amplification standard curve of DNA from pure culture resulted the equation CT = 37.9 – 3.11 C with r value -0.990 and limit of detection
(LOD) 3.2 x 103 CFU / mL. Amplification standard curve of DNA from spiked pempek sample resulted the equation CT = 41.03 - 3.69 C with r value -0.997 and
limit of detection (LOD) 6.3 x 103 CFU / g. In summary, rt-PCR using primer DG69 and DG74 should be considered as reliable method for specific and sensitive detection of L. monocytogenes in pempek.
Keywords: Detection method, fish based snack, Listeria monocytogenes, primer, real-time PCR
vi
© Hak Cipta Milik IPB, Tahun 2016 Hak Cipta Dilindungi Undang-Undang
Dilarang mengutip sebagian atau seluruh karya tulis ini tanpa mencantumkan atau menyebutkan sumbernya. Pengutipan hanya untuk kepentingan pendidikan, penelitian, penulisan karya ilmiah, penyusunan laporan, penulisan kritik, atau tinjuan suatu masalah; dan pengutipan tersebut tidak merugikan kepentingan IPB Dilarang mengumumkan dan memperbanyak sebagian atau seluruh karya tulis ini dalam bentuk apa pun tanpa izin IPB
vii
PENGEMBANGAN ANALISIS LISTERIA MONOCYTOGENES UNTUK JAJANAN PEMPEK DENGAN REAL-TIME
POLYMERASE CHAIN REACTION (rt-PCR)
ARIF MURTAQI AKHMAD MS
Tesis
sebagai salah satu syarat untuk memperoleh gelar Magister Sains
pada
Program Studi Ilmu Pangan
SEKOLAH PASCASARJANA INSTITUT PERTANIAN BOGOR
BOGOR 2016
viii
ix
Judul Tesis : Pengembangan Analisis Listeria monocytogenes untuk Jajanan Pempek dengan Real-time Polymerase Chain
Reaction (rt-PCR)
Nama : Arif Murtaqi Akhmad MS
NIM : F251120181
Disetujui oleh Komisi Pembimbing
Prof. Dr. Winiati P Rahayu Dr. Ir. Siti Nurjanah, M.Si Ketua Anggota
Diketahui oleh
Ketua Program Studi Ilmu Pangan
Dekan Sekolah Pascasarjana
Prof. Dr. Ir. Ratih Dewanti-Hariyadi, M.Sc Dr Ir Dahrul Syah, M.Sc.Agr
Tanggal Ujian : 20 November 2015 Tanggal Lulus :
x
PRAKATA
Puji dan syukur penulis panjatkan kepada Allah subhanahu wa ta’ala atas segala karunia-Nya sehingga karya ilmiah ini berhasil diselesaikan. Tema yang dipilih dalam penelitian yang dilaksanakan sejak bulan Mei 2014 sampai Mei 2015 ini ialah pengembangan metode deteksi mikroba patogen, dengan judul Pengembangan Analisis Listeria monocytogenes untuk Jajanan Pempek dengan
Real-time Polymerase Chain Reaction (rt-PCR).
Penulis menyampaikan penghargaan dan ucapan terima kasih yang tidak terhingga kepada :
1. Ibu Prof. Dr. Winiati P Rahayu dan Ibu Dr. Siti Nurjanah yang telah memberikan bimbingan, waktu dan perhatian selama penelitian dan penulisan tesis ini.
2. Bapak Dr. Puspo Edi Giriwono selaku penguji yang telah memberikan saran-saran guna menyempurnakan penulisan tesis ini.
3. Direktorat Jenderal Pendidikan Tinggi yang telah membiayai penelitian ini melalui Program Hibah Kompetensi.
4. Departemen ITP sebagai homebase IPN.
5. Program pascasarjana IPB yang telah membantu selama proses studi.
6. Pimpinan SEAFAST Center dan Departemen ITP yang telah memberikan ijin penggunaan fasilitas di laboratorium.
7. Seluruh pengajar di Program Studi Ilmu Pangan yang telah memberikan bekal ilmu bagi penulis.
8. Politeknik Gorontalo yang telah memberikan kesempatan dan dukungan dana selama menempuh studi di program studi Ilmu Pangan, Sekolah Pascasarjana Institut Pertanian Bogor.
9. Para teknisi di laboratorium SEAFAST Center dan di laboratorium ITP yang telah memberikan bantuan selama penulis melakukan penelitian.
10.Rifina Murtialmira dan Muhammad Afif Naufal yang telah memberikan semangat dan doa kepada penulis.
11.Ibu dan Bapak serta kakak-kakak dan keponakan-keponakan yang memberikan doa dan dukungan kepada penulis.
12.Teman-teman yang telah membantu selama penelitian. Penulis berharap tesis ini dapat bermanfaat.
Bogor, Januari 2016
xi
DATAR ISI
DAFTAR TABEL xii
DAFTAR GAMBAR xii
DAFTAR LAMPIRAN xii
1 PENDAHULUAN 1 Latar Belakang 1 Perumusan Masalah 1 Tujuan Penelitian 2 Manfaat Penelitian 2 2 TINJAUAN PUSTAKA 2 Karakteristik L. monocytogenes 2
Metode Analisis Polymerase Chain Reaction (PCR) 3
3 METODE 5
Bahan 5
Alat 6
Tahapan Penelitian 6
4 HASIL DAN PEMBAHASAN 11
Pemilihan Metode Ekstraksi DNA 11
Konfirmasi Spesifisitas Primer 13
Penentuan Limit Deteksi DNA L. monocytogenes 16
SIMPULAN 18
SARAN 19
DAFTAR PUSTAKA 19
LAMPIRAN 22
xii
DAFTAR TABEL
1. Penelitian tentang pengembangan deteksi L. monocytogenes 4
2. Sitotoksisitas mikroba yang digunakan 6
3. Kemurnian dan konsentrasi DNA 12
4. Hasil analisis kurva melting 15
5. Hasil analisis rt-PCR DNA L. monocytogenes 16 6. Hubungan nilai CT terhadap sampel yang diuji 18
DAFTAR GAMBAR
1. L. monocytogenes 2
2. Grafik sigmoidal proses amplifikasi dengan rt-PCR 5
3. Skema penelitian 7
4. Hasil elektroforesis pada tahap ekstraksi DNA 12 5. Hasil elektroforesis produk PCR dengan primer (a) LIM 2 /LIMRE dan
(b) DG69/DG74 14
6. Kurva amplifikasi uji spesifisitas primer DG69/DG74 dengan rt-PCR 15 7. Kurva standar hubungan log konsentrasi kultur murni L.
monocytogenes dan nilai CT 17
8. Kurva standar hubungan log konsentrasi L. monocytogenes padasampel
pangan spike dan nilai CT 17
DAFTAR LAMPIRAN
1. Komposisi dalam PCR standar dengan Primer LIM 2 / LIMRE 22 2. Kondisi running PCR standar dengan Primer LIM 2 / LIMRE 22 3. Komposisi bahan dalam PCR standar dengan Primer DG69 / DG74 22 4. Kondisi running PCR standar dengan Primer DG69 / DG74 22 5. Komposisi bahan untuk rt-PCR dengan Primer LIM 2 / LIMRE 23 6. Kondisi running rt-PCR dengan Primer LIM 2 / LIMRE 23 7. Komposisi bahan untuk rt-PCR dengan Primer DG69 / DG74 23 8. Kondisi running rt-PCR dengan Primer DG69 / DG74 23
9. Analisis BLAST primer LIM 2 24
10. Analisis BLAST primer LIMRE 24
11. Analisis BLAST primer DG69 25
1
PENDAHULUAN
Latar BelakangListeria monocytogenes sering mengontaminasi produk-produk pangan
segar maupun pangan dengan minimal proses, seperti produk susu, ikan atau sayuran/buah. L. monocytogenes dapat menyebabkan keracunan darah dan komplikasi meningitis. L. monocytogenes memiliki kemampuan hidup yang luas antara suhu 1-45 °C. Oleh sebab itu, L. monocytogenes sering dijumpai pada pangan siap saji dan pangan yang disimpan pada suhu rendah (Moreno et al. 2012).
Perkembangan teknologi identifikasi DNA yang pesat saat ini dapat membantu untuk mengetahui pencemaran pangan terutama cemaran biologi dan mikrobiologi. Salah satu metode cepat deteksi mikroba yaitu Polymerase Chain
Reaction (PCR). Salah satu kelebihan metode PCR yaitu mampu mendeteksi
L. monocytogenes yang masih hidup namun tidak terdeteksi dengan metode
pencawanan (viable but not culturable). Aplikasi PCR dalam deteksi bakteri
L. monocytogenes telah banyak diaplikasikan contohnya pada sampel susu dan
produk turunannya (Aurora et al. 2008), daging dan produk turunannya (Meyer et al. 2011; Marian et al. 2012), daging ikan dan produk turunannya (Marian et al. 2012), sayuran (Moreno et al. 2012), dan beberapa produk pangan siap saji lainnya (Jamali et al. 2013). Teknologi deteksi DNA dengan real-time PCR (rt-PCR) merupakan pengembangan metode PCR yang dapat mendeteksi dan mengkuantifikasi DNA bakteri secara aktual selama proses berjalan. Oleh karena itu, metode rt-PCR ini relatif lebih cepat dibandingkan dengan PCR standar.
Pangan jajanan anak sekolah (PJAS), khususnya yang berbahan dasar daging ikan, perlu mendapat perhatian lebih. Hal ini disebabkan komposisi daging ikan yang banyak mengandung nutrisi bagi pertumbuhan mikroba dan ditambah lagi praktik penanganan para penjual PJAS yang masih jauh dari higienis. Salah satu contoh PJAS berbasis ikan yang cukup terkenal yaitu pempek. Pempek merupakan makanan khas dari Palembang, Sumatra Selatan yang terbuat dari adonan ikan dan tapioka. Pempek sudah banyak ditemui di tiap-tiap sekolah, perkantoran, pusat perbelanjaan, dan tempat-tempat lainnya di seluruh Indonesia. Pengembangan deteksi L. monocytogenes untuk jajanan pempek dengan rt-PCR perlu dilakukan mengingat masih belum adanya penelitian tersebut baik di dalam maupun luar negeri.
Perumusan Masalah
Penelitian tentang pengembangan metode deteksi L. monocytogenes secara kuantitatif pada PJAS berbasis ikan, khususnya pempek, sangat penting dilakukan mengingat belum adanya penelitian tersebut sampai saat ini. Pengembangan metode ini dimaksudkan untuk memberi informasi metode ekstraksi DNA, penggunaan primer, kondisi running PCR standar dan rt-PCR, sehingga diperoleh
serangkaian metode yang optimal untuk deteksi secara kuantitatif
L. monocytogenes untuk jajanan pempek. Deteksi secara kuantitatif diperlukan
2
Tujuan Penelitian Tujuan dari penelitian ini adalah:
1. Menentukan metode ekstraksi DNA optimum untuk L. monocytogenes pada PJAS berbasis ikan, khususnya pempek
2. Menentukan primer yang spesifik untuk pengujian L. monocytogenes
3. Menguji limit deteksi rt-PCR ESCO Swift 48 untuk kultur murni
L. monocytogenes dan sampel pempek yang di-spike dengan
L. monocytogenes
Manfaat Penelitian
Manfaat dari penelitian ini yaitu tersedianya metode analisis yang valid untuk mengidentifikasi dan mengkuantifikasi L. monocytogenes pada sampel PJAS berbasis ikan, khususnya pempek, secara cepat sehingga dapat dimanfaatkan oleh berbagai pihak khususnya pemerintah dan peneliti yang terkait dalam bidang keamanan pangan.
2
TINJAUAN PUSTAKA
Karakteristik L. monocytogenes
L. monocytogenes termasuk bakteri berbentuk batang (Gambar 1), Gram
positif, psikotropik, fakultatif anaerob, katalase positif, oksidase-negatif (Campos
et al. 2011). L. monocytogenes umum ditemukan di lingkungan dan berbagai
pangan mentah. L. monocytogenes tergolong bakteri yang mampu beradaptasi pada kondisi lingkungan yang luas (Acciari et al. 2011). L. monocytogenes dapat
bertahan pada pH sekitar 4.4 dan konsentrasi garam di atas 14 %.
L. monocytogenes mampu hidup pada suhu 1-45 °C, sehingga makanan yang
disimpan di refrigerator justru memberi kesempatan hidup bagi
L. monocytogenes yang bagi bakteri lain tidak dapat tumbuh (Kramarenko et al.
2013)
3
Listeria monocytogenes dapat menyebabkan penyakit pada manusia melalui
infeksi yang disebut listeriosis. Listeriosis dapat menyebabkan penyakit pada orang sehat, terlebih lagi pada balita, penderita immunocompromised, golongan wanita hamil, dan orang yang kehilangan imunitas akibat suatu penyakit (Todd et al. 2011). Listeria monocytogenes sering menyebabkan infeksi fatal seperti meningitis, sepsis, atau infeksi pada janin dan keguguran. Bakteri ini dapat menyebabkan penyakit pada orang sehat seperti gastroenteritis dan demam yang akan sembuh dengan sendirinya (Schuppler et al. 2010). Identifikasi dan kuantifikasi L. monocytogenes pada pangan sangat perlu dilakukan mengingat adanya regulasi dari Eropa yang mensyaratkan jumlah cemaran kurang dari 100 CFU/g L. monocytogenes pada pangan siap saji terutama yang ditujukan pada konsumsi anak kecil dan keperluan medis (Moreno et al. 2012).
Metode Analisis Polymerase Chain Reaction (PCR)
Metode klasik dalam deteksi cemaran mikroba di dalam produk pangan awalnya menggunakan isolasi di media dan konfirmasi melalui tes biokimia dan serologi. Pengembangan metode deteksi mikroba selanjutnya yaitu menggunakan PCR yang menghasilkan data yang lebih akurat dan relatif cepat. Metode konvensional meliputi persiapan media kultur (pengayaan, pengayaan selektif, dan penumbuhan pada agar selektif), penghitungan koloni, pengkarakterisasian dengan uji biokimia, yang dapat dilanjutkan dengan penetapan serotipe (serovar) dengan uji serologi. Hasil uji biokimia yang sama dari genus mikroba yang sama, tidak menjamin bahwa mikroba-mikroba tersebut berasal dari serovar yang sama. Untuk mengatasinya, dilakukan uji serologi untuk mengonfirmasi koloni tipikal yang telah diperoleh dari uji morfologi dan uji biokimia. Cara ini juga digunakan untuk mengidentifikasi mikroba pada tingkat subspesies. Kekurangan dari metode ini ialah memerlukan keahlian yang baik, waktu yang lama, dan mahal. Kekurangan tersebut dapat diatasi dengan metode deteksi cepat terhadap mikroba cemaran yaitu teknik PCR.
Metode PCR dapat melipatgandakan sekuens DNA atau RNA menjadi ribuan sampai jutaan copy, sehingga PCR dapat membantu mendeteksi cemaran mikroba pada pangan (Pochop et al. 2013). Pada mulanya, PCR memiliki kelemahan yaitu tidak dapat membedakan DNA dari organisme hidup dan organisme mati. Tahap penting sebelum dilakukannya metode PCR adalah tahap ekstraksi DNA. Hal ini disebabkan pada matriks sampel pangan, khususnya yang berbasis daging, mengandung beberapa komponen seperti produk reaksi mailard, glikogen, lemak, kolagen, asam fulvat, dan besi yang dapat ikut terpurifikasi bersama DNA target. Keberadaan komponen-komponen tersebut memungkinkan terjadinya penghambatan amplifikasi asam nukleat dengan PCR (Camma et al. 2012). Penelitian tentang deteksi L. monocytogenes dengan PCR sudah cukup banyak dilakukan terutama pada jenis pangan siap saji, susu dan produk hasil susu, sayuran, daging dan produk hasil daging, serta produk ikan (Tabel 1).
Rt-PCR merupakan pengembangan teknologi dari PCR standar. Rt-PCR mengamplifikasi dan mendeteksi DNA dalam satu proses secara real-time. Deteksi menggunakan rt-PCR dapat berupa data kualitatif maupun kuantitatif. Hal ini berbeda dengan PCR standar yang hanya dapat mendeteksi mikroba secara kualitatif dari pengamatan dalam gel agarose terhadap amplikon dengan panjang
4
basa tertentu. Kelebihan rt-PCR dibanding PCR standar antara lain: lebih mudah, akurat, cepat, dan sensitif.
Kuantifikasi rt-PCR secara umum dapat dilakukan dengan dua cara: (1) menggunakan dye fluoresence yang berikatan dengan amplikon, dan (2) menggunakan probe oligonukleotida yang dimodifikasi sedemikian rupa
sehingga akan berpendar ketika terhibridisasi dengan DNA komplementer. Jenis
dye fluorescence yang banyak digunakan sampai saat ini yaitu SYBR® Green
sedangkan jenis probe yang digunakan sampai saat ini yaitu TaqMan®, molecular
beacons, dan Scorpions® (Dharmaraj 2009). Penggunaan dye fluorescence relatif
lebih ekonomis namun spesifisitasnya lebih rendah dibanding probe. Tabel 1. Penelitian tentang pengembangan deteksi L. monocytogenes
Tujuan Penelitian Sampel Pustaka
Deteksi L. monocytogenes dengan metode PCR kompetitif
Susu Choi dan Hong 2003
Deteksi gen multi asosiasi virulen L. monocytogenes
Darah, feses, dan susu sapi
Rawool et al. 2007
Perbandingan antara uji PI-PLC dan PCR dalam deteksi patogenisitas L. monocytogenes
Susu dan produk susu
Aurora et al. 2008
Deteksi L. monocytogenes dengan metode immunoassay VIDAS LMO2
Daging babi dan sapi mentah
Meyer et al. 2011
Perbandingan antara deteksi real-time PCR
L. monocytogenes menggunakan SYBR
Green I dan TaqMan
Nutrient broth dan susu
Dadkhah et al, 2012
Deteksi L. monocytogenes dengan metode MPN-PCR Sosis, burger, ikan kaleng, daging cincang, ikan, ayam, daging mentah Marian et al. 2012
Deteksi dan penghitungan L. monocytogenes viabel pada pangan nabati siap saji dengan metode pengkulturan, PCR, dan DVC-FISH
Sayuran Moreno et al. 2012
Deteksi L. monocytogenes viabel dengan real-time Reverse Transcriptase PCR
Daging babi Ye et al, 2012 Deteksi dan isolasi Listeria spp dan
L. monocytogenes dalam berbagai jenis
media agar selektif
396 jenis pangan siap saji dari supermarket Jamali et al. 2013
Deteksi L. monocytogenes dengan metode biosensor multiplex fiber optic
Daging siap saji
Ohk dan Bhunia. 2013
Proses amplifikasi DNA menggunakan rt-PCR ditampilkan secara terkini dalam bentuk grafik pada layar monitor. Grafik-grafik yang diperoleh dapat berupa grafik amplifikasi, kurva standar, dan atau kurva melting. Grafik amplifikasi merupakan grafik utama yang terbentuk selama siklus amplifikasi.
5
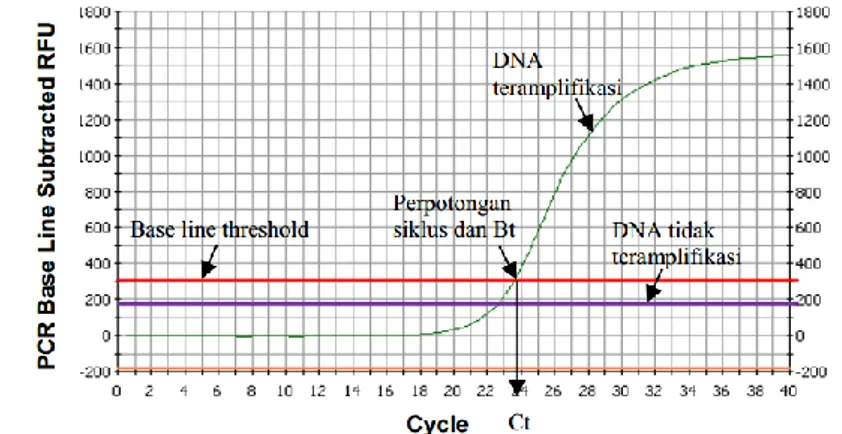
Grafik amplifikasi menunjukkan ada tidaknya DNA target dengan terbentuknya kurva sigmoidal (Gambar 2). Semakin banyak amplikon yang terbentuk maka akan semakin meningkatkan akumulasi flouresen yang terbaca. Akumulasi fluoresen tersebut akan membentuk kurva sigmoidal. Analisis kurva amplifikasi terhadap base line threshold akan menghasilkan nilai yang disebut threshold cycle (CT) (Edwards et al. 2004).
Kurva standar merupakan hubungan antara log konsentrasi bakteri dan CT. Kurva ini dapat digunakan dalam penetapan limit deteksi dan menghitung
konsentrasi bakteri yang belum diketahui pada suatu sampel pangan. Sampel yang digunakan untuk pembuatan kurva standar dibuat serial pengenceran dari pengenceran tertinggi sampai terendah yang masih dapat dideteksi, dengan konsentrasi bakteri dalam kultur antara 100 - 109 CFU/mL. Masing-masing pengenceran diekstrak DNA nya untuk ditentukan nilai CT nya menggunakan
rt-PCR. Pembuatan kurva standar hanya dilakukan untuk deteksi mikroba secara kuantitatif.
Gambar 2. Grafik sigmoidal proses amplifikasi dengan rt-PCR
Kurva melting (peleburan) merupakan kurva turunan dari hubungan antara fluoresen dan suhu (dF/dT). Suhu melting (Tm) merupakan suhu saat 50 %
amplikon DNA untai ganda terpisah menjadi dua untai tunggal. Nilai Tm diketahui
sebagai suhu pada saat munculnya puncak pada kurva melting. Analisis kurva
melting sangat disarankan untuk metode rt-PCR menggunakan SYBR Green®
karena dapat membantu mengidentifikasi adanya produk non-spesifik maupun primer dimer (Brisson et al. 2000).
3
METODE
Bahan
Mikroba standar yang digunakan adalah L. monocytogenes ATCC 7644.
Sedangkan untuk penentuan spesifisitas primer digunakan kontrol
L. monocytogenes ATCC 13932, Escherichia coli ATCC 25922, Staphylococcus
aureus ATCC 25923, Lactobacillus plantarum ATCC 8014, dan Cronobacter
sakazakii ATCC 29544 (Tabel 2). Media yang digunakan untuk penyegaran kultur
6
Tabel 2. Sitotoksisitas mikroba yang digunakan
Spesies Sitotoksisitas Pustaka
L. monocytogenes
ATCC 7644; ATCC 13932
α-hemolitik Choi dan Hong (2003)
E. coli ATCC 25922 β-hemolitik Snyder dan Carson (2010)
S. aureus ATCC 25923 β-hemolitik Mak et al. (2008)
C. sakazakii ATCC 29544 β-hemolitik Fakruddin et al. (2014)
L. plantarum ATCC 8014 - -
Bahan kimia yang digunakan adalah lisozim ( ≥ 20000 unit/mg protein) (Bio Basic, Canada), proteinase K ( > 30 unit/mg protein) (Peqlab, Jerman), cetyl trimethylammonium bromide (CTAB) (Bio Basic, Kanada), ethylenediaminetetraacetic acid (EDTA) (Amersham BioSciences, UK), QIAamp DNA Blood Mini Kit (Qiagen, Jerman), agarose (Bio Basic, Kanada), larutan Tris-HCl (Promega, US), buffer tris-EDTA (TE), larutan sodium dodecyl sulfate (SDS) (Merck, Jerman), isopropanol (Merck, Jerman), SYBR® Green (Thermo Fisher Scientific, US), amonium asetat (Merck, Jerman), fenol (MP Bio, US), kloroform (J.T. Baker, Meksiko), NaCl (Merck, Jerman), dan etanol (Merck, Jerman). Primer yang digunakan yaitu primer forward LIM 2
(CTAAAGCGGGAATCTCCCTT) / primer reverse LIMRE
(CCATTGTCTTGCGCGTTAAT) (Alpha DNA, US) untuk gen iap serta primer forward DG69 (GTGCCGCCAAGAAAAGGTTA) / primer reverse DG74 (CGCCACACTTGAGATAT) (Alpha DNA, US) untuk gen hlyA. Bahan PJAS berbasis ikan yang digunakan sebagai contoh adalah pempek.
Alat
Alat-alat yang digunakan antara lain adalah rt-PCR Swift™ Spectrum 48 (ESCO, Singapura), AB 2720 PCR Thermal Cycler (Applied Biosystems, US), Bio-Rad Universal Hood II Gel Doc (Bio-Rad, US), Gel Elektroforesis PowerPac® Basic (Bio-Rad, US), kamera digital Canon® EOS 600D (Canon, Jepang), Spektofotometer UV 2450 (Shimadzu, Jepang), magnetic stirrer,
stomacher, inkubator, bunsen, pipet mikro, water bath, laminar air flow, hot
plate, autoklaf, tabung mikrosentrifus, jangka sorong, dan alat gelas lainnya.
Tahapan Penelitian
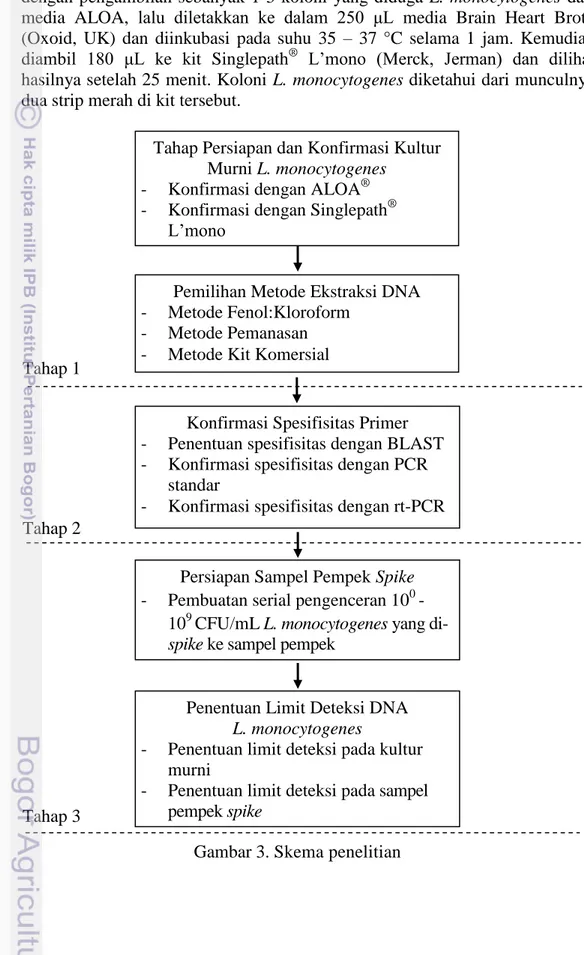
Tahapan penelitian utama pada penelitian ini meliputi 3 tahap utama (Gambar 3). Tahap-tahap utama tersebut adalah; (1) pemilihan metode ekstraksi terbaik, (2) konfirmasi spesifisitas primer, (3) penentuan limit deteksi DNA
L. monocytogenes.
1. Persiapan dan Konfirmasi Kultur Murni L. monocytogenes
Konfirmasi L. monocytogenes dilakukan dengan media selektif Agar
Listeria Ottaviani Agosti (ALOA) (Merck, Jerman) dan kit immuno assay
GLISA Singlepath® L’mono (Merck, Jerman). Konfirmasi dengan media ALOA diawali dengan inokulasi kultur L. monocytogenes pada media TSB
7
selama 18 jam pada suhu 37 °C. Selanjutnya kultur murni diambil 0.1 mL untuk ditumbuhkan pada media ALOA, kemudian diinkubasi pada suhu 37 °C selama 24 jam. Koloni L. monocytogenes ditandai dengan warna hijau dengan lingkaran (halo) putih keruh.
Konfirmasi dengan kit Singlepath® L’mono (Merck, Jerman) diawali dengan pengambilan sebanyak 1-3 koloni yang diduga L. monocytogenes dari media ALOA, lalu diletakkan ke dalam 250 μL media Brain Heart Broth (Oxoid, UK) dan diinkubasi pada suhu 35 – 37 °C selama 1 jam. Kemudian diambil 180 μL ke kit Singlepath®
L’mono (Merck, Jerman) dan dilihat hasilnya setelah 25 menit. Koloni L. monocytogenes diketahui dari munculnya dua strip merah di kit tersebut.
Tahap 1
Tahap 2
Tahap 3
Gambar 3. Skema penelitian Tahap Persiapan dan Konfirmasi Kultur
Murni L. monocytogenes - Konfirmasi dengan ALOA® - Konfirmasi dengan Singlepath®
L’mono
Pemilihan Metode Ekstraksi DNA - Metode Fenol:Kloroform
- Metode Pemanasan - Metode Kit Komersial
Konfirmasi Spesifisitas Primer - Penentuan spesifisitas dengan BLAST - Konfirmasi spesifisitas dengan PCR
standar
- Konfirmasi spesifisitas dengan rt-PCR
Persiapan Sampel Pempek Spike - Pembuatan serial pengenceran 100 -
109 CFU/mL L. monocytogenes yang
di-spike ke sampel pempek
Penentuan Limit Deteksi DNA L. monocytogenes
- Penentuan limit deteksi pada kultur murni
- Penentuan limit deteksi pada sampel pempek spike
8
2. Pemilihan Metode Ekstraksi DNA untuk Kultur Murni
Metode ekstraksi yang digunakan sebagai perlakuan terdiri dari tiga metode, yaitu 1) metode fenol-kloroform, 2) metode pemanasan 3) metode
kit komersial. Penentuan metode terbaik diawali dengan inokulasi
L. monocytogenes pada media TSB selama 18 jam pada suhu 37°C.
Selanjutnya kultur murni overnight dalam TSB diisolasi DNA-nya dengan tiga metode tersebut.
1. Metode fenol:kloroform oleh Sambrook dan Russel (2001) yang dimodifikasi
Modifikasi yang dilakukan terhadap metode fenol:kloroform oleh Sambrook dan Russel (2001) adalah penggunaan buffer TE yang digunakan pada 1 mL kultur bakteri untuk menggantikan buffer lisis. Campuran buffer lisis dan 0.2 mg/mL proteinase K diganti dengan campuran antara 500 µL buffer TE 1X dan 100 µL lisozim. Pelarut fenol:kloroform (1:1 v/v) dipakai sebagai ganti dari pelarut fenol:kloroform:isoamil alkohol (25:24:1 v/v/v). Garam 10 M ammonium asetat pH 7.4 digunakan sebagai pengganti 0.3 M natrium asetat pH 4.8. Selain itu, etanol absolut diganti dengan isopropanol. Cara pengeringvakuman pelet diganti dengan cara pengeringudaraan.
Sampel diambil 1 mL, lalu disentrifus dan pelet diresuspensi dengan 500 μL buffer TE 1X sebanyak 2 kali. Setelah disentrifus, pelet diresuspensi dengan 500 μL buffer TE 1X dan 100 μL lisozim, diinkubasi pada suhu 4 °C selama 5 menit. Kemudian ditambahkan 25 μL SDS 10 %, 50 μL NaCl 5M, dan 100 μL proteinase K (20 mg/mL), divortex dan diinkubasi pada suhu 65 °C selama 2 jam. Selanjutnya ditambahkan 500 μL fenol:kloroform (1:1 v/v), divortex dan diinkubasi -20 °C selama 30 menit, lalu disentrifus pada suhu 4 °C, 12000 rpm selama 10 menit. Supernatan diambil 500 μL dan dipindahkan ke tabung eppendorf baru, ditambahkan 500 μL fenol:kloroform (1:1 v/v), divortex dan diinkubasi -20 °C selama 30 menit, lalu disentrifus pada suhu 4°C, 12000 rpm selama 10 menit. Supernatan diambil 500 μL dan dipindahkan ke tabung eppendorf baru, ditambahkan 500 μL kloroform, disentrifus pada suhu 4 °C, 12000 rpm selama 10 menit. Supernatan diambil 500 μL dipindahkan ke tabung eppendorf baru, ditambahkan ammonium asetat 10 M pH 7.4 sebanyak 150 μL volume dan isopropanol dingin 500 μL, diinkubasikan -20 °C selama semalam, kemudian disentrifus 14000 rpm selama 30 menit. Selanjutnya pelet ditambah dengan 500 μL etanol 70 % dan disentrifus 12000 rpm selama 5 menit. Supernatan dibuang dan pelet dikeringudarakan, kemudian dilarutkan dalam 50 μL buffer TE 1X.
2. Metode pemanasan oleh Rahayu et al. (2009) yang dimodifikasi
Metode ini merupakan metode dari Rahayu et al. (2009) yang mengacu pada metode dari Hein et al. (2001). Modifikasi yang dilakukan pada metode yang digunakan oleh Rahayu et al. (2009) adalah penurunan suhu pemanasan dari 100 menjadi 95 °C.
Masing-masing kultur murni dimasukkan ke dalam tabung mikrosentrifus. Suspensi tersebut disentrifus dalam mikrosentrifus selama 10 menit 14.000 rpm dengan suhu 20 °C dengan tujuan untuk mengendapkan sel bakteri. Supernatan yang dihasilkan dibuang dan pelet
9
diresuspensi dengan 500 μL buffer TE lalu dihomogenkan dengan cara divortex. Suspensi tersebut disentrifus kembali pada mikrosentrifus selama 3 menit 14.000 rpm pada suhu 20 °C. Pelet yang dihasilkan diresuspensi dengan 100 μL lisozim dan dihomogenisasi dengan cara divortex. Kemudian suspensi tersebut diinkubasi pada suhu 25 °C selama 15 menit. Setelah diinkubasi, sebanyak 100 μL proteinase K ditambahkan ke dalam suspensi dan divortex sampai homogen. Selanjutnya suspensi diinkubasi pada suhu 65 °C selama 1 jam dan dinkubasi kembali pada suhu 95 °C selama 15 menit. Proses tersebut bertujuan untuk melisiskan sel bakteri dan menghilangkan protein yang terkandung di dalamnya. Penambahan buffer TE berfungsi untuk menjaga kestabilan DNA pada saat melisiskan sel. Suspensi yang telah diinkubasi didinginkan dalam es sampai membeku dan
di-thawing dan disentrifus selama 5 menit 14.000 rpm pada suhu 20 °C.
Supernatan yang dihasilkan dipindahkan pada tabung mikrosentrifus baru, disimpan pada suhu -20 °C sebagai DNA target.
3. Metode Kit Komersial QIAamp® DNA Blood Mini Kit yang dimodifikasi Modifikasi yang dilakukan terhadap metode dari produsen yaitu penggunaan 1 mL CTAB untuk preparasi awal sampel, kemudian divortex hingga homogen. Suspensi tersebut disentrifus selama 1 menit dengan kecepatan 13000 rpm dan supernatan yang dihasilkan dibuang sehingga menyisakan pelet pada tabung mikrosentrifus. Tahap selanjutnya sesuai dengan petunjuk dari produsen.
QIAamp® DNA Blood Mini Kit tersebut terdiri dari empat jenis buffer (AW1, AW2, AE, dan AL), collection tube, dan kolom mini dimana di dalamnya terdapat filter putih. Teknis pengisolasian DNA dengan kit komersial pertama-tama adalah masing-masing satu mL suspensi kultur murni mikroba spesifik yang terdapat dalam media NB dimasukkan ke dalam tabung mikrosentrifus. Kemudian ditambahkan dengan 1 mL CTAB sebagai suatu modifikasi metode dari prosedur yang ditunjukkan pada
handbook produsen kit, kemudian divortex hingga homogen. Suspensi
tersebut disentrifus selama 1 menit dengan kecepatan 13000 rpm dan supernatan yang dihasilkan dibuang sehingga menyisakan pelet pada tabung mikrosentrifus. Jika tidak dihasilkan pelet, maka suspensi disisakan sebanyak 200 μL supernatan pada tabung mikrosentrifus. Kemudian ditambahkan proteinase K sebanyak 30 μL ke dalam tabung mikrosentrifus, dan dihomogenkan dengan vortex kemudian diinkubasi selama 20 menit pada suhu 65 °C.
Setelah itu, ditambahkan buffer AL sebanyak 300 μL dan dihomogenkan dengan vortex. Suspensi diinkubasi selama 10 menit di dalam water bath pada suhu 65 °C. Setelah itu, ditambahkan 500 μL etanol (100 %), dihomogenkan dengan vortex dan disentrifus selama 2 menit dengan kecepatan 10000 rpm. Supernatan yang dihasilkan dipipet dan dimasukkan ke dalam kolom mini yang sudah dipasang pada collection tube, kemudian disentrifus 8000 rpm selama 1 menit. Sebanyak 500 μL buffer AW1 ditambahkan ke dalam kolom mini yang masih terpasang pada
collection tube dan disentrifus 8000 rpm selama 1 menit.
Tahap selanjutnya adalah kolom mini dipindahkan ke dalam collection
10
kolom mini, kemudian disentrifus 13000 rpm selama 3 menit. Kolom mini dipindahkan kembali ke dalam collection tube dan ditambah 500 μL buffer AW2 dan disentrifus kembali selama 1 menit 13000 rpm. Kolom mini dipindahkan ke dalam tabung mikrosentrifus steril dan ditambahkan ke dalamnya sebanyak 80 μL buffer AE yang kemudian disentrifus 8000 rpm selama 1 menit. Cairan yang diperoleh pada tabung mikrosentrifus disimpan pada freezer dengan suhu -20 °C sebagai DNA target.
Visualisasi Hasil Ekstraksi DNA
Hasil ekstraksi DNA dari tiga metode tersebut dinilai melalui penentuan kualitas dan kemurnian DNA. Tahap ini diawali dengan elektroforesis DNA menggunakan 1.5 % gel agarose pada tegangan 100 volts. Setelah dimigrasi, gel berisi DNA direndam dalam larutan Ethidium Bromida (EtBr) kemudian dicuci menggunakan air selama kurang lebih 10 menit. Setelah itu, gel dimasukkan ke dalam Bio-RAD gelldoc untuk melihat pita DNA genom. Teknis pengukurannya adalah dengan membandingkan nilai absorbansi pada panjang gelombang 260 dan 280 nm (A260/A280). Metode ekstraksi terpilih didasarkan pada jumlah konsentrasi DNA genom yang paling tinggi.
3. Konfirmasi Spesifisitas Primer
Tahap awal yang dilakukan sebelum pemilihan primer yaitu penentuan spesifisitas masing-masing primer menggunakan BLAST. Pemilihan primer dilakukan dengan prosedur kerja PCR standar (Lampiran 1, 2, 3 dan 4). Amplikon yang dihasilkan dari PCR standar dinilai dengan pengamatan menggunakan gel elektroforesis. Primer yang dijadikan perlakuan adalah : a. Primer LIM 2 dan LIMRE yang merupakan sekuens dari gen iap (Dadkhah
et al. 2012).
b. Primer DG69 dan DG74 yang merupakan sekuens dari gen hlyA (Choi dan Hong 2003).
DNA yang digunakan yaitu DNA L. monocytogenes sebagai DNA target, dibandingkan dengan DNA S. aureus dan C. sakazakii sebagai DNA bakteri lain. Primer terbaik didasarkan pada spesifisitasnya menggunakan PCR standar. Primer terpilih digunakan untuk tahap-tahap selanjutnya.
Spesifisitas primer dinilai menggunakan prosedur kerja rt-PCR (Lampiran 5, 6, 7 dan 8). DNA yang digunakan yaitu DNA L. monocytogenes
sebagai DNA target, dibandingkan dengan DNA E. coli, S. aureus,
L. plantarum, dan C. sakazakii sebagai DNA bakteri lain. Parameter yang
diamati adalah pola kurva amplifikasi dan kurva melting. Spesifisitas primer ditunjukkan oleh tidak adanya amplifikasi dari DNA lain. Spesifisitas primer pada kurva melting ditunjukkan dengan nilai puncak kurva melting (Tm) yang
berbeda antara DNA target, kontrol negatif dan kontrol DNA bakteri lain. 4. Persiapan Sampel Pempek Spike
Sampel yang digunakan yaitu pempek yang telah direbus dan belum digoreng. Langkah pertama dilakukan pengambilan sampel pempek sebanyak 25 g untuk dihomogenkan dengan 75 mL BPW yang mengandung konsentrasi
11
yang telah di-spike digunakan sebagai sampel untuk penentuan limit deteksi
DNA L. monocytogenes pada sampel pangan spike.
5. Penentuan Limit Deteksi DNA L. monocytogenes
Pengujian dilakukan untuk deteksi DNA L. monocytogenes kultur murni dan sampel pangan spike (SPS). Sensitivitas dari realtime PCR untuk deteksi
L. monocytogenes ditentukan dengan pembuatan kurva standar hubungan nilai
threshold cycle (CT) dan log konsentrasi kultur murni. Kurva standar
memberikan persamaan linear yang digunakan untuk mengukur limit deteksi
L. monocytogenes dalam sampel pangan. Persamaan linear kurva standar
digunakan untuk menghitung konsentrasi bakteri yang belum diketahui dalam suatu kultur. Konsentrasi kultur L. monocytogenes dapat dihitung dengan memasukkan nilai CT hasil amplifikasi sebagai nilai y pada persamaan linear,
kemudian nilai x yang diperoleh dicari nilai antilog-nya.
Tahapan pertama persiapan sampel untuk kultur murni diawali dengan membuat serial pengenceran dari pengenceran tertinggi sampai terendah yang
masih dapat dideteksi, dengan konsentrasi bakteri dalam kultur antara 100 - 109 CFU/mL. Langkah selanjutnya dilakukan pengambilan 1 mL untuk
ekstrasi DNA dan 1 mL lainnya untuk dihitung konsentrasi bakterinya dengan metode pencawanan (FDA 2001). DNA y a n g diekstrak dari beberapa tingkat pengenceran tersebut kemudian diamplifikasi dengan rt-PCR. Perpotongan kurva amplifikasi yang dihasilkan dengan basement-threshold menghasilkan nilai CT. Sampel yang dipakai untuk pembuatan kurva standar L.
monocytogenes ditentukan berdasarkan :
a. Sampel yang masih terdeteksi jumlah koloninya pada saat pencawanan b. Sampel yang nilai CT nya di bawah nilai siklus tertinggi yang masih
menunjukkan spesifisitas primer (berdasarkan uji spesifisitas primer)
4
HASIL DAN PEMBAHASAN
Pemilihan Metode Ekstraksi DNA
Salah satu tahap penting dalam analisis DNA dengan metode PCR yaitu tahap ekstraksi DNA. Ekstraksi DNA menjadi titik kritis yang sangat mempengaruhi hasil analisis. Jumlah dan mutu DNA hasil ekstraksi akan mempengaruhi analisis lebih lanjut dengan PCR. Hasil ekstraksi DNA menggunakan tiga metode pada penelitian ini dievaluasi melalui tahap elektroforesis menggunakan gel agarose. Hasil visualisasi DNA dengan gel elektroforesis berupa pita DNA genom dari masing-masing metode ekstraksi (Gambar 4).
Hasil elektroforesis terhadap DNA L. monocytogenes (Gambar 4) menunjukkan bahwa metode ekstraksi fenol: kloroform yang dimodifikasi dari Sambrook dan Russel (2001) menghasilkan pita yang paling jelas dibanding dua metode lainnya. Hal ini dimungkinkan karena konsentrasi yang dihasilkan dari ekstraksi fenol:kloroform yang lebih tinggi dibandingkan metode lainnya (Tabel 3). Pita yang dihasilkan dengan metode fenol:kloroform ada 3 pita, yaitu
12
pita yang terdapat di antara marker 500 bp dan 1000 bp, di antara marker 1000 bp dan 1500 bp, serta di atas 10000 bp.
Keterangan : a. Kontrol
b. DNA hasil isolasi metode fenol:kloroform
c. DNA hasil isolasi metode pemanasan
d. DNA hasil isolasi metode kit komersial
e. DNA ladder 1kb
Gambar 4. Hasil elektroforesis pada tahap ekstraksi DNA Tabel 3. Kemurnian dan konsentrasi DNA
Metode Ekstraksi
Kultur murni Sampel pempek spike A260/A280 Konsentrasi DNA (ng/µL) A260/A280 Konsentrasi DNA (ng/µL) Fenol:kloroform 1.24 1857 1.28 1406 Pemanasan 0.40 1027 - - Kit komersial 0.76 915 0.70 656
DNA dikatakan murni jika rasio diantara kedua nilai absorbansi 260 dan 280 berada di kisaran 1.8 (Nolan et al. 2007). Pengukuran kemurnian DNA (Tabel 3) menunjukkan bahwa DNA yang dihasilkan dari metode fenol:kloroform untuk sampel kultur murni dan sampel pempek spike masih terdapat pengotor lain ditunjukkan oleh nilai rasio A260 dan A280 di bawah 1.8. Pengotor yang diduga menyebabkan hal tersebut diduga adalah protein. Hal ini dapat disebabkan karena kurangnya proses pencucian menggunakan isopropanol. Bettelheim et al. (2013) menjelaskan bahwa isopropanol dapat meningkatkan kemurnian DNA dengan mengoptimalkan proses pembuangan residu protein.
Hasil ekstraksi metode pemanasan menghasilkan konsentrasi DNA dari kultur murni yang lebih kecil dibanding metode fenol:kloroform, hal ini dapat disebabkan karena rusaknya sebagian DNA karena pengaruh suhu 95 °C. Menurut Sambrook dan Russel (2001) pendidihan dapat memiliki potensi mendenaturasi DNA menjadi irreversibel. Metode pemanasan tidak dapat diaplikasikan untuk sampel pempek dikarenakan perlakuan pemanasan ternyata menggumpalkan pati dari sampel pempek yang terbuat dari campuran ikan dan tapioka. Coral et al. (2009) menjelaskan bahwa, pati secara umum akan mulai tergelatinisasi ketika mencapai suhu di atas 70 °C.
13
Metode kit komersial QIAmp DNA Blood Mini Kit telah digunakan sebelumnya oleh Dauphin et al. (2009), Nur’utami et al. (2011) dan Dibbern et al. (2015). Metode kit komersial oleh Dauphin et al. (2009) yang digunakan untuk bakteri Brucella menghasilkan DNA yang memiliki kemurnian yang kurang baik dimana nilai rasio A260/A280 lebih kecil dari 1.8, sedangkan penelitian Dibbern et al. (2015) pada bakteri S. aureus justru menunjukkan bahwa metode kit QIAmp DNA Blood Mini Kit justru menghasilkan kemurnian DNA yang lebih baik dari metode lainnya pada rasio A260/A280 sebesar 1.76. Metode yang dikembangkan oleh Nur’utami et al. (2011) dengan modifikasi penggunaan CTAB untuk pelisisan awal menghasilkan kemurnian DNA
Salmonella Typhimurium yang baik sekitar 2.0. Hal ini berbeda dengan hasil
penelitian kali ini yang menghasilkan kemurnian DNA di bawah 1.0. Konsentrasi yang dihasilkan metode kit juga lebih rendah dibanding metode fenol:kloroform baik untuk sampel kultur murni maupun sampel pempek spike. Hal ini dimungkinkan karena CTAB yang digunakan sebagai pengganti tissue lysis
buffer (buffer ATL) kurang optimal jika digunakan untuk bakteri Gram positif.
Buffer ATL direkomendasikan oleh produsen sebagai buffer yang digunakan
dalam pelisisan jaringan sel. Ekstraksi DNA selanjutnya dilakukan dengan metode fenol:kloroform.
Konfirmasi Spesifisitas Primer
Tahapan awal sebelum penentuan spesifisitas primer yaitu pemilihan primer yang sesuai untuk deteksi L. monocytogenes. Penelitian ini menggunakan dua pasang primer berbeda. Masing-masing pasangan primer diuji menggunakan PCR standar. Masing-masing primer dianalisis dengan menggunakan Basic Local
Alignment Search Tool (NCBI) dan ditentukan data spesies yang memiliki
kecocokan 100% terhadap urutan sekuens primer yang dianalisis (Lampiran 9, 10, 11 dan 12). Untuk mengkonfirmasi bahwa kedua primer tersebut spesifik terhadap L. monocytogenes maka dilakukan pengujian dengan PCR standar dan rt-PCR.
Primer LIM 2/LIMRE digunakan untuk mengamplifikasi gen iap. Gen iap
(invasion associated secreted endopeptidase) merupakan gen yang memegang
peranan penting pada viabilitas sel L. monocytogenes. Gen iap memproduksi protein p60 yang mempunyai aktivitas bakteriolitik (Kohler et al. 1991). Gen iap hanya terdapat pada L. monocytogenes dan tidak terdapat pada bakteri lain yang digunakan sebagai kontrol. Hal ini sesuai dengan hasil BLAST yang menunjukkan bahwa kecocokan 100% dari primer forward (LIM 2) dan reverse (LIMRE) hanya berasal dari spesies Listeria saja (Lampiran 9 dan 10). Hasil PCR standar dengan primer LIM 2/LIMRE menunjukkan adanya pita (band) yang terbentuk pada hasil elektroforesis produk amplifikasi PCR dengan ukuran di bawah 200 bp pada semua sampel (Gambar 5). Hal ini menunjukkan bahwa kondisi running untuk primer LIM 2/LIMRE belum baik karena masih menghasilkan produk non spesifik.
Primer DG69/DG74 mengamplifikasi gen hlyA yang berperan dalam produksi 58 kDa listeriolysin O yang menjadi penentu virulensi dan banyak digunakan untuk mendeteksi L. monocytogenes (Choi dan Hong 2003). Listeriolysin O ini merupakan toksin penyebab terjadinya hemolisis (kerusakan sel darah merah) yang masuk
14
kategori α-hemolitik. Bakteri lain yang digunakan sebagai kontrol juga mempunyai kemampuan hemolitik, antara lain E. coli ATCC 25922 (Snyder dan Carson 2010),
S. aureus ATCC 25923 (Mak et al. 2008) dan C. sakazakii ATCC 29544
(Fakruddin et al. 2014) yang ketiga-tiganya termasuk kategori β-hemolitik. Sedangkan L. plantarum ATCC 8014 tidak mempunyai sifat hemolitik. Hasil PCR standar dengan primer DG69/DG74 menunjukkan adanya pita 635 bp hanya pada DNA L. monocytogenes (Gambar 5). Hal ini sesuai dengan hasil BLAST yang menunjukkan bahwa kecocokan 100% dari primer forward (DG69)
dan reverse (DG74) hanya berasal dari spesies Listeria saja (Lampiran 11 dan
12).
Perbandingan hasil kedua primer (Gambar 5) menunjukkan bahwa primer DG69/DG74 lebih cocok digunakan untuk deteksi L. monocytogenes. Selain itu, primer DG69/DG74 lebih banyak dipakai pada penelitian-penelitian sebelumnya untuk deteksi L. monocytogenes dibanding primer LIM 2/LIMRE. Amplikon primer DG69/DG74 berada pada ukuran 635 bp sehingga lebih mudah dibedakan dari produk non spesifik maupun primer dimer.
Keterangan: a. Ladder 100bp b.Kontrol tanpa DNA
c. DNA L. monocytogenes
ATCC 7644
d. DNA S. aureus ATCC
25923
e. DNA C. sakazakii ATCC
29544 (a) (b)
Gambar 5. Hasil elektroforesis produk PCR dengan primer (a) LIM 2/LIMRE dan (b) DG69/DG74
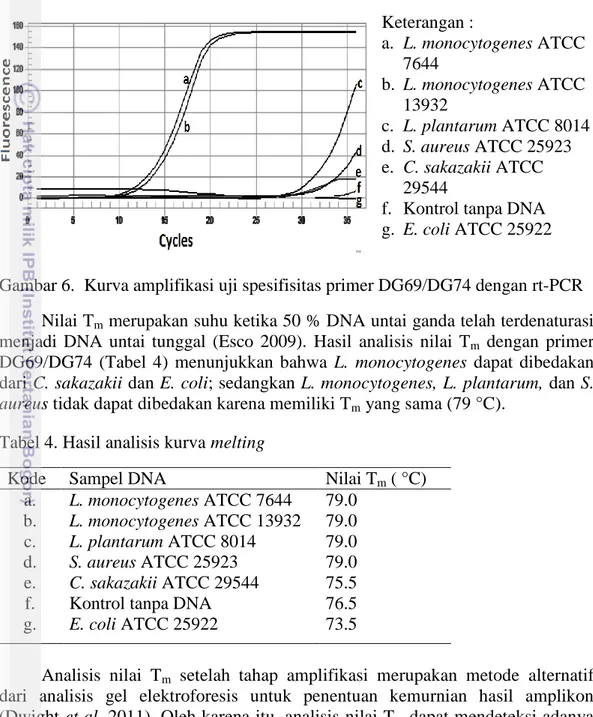
Pengujian spesifisitas primer dilakukan untuk menentukan kemampuan primer dalam membedakan DNA bakteri target dengan bakteri lain. Hasil uji spesifisitas primer DG69/DG74 dengan rt-PCR menunjukkan bahwa kondisi
running yang dipakai dapat mendeteksi DNA L. monocytogenes dengan hasil
kurva amplikasi yang baik sampai siklus 27 (Gambar 6). Setelah siklus ke-27, kondisi running tersebut tidak spesifik lagi karena dapat menghasilkan produk non-spesifik pada sampel L. plantarum, S. aureus, C. sakazakii, E. coli, dan kontrol tanpa DNA.
Berdasarkan hasil BLAST, kontrol bakteri lain yang digunakan tidak satupun yang memiliki kecocokan 100 % terhadap sekuens primer yang digunakan. Sehingga, adanya produk hasil amplifikasi pada DNA kontrol bakteri lain setelah siklus ke-27 menunjukkan munculnya produk non spesifik atau primer dimer. Penggunaan SYBR Green secara umum kurang spesifik dibanding metode lain seperti Taqman. Hal ini disebabkan karena SYBR green dapat berikatan dengan DNA rantai ganda non-spesifik ataupun terjadinya primer dimer (Jenie 2013). Primer-dimer merupakan proses saling berikatannya primer
15
yang teramplifikasi dan terkuantifikasi sehingga menghasilkan data
false-positive (Pestana et al. 2010). Menurut Altshuler (2006), munculnya primer
dimer dan produk non-spesifik dapat dikurangi dengan optimasi kondisi running. Oleh karena itu, metode rt-PCR menggunakan SYBR Green biasanya diikuti dengan analisis kurva melting untuk melihat suhu pelelehan (Tm) dari
masing-masing sampel. Keterangan : a. L. monocytogenes ATCC 7644 b. L. monocytogenes ATCC 13932 c. L. plantarum ATCC 8014 d. S. aureus ATCC 25923 e. C. sakazakii ATCC 29544
f. Kontrol tanpa DNA
g. E. coli ATCC 25922
Gambar 6. Kurva amplifikasi uji spesifisitas primer DG69/DG74 dengan rt-PCR Nilai Tm merupakan suhu ketika 50 % DNA untai ganda telah terdenaturasi
menjadi DNA untai tunggal (Esco 2009). Hasil analisis nilai Tm dengan primer
DG69/DG74 (Tabel 4) menunjukkan bahwa L. monocytogenes dapat dibedakan dari C. sakazakii dan E. coli; sedangkan L. monocytogenes, L. plantarum, dan S.
aureus tidak dapat dibedakan karena memiliki Tm yang sama (79 °C).
Tabel 4. Hasil analisis kurva melting
Kode Sampel DNA Nilai Tm ( °C)
a. L. monocytogenes ATCC 7644 79.0
b. L. monocytogenes ATCC 13932 79.0
c. L. plantarum ATCC 8014 79.0
d. S. aureus ATCC 25923 79.0
e. C. sakazakii ATCC 29544 75.5
f. Kontrol tanpa DNA 76.5
g. E. coli ATCC 25922 73.5
Analisis nilai Tm setelah tahap amplifikasi merupakan metode alternatif
dari analisis gel elektroforesis untuk penentuan kemurnian hasil amplikon (Dwight et al. 2011). Oleh karena itu, analisis nilai Tm dapat mendeteksi adanya
mispriming ataupun primer dimer lebih cepat dibanding analisis gel
elektroforesis. Manfaat lain dari analisis nilai Tm yaitu penentuan variasi genetik
dengan mudah tanpa proses sekuensi (Erali dan Wittwer 2010; Montgomery et al. 2010).
16
Penentuan Limit Deteksi DNA L. monocytogenes
Limit deteksi merupakan nilai pengenceran tertinggi yang masih dapat teramplifikasi yang menunjukkan sensitivitas deteksi suatu metode. Limit deteksi rt-PCR menggunakan primer DG69/DG74 ditentukan dari konsentrasi saat nilai CT = 27 (berdasarkan tahap konfirmasi spesifisitas primer). Hasil analisis limit
deteksi pada sampel kultur murni dan sampel pempek spike dengan rt-PCR
menghasilkan data nilai CT dan nilai Tm dari masing-masing konsentrasi
L. monocytogenes (Tabel 5).
Tabel 5. Hasil analisis rt-PCR DNA L. monocytogenes
Kultur murni Sampel pempek spike
Konsentrasi (CFU/mL) CT Tm ( °C) Jumlah koloni (CFU/g) CT Tm ( °C) 4.3 x 109 8.06 78.5 4.1 x 108 9.32 79.0 5.7 x 108 11.59 78.5 4.1 x 107 12.60 79.0 5.6 x 107 12.67 78.5 4.4 x 106 16.04 79.0 5.8 x 106 16.17 78.5 4.4 x 105 20.94 79.0 4.9 x 105 21.07 78.5 4.8 x 104 24.28 79.0 5.3 x 104 23.23 79.0 4.4 x 103 26.93 79.0 4.2 x 103 27.13 79.0 < 1.0 x 103 27.27 79.5 5.8 x 102 33.86 78.5
Hasil nilai CT pada kultur murni dan sampel pempek spike (Tabel 5)
menunjukkan bahwa semakin besar konsentrasi L. monocytogenes maka nilai CT
akan semakin kecil. Hal ini terjadi karena konsentrasi bakteri yang semakin tinggi akan menghasilkan DNA terekstrak yang lebih banyak, sehingga semakin tinggi DNA yang terekstrak maka semakin cepat mencapai garis basement-threshold saat proses amplifikasi berjalan (Applied Biosystems 2015). Hasil analisis nilai Tm pada kultur murni dan sampel pempek spike yang berada pada nilai 79 ± 0.5
menunjukkan bahwa bahwa kondisi running yang diterapkan telah spesifik dan tidak terjadi mispriming.
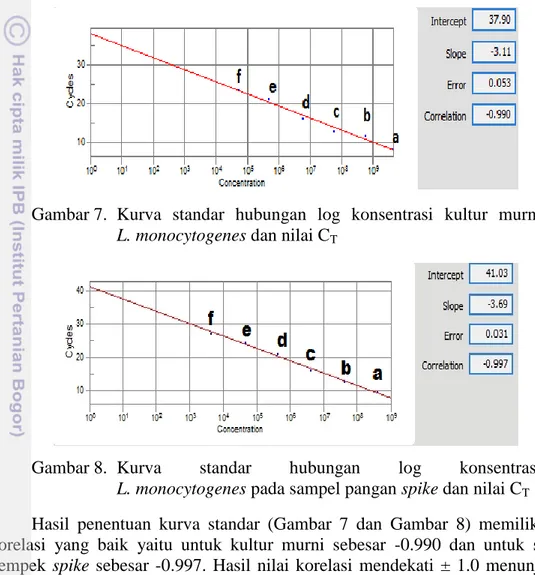
Hasil analisis deteksi L. monocytogenes dengan rt-PCR (Tabel 5) dapat dibuat kurva standar hubungan nilai CT dan konsentrasi L. monocytogenes. Kurva
standar yang dihasilkan pada kultur murni (Gambar 7) memiliki persamaan: CT : Nilai CT
C : Nilai Log Konsentrasi bakteri (CFU/mL)
Sedangkan kurva standar yang dihasilkan pada sampel pempek spike (Gambar 8) memiliki persamaan :
CT : Nilai CT
C : Nilai log konsentrasi bakteri pada SPS
Nilai konstanta persamaan kurva standar kultur murni (37.9) lebih kecil dibanding nilai konstanta SPS (41.03). Sedangkan nilai koefisien persamaan
CT = 37.9 – 3.11 C
17
kurva standar kultur murni (3.11) sedikit lebih rendah dibanding nilai koefisien SPS (3.69). Ini menunjukkan bahwa pada konsentrasi L. monocytogenes yang rendah, sampel kultur murni lebih dulu teramplifikasi dibanding SPS. Sedangkan pada konsentrasi L. monocytogenes yang semakin tinggi, SPS justru lebih cepat teramplifikasi. Hal ini menunjukkan bahwa matriks pangan pempek mempengaruhi hasil amplifikasi di rt-PCR. Menurut Dadkhah et al. (2012) dan Camma et al. (2012), adanya komponen lain dari pangan dapat menjadi sebab terjadinya perbedaan hasil amplifikasi, baik yang disebabkan karena pengaruh saat ekstraksi maupun pengaruh saat proses amplifikasi rt-PCR.
Gambar 7. Kurva standar hubungan log konsentrasi kultur murni
L. monocytogenes dan nilai CT
Gambar 8. Kurva standar hubungan log konsentrasi
L. monocytogenes padasampel pangan spike dan nilai CT
Hasil penentuan kurva standar (Gambar 7 dan Gambar 8) memiliki nilai korelasi yang baik yaitu untuk kultur murni sebesar -0.990 dan untuk sampel pempek spike sebesar -0.997. Hasil nilai korelasi mendekati ± 1.0 menunjukkan hubungan yang sangat kuat antara konsentrasi L. monocytogenes terhadap nilai CT
yang dihasilkan (Taylor 1990). Nilai korelasi yang negatif menunjukkan bahwa semakin besar nilai pada sumbu x (konsentrasi L. monocytogenes) maka semakin kecil nilai pada sumbu y (nilai CT).
Persamaan kurva standar yang telah dihasilkan dapat digunakan untuk kuantifikasi sampel yang belum diketahui konsentrasi L. monocytogenes-nya. Selain itu, penentuan limit deteksi dari sampel kultur murni dan sampel pempek
spike juga dapat ditentukan dengan persamaan kurva standar yang telah didapat.
Jika dihubungkan dengan hasil tahap konfirmasi spesifisitas bahwa metode ini spesifik hanya sampai siklus 27, maka limit deteksi didapat dengan memasukkan nilai 27 ke persamaan kurva standar sebagai nilai CT.Sehingga, limit deteksinya
18
didapat dari nilai C dari persamaan kurva standar tersebut. Hasil limit deteksi pada sampel kultur murni dan sampel pempek spike masing-masing adalah 3.2 x 103 CFU/mL dan 6.3 x 103 CFU/g. Hasil limit deteksi ini mendekati hasil dari metode yang digunakan oleh Dadkhah et al. (2012) yang memiliki limit deteksi pada sampel kultur murni sebesar 1.2 x 103 CFU/mL dan limit deteksi pada sampel susu sebesar 2.1 x 103 CFU/mL.
Hasil penentuan limit deteksi untuk kultur murni dan sampel pempek spike memiliki kepekaan 3 kali lebih rendah dibanding hasil penelitian Dadkhah et al. (2012). Salah satu penyebabnya yaitu nilai kemurnian DNA hasil ekstraksi yang tidak baik (Siebert 1999). Selain itu, perbedaan kehomogenan sampel pempek
spike yang lebih rendah dibanding sampel susu spike pada penelitian Dadkhah et
al. (2012) merupakan penyebab menurunnya sensitivitas deteksi penelitian ini. Aplikasi penerapan deteksi sampel dengan rt-PCR antara kultur murni dan sampel pempek spike akan berbeda ketika nilai CT berada di atas 27, sedangkan
jika nilai CT di bawah angka 27 dapat dinyatakan bahwa sampel positif terdapat
L. monocytogenes (Tabel 6). Sampel kultur murni yang memiliki nilai CT di atas
27 dapat dinyatakan negatif L. monocytogenes berdasarkan hasil konfirmasi spesifisitas sebelumnya, sedangkan untuk sampel pempek spike yang memiliki nilai CT di atas 27 masih perlu adanya uji rt-PCR lanjut dengan perlakuan
enrichment sampel. Enrichment sampel pempek spike tersebut dimaksudkan
untuk meningkatkan jumlah konsentrasi awal L. monocytogenes sehingga memungkinkan pengujian kualitatif dengan rt-PCR pada sampel spike tersebut. Tabel 6. Hubungan nilai CT terhadap sampel yang diuji
Nilai CT
Kultur murni Sampel pempek spike
Status Status Langkah lanjut
≤ 27 Positif L. monocytogenes Positif L. monocytogenes Tidak ada
> 27 Negatif Perlu uji lanjut Uji rt-PCR dengan
enrichment sampel
SIMPULAN
Metode ekstraksi yang disarankan untuk analisis L. monocytogenes adalah metode fenol:kloroform dengan hasil DNA genom yang lebih tinggi dibanding metode pemanasan dan metode kit komersial. Primer yang sesuai untuk analisis
L. monocytogenes yaitu primer DG69/DG74 dengan hasil kespesifikan lebih baik
dibanding primer LMS 2/LIMRE. Kondisi running yang dipakai mendeteksi
DNA L. monocytogenes menghasilkan kurva amplikasi yang baik sampai siklus
ke-27. Deteksi DNA kultur murni L. monocytogenes menghasilkan kurva standar CT = 37.9 – 3.11 C dengan sensitivitas 3.2 x 103 CFU/mL, sedangkan pada SPS
menghasilkan kurva standar CT = 41.03 – 3.69 C dengan sensitivitas
19
SARAN
Pengembangan metode ekstraksi perlu dilakukan untuk mendapatkan metode ekstraksi yang memiliki kemurnian dan kualitas DNA yang lebih baik. Hasil pengembangan metode menunjukkan adanya perbedaan kurva standar antara kultur murni dan sampel pangan, sehingga perlu ditentukan kurva standar baru untuk penelitian tentang deteksi L. monocytogenes pada sampel pangan yang berbeda. Selain itu, pengembangan rt-PCR juga perlu dilakukan untuk deteksi
L. monocytogenes pada jajanan pempek dengan metode lain seperti TaqMan®.
DAFTAR PUSTAKA
Acciari VA, Torresi M, Migliorati G, Giannatale ED, Semprini P, Prencipe P. 2011. Characterization of L. monocytogenes strains isolated from soft and semi-soft cheese sampled in a region of Italy. Veterinaria Italiana. 47(1): 15-23 Altshuler ML. 2006. PCR Troubleshooting: The Essential Guide. Caister
Academic Press. Moscow
Applied Biosystems (AB). 2015. Essentials of Real Time PCR. US
Aurora R, Prakash A, Prakash S, Rawool DB, Barbuddhe SB. 2008. Comparison of PI-PLC based assay and PCR along with in vivo pathogenicity test for rapid detection of pathogenic L. monocytogenes. Food Control. 19: 641-647
Bettelheim FA, Brown WH, Campbell MK, Farrell SO, Torres OJ. 2013.
Introduction to General, Organic, and Biochemistry. Brooks/Cole. Australia
Brisson M, Tan L, Park R, Hamby K. 2000. Identification of nonspecific products using melt-curve analysis on the iCycler iQ™ detection system. Bio-Rad Laboratories, Inc
Camma C, Domenico MD, Monaco F. 2012 Development and validation of fast real-time PCR assays for species identification in raw and cooked meat mixtures. Food Control. 23: 400-404
Campos CA, Castro MP, Gliemmo MF, Schelegueda LI. 2011. Use of natural antimicrobials for the control of Listeria monocytogenes in foods. Formatex. 1111-1123
Coral D - P, Rosales-Rivera A, Rodriguez-Garcia ME. 2009. Determination of the gelatinization temperature of starch presented in maize flours. Journal of Physics. 167: 1-5
Choi WS, and Hong CH. 2003. Rapid enumeration of Listeria monocytogenes in milk using competitive PCR. Int. J. Food Microbiol. 84: 79-85
Dadkhah H, Bassami MR, Hashemi S, Shahraz F, Hosseini H, Karatzas KAG, Khaksar R. 2012. Evaluation and comparison of SYBR Green I real-time PCR dan TaqMan real-time PCR methods for quantitative assay of Listeria
monocytogenes in nutrient broth and milk. African Journal of Microbiology
Research. 69: 1908-1917
Dharmaraj, S. 2015. RT-PCR: The Basics. http://www.lifetechnologies.com [12 Juni 2015]
Dauphin LA, Hutchins RJ, Bost LA, Bowen MD. 2009. Evaluation of automated and manual commercial DNA extraction methods for recovery of Brucella DNA from suspensions and spiked swabs. J of Clinical Microbiology 47(12): 3920-3926.
20
Dibbern AG, Botaro BG, Viziack MP, Silva LFP, Santos MV. 2015. Evaluation of methods of DNA extraction from Staphylococcus aureus in milk for use in real-time PCR. Genet. Mol. Res. 14(1): 227-233
Dwight Z, Palais R, Wittwer CT. 2011. uMELT: prediction of high-resolution melting curves and dynamic melting profiles of PCR products in a rich web application. Bioinformatics. 27(7):1019–1020
Esco. 2009. User Manual Swift Spectrum 48 Fluorescence Quantitative PCR
Detection System. Esco Healthcare Pte. Ltd.
Erali M, Wittwer CT. 2010. High resolution melting analysis for gene scanning. Methods. 50: 250-261
Fakruddin M, Rahaman M, Ahmed MM, Hoque MM. 2014. Stress tolerant virulent strains of Cronobacter sakazakii from food. Biological Research. 47(1): 63
Food and Drug Admission (FDA). 2001. Bacteriological Analytical Manual. Food and Drug Administration.
Hein I, Lehner A, Rieck P, Klein K, Brandl E, Wagner M. 2001. Comparison of different approaches to quantify Staphylococcus aureus cells by real-time quantitative pcr and application of this technique for examination of cheese. Applied and Environmental Microbiology. 67(7): 3122–3126.
Jamali H, Chai LC, Thong KL. 2013. Detection and isolation of Listeria
monocytogenes in ready to eat foods with various selective culture agar. Food
Control. 32: 19-24
Jenie BSL, Kusumaningrum HD, Nurjanah S. 2013. Deteksi bakteri patogen dan fermentatif dari pangan menggunakan real-time polymerase chain reaction. Prosiding Seminar Hasil-Hasil PPM IPB. 1: 292- 308
Kohler S, Bubert A, Vogel M, Goebel W. 1991. Expression of the iap gene coding for protein p60 of Listeria monocytogenes is controlled on the posttranscriptional level. Journal of Bacteriology. 173: 4668-4674
Kramarenko T, Roasto M, Meremae K, Kuningas M, Poltsama P, Elias T. 2013.
Lysteria monocytogenes prevalence and serotype diversity in various foods.
Food Control. 30: 24-29
Kunkel D. 2013. Listeria monocytogenes - rod prokaryote (bacterium). http://www.denniskunkel.com/DK/Bacteria/20229A.html [12 November 2013] Mak P, Maszewska A, Rozalska M. 2008. The amino acid sequences and
activities of synergistic hemolysins from Staphylococcus cohnii. FEMS Microbiology Letters. 287: 230-235
Marian MN, Aminah SMS, Zuraini MI, Son R, Maimunah M, Lee YH, Wong WC. Elexson N. 2012. MPN-PCR detection and antimicrobial resistance of
Listeria monocytogenes isolated from raw and ready to eat foods in Malaysia.
Food Control. 28: 309-314
Meyer C, Ahomaa MF, Sperner B, Martlbauer E. 2011. Detection of Listeria
monocytogenes in pork and beef using the VIDAS LMO2 automated enzyme
linked immunoassay method. Meat Science. 88: 594-596
Montgomery JL, Sanford LN, Wittwer CT. 2010. High-resolution DNA melting analysis in clinical research and diagnostics. Expert Rev Mol Diagn. 10: 219-240
Moreno Y, Contreras JS, Montes RM, Hernandez JG, Ballesteros L, Ferrus MA. 2012. Detection and enumeration of viable Listeria monocytogenes cells from
21
ready-to-eat and processed vegetable foods by culture and DVC-FISH. Food Control. 27: 374-379
Nolan T, Mueller R, Bustin S. 2007. QPCR: target preparation. In: Mackay IM (ed.). Real-time PCR in Microbiology From Diagnosis to Characterization. UK: Caister Academik Press
Nur’utami DA, Nurwitri CC, Rahayu WP, Panggabean RIL, Yuliangsih S. 2011. Metode analisis isolasi dan identifikasi Salmonella Typhimurium pada susu dengan metode real-time PCR (Polymerase Chain Reaction) [skripsi]. Bogor (ID): Institut Pertanian Bogor.
Ohk SH, Bhunia AK. 2013. Multiplex fiber optic biosensor for detection of
Listeria monocytogenes, Escherichia coli O157:H7 and Salmonella enteritica
from ready to eat meat samples. Food Microbiology. 33: 166-171
Pestana EA, Belak S, Diallo A, Crowther JR, Viljoen GJ. 2010. Early, Rapid, and
Sensitive Veterinary Molecular Diagnostics Real-Time PCR Application.
Springer. Dordrecht
Pochop J, Kacaniova M, Hleba L, Petrova J, Lopasovsky L, Pavelkova A, Bobkova A. 2013. Real-time PCR detection of Listeria monocytogenes in Food samples of animal origin. International Journal of Microbiology, Biotechnology And Food Sciences. 141: 1550-1558
Rahayu WP, Panggabean RIL, Yuliangsih S, Pusparini N, Khotimah K. 2009. Comparison of three extraction methods for identification Bacillus cereus and
Staphylococcus aureus in fried rice. Research Center for Drug and Food.
National Agency of Drug and Food Control.
Rawool DB, Malik SVS, Shakuntala I, Sahare AM, Barbuddhe SB. Detection of multiple virulence-associated genes in Listeria monocytogenes isolated from bovine mastitis cases. International Journal of Microbiology. 113: 201-207 Sambrook J and Russel DW. 2001. Molecular Cloning: A Laboratory Manual.
Cold Spring Harbor Laboratory Press, New York.
Schuppler M, Loessner MJ. 2010. The opportunistic pathogen Listeria
monocytogenes: pathogenicity and interaction with mucosal immune system.
Int. Journal of Inflammation. 2010: 1-12
Siebert PD. 1999. Quantitative RT-PCR. In: Kochanowski B, Reischl U (eds.).
Methods in Molecular Medicine: Quantitative PCR Protocols. Humana Press.
New Jersey
Snyder RO, Carson KL. 2010. New viral gene vector reference material. ATCC Connection. 30(1): 3-5
Taylor R. 1990. Interpretation of the Correlation Coefficient: A Basic Review. Journal of Diagnostic Medical Sonography. 6: 35-39
Todd ECD, Notermans S. 2011. Surveillance of listeriosis and its causative pathogen, Listeria monocytogenes. Food Control. 22: 1484–1490
Ye K, Zhang Q, Jiang Y, Xu X, Cao J, Zhou G. 2012. Rapid detection of viable
Listeria monocytogenes in chilled pork by real-time reverse-transcriptase PCR.