Vol 8, No 3, July - September 1999 P rio n in neurode generativ e di seas e s 149
Prion:
the infectious
protein in
neurodegenerative diseases
M.M. Vita Kumiati,
SepteliaInawati Wanandi
Abstrak
Prion merupalan suatu partikel infeksius yang menyebabkan beberapa penyakit neurodegeneratif, seperti penyakit Creutqfeldt-Jakob, penyakit kuru, sindrom Gerstmann-Striiussler-Scheinker dan insomnia familial fatal pada manusia, serta penyakit sapi gila, scrapie, feline
Protein prion pascatranslasi menjadilemba
suatu chaperone. Dalam dua dekade terakhir ini, banyak lcarakteristik prion yang telah terungkap, seperti karakteristikfisik, kimiawi, snain, biologi molekuler dan imunologik Beberapa peneliti juga telah menemulcan bentuk topologis protein prion dalam membran retikulum endoplasma serta peran bentuk-bentuk lopologis tersebut dalam proses patofisiologis penyakit. Namun demikian, masih banyak pertanyaan mengenai penyakit prion yang belum terjawab. Salah satu aspek prion yang belum terungkap adalah multiplikasi
prion.
Dai
gdiajukanolehparapeneliti, terdapat satuhalyangselalud
itubahwareplikasi
pr
Penelitian mengenai patogenesis penyakit prion juga perlum
tian untuk mengembangkan terapi yang efektif untuk penyakit neurodegeneratif ini. Diagnosis penyakit prion ditegakkan berdasarkan temuan klinis, histopatologis dan uji imunokimia dari beberapa protein dalam cairan serebrospinal. Uji imunokimia dari protein dalam cairan serebrospinal ini penting untuk menegakkan diagnosis pramortem dari penyakitpion.
Abstract
Prion particle is an infectious agent causing neurodegenerative diseases in humans and animals, such as Creut4feldt-Jakob disease, kuru, Gerstmann-Striiussler-Scheinker syndrome, and
fatal
familial
insomniain
humans; mad cow disease, scrapie, and felinespongiform encephalopathy in animals. This particle is devoid of
n
Theprotein
(PrP-) is
convertedinto
its abnormal isoform (PrP-")p
PrPconformational change whereby the a-helical content decreases
il
of PanunknownproteinXwhichmightfunctionasamolecularchaperone. Manyprioncharacteristicshavebeenrevealedintheselasttvvo decades, such as physical, chemical, sftain, molecular biology, and immunological characteristics. Some investigators revealed the topologicalforms ofprionprotein in the endoplasmic reticulum membrane and theirrole inthe pathophysiologicalprocess of the disease. But still prion diseases continue 1o raise many unanswerable questions to be investigated. One of the unanswerable questions is prion muhiplication. Many prion multiplication models are
sug
constantftntlitry is that prion replication requires the interaction ofPrPt-PrP".
The studies ofpathogenes
ch more attention to develop an effective therapyfor
these neurodegenerative dise.ases. Diagnosis ofprion
diseasesis
basedon
theclinical
and histopathological findings, and immunochemical testsof
some proteins in cerebrospinalfluid.
Immunochemical tests of proteins in cerebrospinal Jluid are important in developing premortem diagnosis of prion di.seases.Keywords : scrapie, kuru, Creut4feldt-J akob disease, conformational change
During
the past two decades, atransmissible
pathogencausing
agroup
of
human
andanimal
neurodegenera-tive diseases has been found. This infectious
agentis
different
from both viroids and
viruses.
Someinves-tigators had indicated that this
agent could not
beinactivated
by procedures modifying or hydrolyzing
Department of Biochemistry, Faculty of Medicine University of Indonesia, Jakarta, Indonesia
nucleic
acid, such as nucleasedigestion,
or UVinadia-tion.l'2
Otherevidence
showsthat
the agentcontains
a
protein
that is required
for infectivity,
and itsinfec-tivity
is lost upon
inactivation by
proteinase
K,
deter-gent
(sodium
dodecylsulqhate), guanidinium
thiocyanate,
or urea
(Table
l).'
On
the basisof these
evidence, the
term "prion"
was
introduced to
distin-guish
this
infectious pathogen
from
those responsiblefor viral
illness, i.e. viruses
andviroids.'
Prion
has aninfec-150
Kumiati andWananditious particles
which resist inactivation by procedures
that modify nucleic acids.
The term prion emphasizes
that theinfectivity of this
transmissible
pathogen of theneurodegenerative
diseases depends on a proteincom-ponent.É
Prion diseases
are neurodegenerative disorders
of
humans and animals causing
severeneurologic
dys-function and
death.
Prions
cause four transmissible
neurodegenerative
diseases
of
humans and
six
of
animals.Human
prion
diseases, canmanifest
asinfec-tious, sporadic, and inherited forms, include kuru,
Creutzfeldt-Jakob
disease(CJD),
fatal familial
insom-nia (FFI), and Gerstmann-Strâussler-Scheinker
syndrome (GSS).
The animal prion diseases
include
the following:
scrapie
of sheep, transmissible mink
encephalopathy
(TME),
chronic wasting disease
(CWD) of mule,
deer and elk, bovine spongiformence-phalopathy
(BSE), feline spongiform encephalopathy
(FSE), and exotic ungulate encephalopathy.
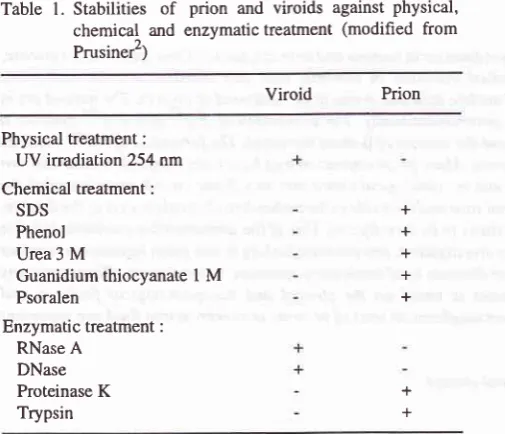
Table
l. Stabilities of
prion and viroids against physical, chemical and enzymatic treatrnent (modified from Prusiner2)Viroid
PrionPhysical treatment: UV irradiation 254 nm Chemical treatment :
SDS Phenol Urea 3
M
Guanidium thiocyanate
I M
PsoralenEnzymatic treatment :
RNase A DNase Proteinase
K
Trypsin+ + + + +
+ +
+ = inactivated; - = no change in infectivity
The
mal
The
ally modified process
that involves a change
in
theconformation
without evidence
for chemical
modifica-tion. Both
PrPisoforms
are encoded by a chromosomal gene. The human PrP genemaps to the short arm
of
chromosome
20 and
is designated PRNP;
the mouse
PrP gene maps
to chromosome
2 and
is
designatedP*-p.'
Med J Indones
Characteristic of
the cellular
prion
protein
and
scrapie prion protein
The PrPc and PrPS" have a molecular
weight (Mr)
of
33to 35 kD.l
Both PrPc
andPrPs"
are encodedby
asingle exon
of the chromosomal
gene asproteins
with
the same
254 amino acid sequence.
The
first
22-ma membrane
by
a
glycosyl-phosphatidylinositol
(GPI) moiety located at the COOH terminus
of
thepolypeptide chain. The anchor is added
post-transla-tionally
in
the endoplasmic reticulum, following
cleavage
of
a 23-residue
COOH-terminal
hydro-phobic
sequencewhich
serves as a signalfor the anchor
attachment.-'The two PrP
isoforms
can be distinguished by their
K
digestion,
only
the firstof irPs"
arehydrolyzed and converted
PrP 33-35s" to P.P 27-30
presence
of PrP amyloid rods in some prion
diseases has led to assumptions that amyloid formation isessen-tial for
the formation of PrPs". However, PrPs" can beformed
in the
absenceof amyloid, and the presence
of
amyloid
plaques is not obligâtory for prion diseases.3The physiological function of PrPc is unknown, but it
appears to be unnecessary sincemice in which
the PrP gene has been deleted develop normally and areheal-thy for more
than 9 months.' Recently, some
inves-tigators suggested
several roles of PrP, such
as:postsynaptic PrP
might be necessary
for
GABA-de-pendent
synapses to befully
functional,
PrP
lacking
animals exhibit
altered sleep patterns andrhythms
of
circadian
activity. PrP also contributes
to
the prion
diseases;the PrP fragment,-PrP
106-126,
is toxic
tocortical and cerebellar cells.'t
[image:2.595.82.335.358.575.2]Vol 8, No 3, July - September 1999
Studies
of
CJD
demonstrate
four distinct
patterns
of
protease-resistant PrP
on
Western
blots after limited
proteolysis. Types
I
and2
are seen in
sporadic CJD
and alsoin some iatrogenic
CJD. Thethird type is
seenin
acquired
prion
diseases that arisefrom
aperipheral
route
of
exposure to prions,
while
central
nervoussystem
(CNS)
exposure
typically
resembles sporadic
CJD. Type
4 is
associated with
a new variant
CJDwhich
arisesfrom dietary
exposure
to
bovine prions.
The bands pattern
of
type
4 is similar
to type 3, but
it
can be distinguishedfrom all
three typesof
CJD
by a
characteristic pattern
of band intensities. Type 1
isalways associated
with
genotype MM, type 2
with all
genotypes
(MM,
MV
or VV).
Type
3 is
seen in
g"notyp"
MV or VV, and type
4in
gênotypeMM.6
The
transmission of prions
from
one species to anotheris generally
accompanied
by
a prolonged incubation
time relative to transmission to which
the host speciesis the same.l'7
Thir is
often referred to
asthe "species
barrier". The
speciesbarrier
concept
is
important in
assessing the
risk for
humansof
developing CJD after
consumption of
scrapie-infected
lamb
or
BSE
beef.There
are
three factors that might contribute
to the
speciesbarrier: (i) the difference
in amino acid
se-quenceof
PrP between
prion
donor
and recipient,(ii)
strain
of prion,
(iii)
the species
specificity
of
protein
X, a factor that binds to
PrP"
and facilitates PrP'"
formation.T PrPc is most
efficiently
converted
toPrPs"
when the amino acids
sequencêsof PrPc
and PrPSt areidentical.s
Studies
of
PrP
genes responsiblefor the prolonged
incubation
time
in mice have
demonstrated
geneticlinkage
between
Prn-p
gene and
a
gene modulating
aps to mouseverv
closelv
cubation
time is
also influencedby
theeË"'ifr::*
When prions
are passagedinto
mice
with
anonmatch-ing
PrP sequennce, the incubation
time
is longer
thanthat
in
micè
with
amatching
PrP sequence.8Another characteristics
of this
agent are that the agentis not
destroyed
by boiling
in water,
andit is not in
activated
by
standard exposurein
an autoclaveto
wet
heat atl2loC for
15min.
Exposureto
134-138 oCfor
18
min
in
porous-load autoclave
is currently
recom-mended but may not be adequatein
all
circumstances.The scrapie agent withstands alcohol
and strongdisin-fectants
such as formaldehyde and
glutaraldehyde.
Formaldehyde may even increase
its heat-stability. It
may be inactivated
by
exposurefor
t
hour
to
sodium
Prion in neurodegenerative
diseases
151hypochlorite
providing
2Voavailable chlorine.
Thein-fectivity
of
scrapie
affected brain
homogenate
can passthrough small-pore filters.e
The
formation of
the
scrapie
prion
protein
The
prion
diseases are dueto
the conversion
of
PrPc
into
PrPs".
PrPs"
formation
is
a
posttranslational
terol-rich
membraneso*".
loTh" formation
the cell surface
andlysosomal
membranes,
releasing hydrolases
into
thecell.
These enzymeswill
causecytoskeletal disruption
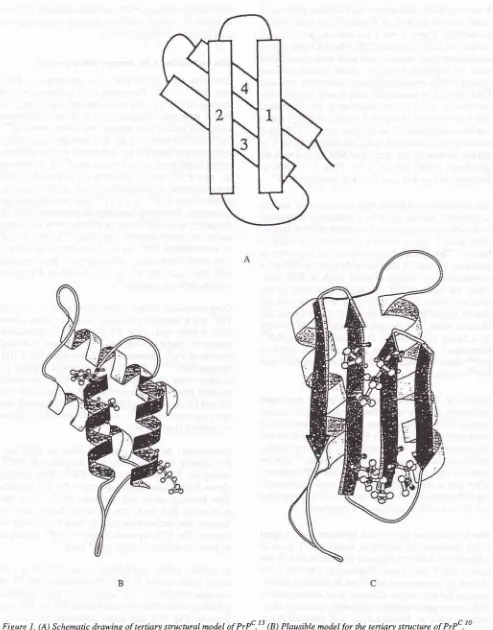
and spongiform changes. l2al studies of prion proteins showed that
condary structure
which
contains
aboutand
only
3vo p-sheet.l0'13
Molecular
modelins
studiesresions
Jt
prpc.
athiough
H4).t'tq''
residues were
identified
asDotential sites that
would
mediate
helix-helix
interacti,on.13It
i,
,ugg"sted
that
Hl
andH2
are convertedinto
p-sheet structuresduring
the formation of
PrPsc,
while
H3 and
H4
remain unchanged(Fig.
1A
and1B).lo
Pre
a central domain
of PrPc
1ap-pro
95 to
170)
that binds
to
PrPscdur
of
nascentPrP-t. This
domain
shows
higher homology
between
cattle
and
humansthan between
sheep
and humans,
which
raises
thepossibility that prion
transmission from
cattle to
humans
than from
sheepto
humans
in
ofPlPc
is thoughtto
form
binds.TIn
contrast to PrPc, the p-sheet content of PrPs"it
43Eoand
a-helix
content
is
30Vo.Furthermore,PrP
27-30
contains 54Vop-sheet
The formation of PrPs" involvesrefoldingof theNH2-terminal
helices(Hl
andH2)
into
p-she"{s
(Fig.
1Ç).The
major conformational
changeof
PrPuinto
PrPrc has been localized to residues 90to
t52
Kurniati and Wanandi Med J Indone.s [image:4.595.81.574.82.712.2]c
Figure I . (A) Schemaric drawing of tertiary stuctural model of PrPc.t 3 (B) Plausibte model
for
the tertiary structure of PrPc .10+
n
Finally,
in
the
caseof
mutations (Â) would
desconversion
into
ÂPrP*
formation.lo
\
svnttresi,ÂPrPc-
APrP'-f
o"g^d^tion
Vol 8, No 3, July - September 1999Mutations
of
PrP
qenecan destabilize the
conforma-tion
of
PrPc
andiacilitate
its
refolding
into
PrPsc.Usually
PrP genemutations occur
at or near thehelix-helix interaction
residues. Thesemutations could
dis-rupt or
destabilize
the
structure
of
PrPC and produce mice or humanswhich
are susceptible toprion
disease.Mutation
atcodon
178in fatal familial
insomnia
andfamilial
CJD
would
eliminate
thenegative
chargethat
interacts
with
the positive
end
of
a
helix dipole
anddestabilizes
H3. Another mutation
at codon
2lO that
also producesfamilial
CJD would disrupt
theH4
-Hl
interàction
and, thus,perturbs
the structureof
PrPc.13Synthesis
of
PrPst probably also involves
anotherprotein which
is notknown. This protein
X,
possibly
a"chaperone", facilitates the refolding
process
of
o,-helicis in
PrPc
into
p-sheetsin PrPs".r'rc
TheCooH-terminal
domain
of
H3 in
PrPc
appearsto contain
thesite
for
protein
X
binding.T
fnJ
Èinaing
of
PrPc
to
protein
X
seemsto exhibit
the highest
affinity
when
ihese
two
proteins
arefrom
the saire
species.lÔPrion protein receptor
The cell-surface form
of
the prion protein, PrPc,
is anchored to the plasma membraneby
aglycosyl-phos-a
neurotoxic region in
humanprion
(residues 106- 126) that contains thebinding
siterecognized by
aputative
cell receptor.
Rieger
et
al.r)
identified the
37
kD
laminin
receptor precursor
(LRP)
asinteracting with
PrP'in
yeast,insect
andmammalian cells. The
37kD
LRP
is located on
thecell
surface, andit
may
act as areceptor or co-receptor
for
theprion
protein.Is
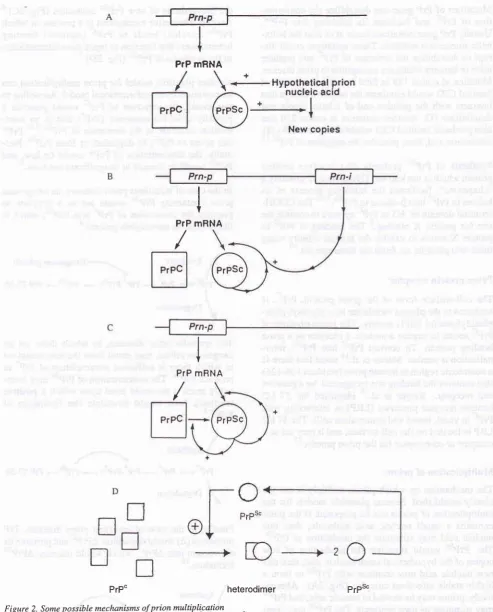
Multiplication
of prions
The
mechanism
by
which prions
multiply is not
yet
clearly
established. Several plausible models
for
themultiplication of
prions
can beproposed.
If
theprion
contains
a
small nucleic
acid molecule, than this
nucleic acid may stimulate the production
of
PrPs".The
PrPùc
would
stimulate
the production
of
new
copies
of
thehypothetical small nucleic
ac_id, thenthis
new nucleic
acid
may
combine
with
PrPscto form
ahighly
stable
infectious
terna-tively,
prions
may bede
PrPs"may stimulate
its own
s
com-bine w
would
2B).
rt
the biosynthesis
of
new
PrPS"molecules
(Fig.
2C).aAnother
alternative mechanism
is
a Drocessin which
PrPsc
lcircles)
binds
to
PrPc (squares) forming
heterodimers thatfunction
asreplication intermediates
in
the synthesisof
PrPsc(Fig.
2b).t
Another
plausible model
for prion multiplication
can nalmodel. According to
PrPc would
generate
a(PrP
)
that
is
an
inter-ation
of
P.C".8'lo P.P*
can revert
to
PrPc, be
degraded,or form
PrPst.
Nor-mally,
the concentration
of
PrP* would
be
low,
and PrPScwould
beformed
in insignificant
amounts.San exogenous
a
template
torPst, which
is
u.-19*ogenous
Prions)*
PrP"-PrÉc-
PrPSc-
Prp27-30The
sporadic
prion
diseases,in
which there are
no exogenousprions, may
result
nsin
which
there
is
sufficient
a
to
produce
PrPs".The concentration
of
PrPst may
even-tually
reach a threshold
level upon which
a positive
feedback
loop
would
stimulate
the formation
of
PrPS".8
Prion in neurodegenerative
diséases
153PrPs"*
PrP27-30t54
Kurniati andWanandi Med J IndonesPrP
mSNA
I
PrP mRNA
/
I
PTP
mBNA
/
[image:6.595.77.571.81.693.2]PrP"
heterodimer
PrPs"Figure 2. Some possible mechanisms of prion multiplication
(Aj A snalt nucieic acid triggers the syithesis o7 Prf'.a
(il Prf
the synthesis of^or"
PrË'.4
Pm-n=
Prn'i
=
ons thatproduce
VoL 8, No 3, July - September 1999
The role of the immune
system
in the
prion
diseaseIt
has beennoted for years that
scrapieinfection fails
to induce
animmune
response.This lack of immune
response
to
the infectious
scrapie particle can be
ex-plained by
thedis
themajor
scrapie
(PrPN)
is
form of
thprotein
(PrPt);
t
sms may
scrapie
particle
also
reasonable to
rugfest
thut
an
to
both PrPc
andPrPt",
because
in
most
tissuesof
uninfected animals
and is evenfound
on the surfâceof
T
lymphocytes,16 and
B
cells.tT
The T and
B
cells'
tolerance to PrP
would explain
the absenceof immune
response
to
scrapieinfection.l6
A
scrapie infection
from
adifferent
speciesor in
thePrP-deficient mice also can
not
induce an immune
response
although the scrapie particle would
becon-sidered foreign.
This
absenceof immune
response isdue to the failure
of
scrapie infection
to
activate
thenonspecific immune
mediatorsthatnormally
signal theinvasion
of
pathogenic microorganisms,
such asinter-ferons, tumor
necrosisfactor,
interleukin
I
(IL-1),
andIL-6.
In
the
absenceof
thesenonspecific
mediators,even
foreign proteins
do notelicit
an irnmune response.This situation
suggeststhat successful
approaches totreating prion
diseases cannot depend onactivating
theimmune
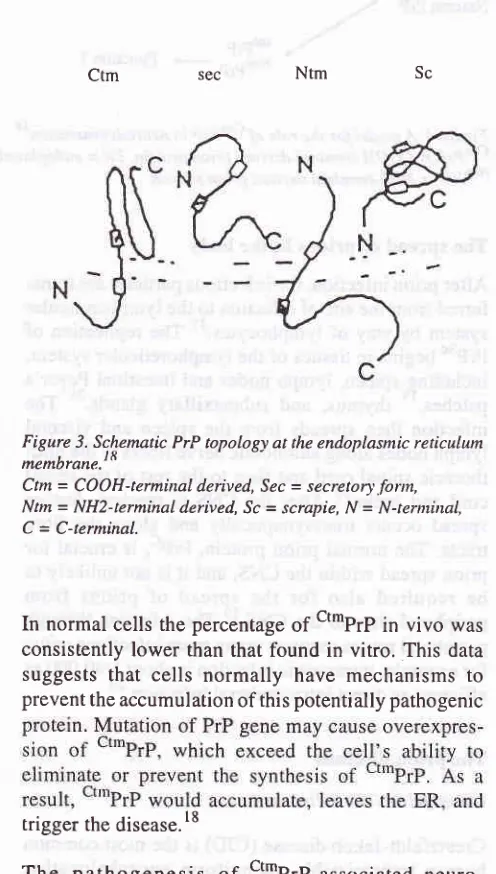
system.toTopology of prion protein
Studies
of PrP topology
at theendoplasmic reticulum
(ER)
membrane have revealed two distinct forms
of
PrP:
onethat is
fully
translocated into
the ER lumen
and is
termed
the secretoryfbrm
1t""PrP;; and onethat
spans the membrane(transmembrane) (ttPrP).
Diges-tion
of
transmembrane form with
proteases addedto
the outside
of
the membrane
yielded two
fragments:
one
is COOH-terminal
derived
andglycosylated
(ctm-PrP), and the
other is NH2-terminal derived
andun-glycosylated
1Nt'PrP;.
These data
indicated
that
transmembrane PrP
chains
spanthe
membranetwice,
with
theNH2-
andCOOH-termini
of
themolecule
in
the
ER lumen
(Fig.
3).18Hegde et al. examined the possible role of PrP
topology
in neurodegeneration.
Their data showed a
marked increasein
ct*PrP
production
at theER
membraneof
scrapie
infected
hamster,with
aconcomitant
decreasein
the t"tPrP. The amount
of
NttPrP
remained
un-changed. These
findings
suggest that
ctmPrP
is
in-;#::,;T
#:'$""::l1P*ent
or
spontaneous
neuro-sec
Ntm
Sc [image:7.595.332.580.133.570.2]Prion in neurodegenerative
diseases
155Figure 3. Sc.hematic PrP topology at the endoplasmic reticulum
^e^brar".]8
Ctm= CooH-terminal derived, Sec = secretory.fonn,
Ntm = NH2-terminaL derived, Sc = scrapie, N = N-ternùnal,
C = C-terminal. Ctm
In normal cells the
percentageof
ct*PrP in vivo
wasconsistently
lower than that
found in vitro. This
datasuggests
that
cells normally
have mechanisms to
prevent the
accumulation of
thispotentially pathogenic
trigger
the disease.lSThe
pathogenesis
of
ct*PrP-associated
neuro-degenerative disease includes
at
least
three distinct
steps
(Fig.4). First,
nascentPrP is synthesized
in
thet""PrP, NttPrP,
o.
cttprp
form.
Second,
the ct'tprp
form may
berapidly
degradedin the ER or,
in
some cases, may be able to escapedegradation to
apost-ER
compartment.
Finally,
in
the post-ER compartment,
cttPrP is
proposedto
cause disease.How
thect*PrP
can cause
the
t known. The pathway
Vol 8, No 3, July - September 1999
F atal
familial
ins omnia
Fatal
familial
insomnia
(FFI)
exhibits
insomnia,
dysautonomia,
ataxia,
dysarthria,
dysphagia,
myoclonus,
andsigns
of pyramidal
tract dysfunction.
This
FFI
is causedby
amutation
atcodon
178(Asp->
Asn). However,
in
contrast
to
the inherited CJD
with
a
valine
residue at
codon
129,
amethionine
residue wasalways
found
atcodon
129 ofFFI
patients.2oBovine
spongtform
encephalopathy (mad cow disease)Bovine spongiform
encephalopathy
(BSE) was first
recognized
in Britain in
November
1986in
cattle. Thecows
initially
became apprehensive, hyperesthetic,
and
uncoordinated. Then they
became hard to handle,and
in
someca
sed tofrenzy. which led
to the name
"m
.20Th"rn"un
incubation
period is
four
to
five
y"u.r.7'20
The BSE
agent can betransmitted
to
another species,such
as
calves,
sheep, goats, and
mice,
by
the
oral
route,
in
some
casesby
very
high-challenge
dosesonly.
Transmission
orally
to
sheep and goats was pos-sible using 0.5 ginfected bovine brain;
andI
g ofbrain
was effective
in
cows.'
In
humans,
there are
threepossible routes
of infection. First, by implantation
or
injection
of
bovine-derived
materials
or
preparationsassociated
with
bovine products, including
"catgut"
sutures. Second,
workers
in
animal
feed preparation
might
have been
at risk
of
inhaling infected
dust or
acquiring
the agentconjunctivally. Infection
byinges-tion is
the
third
possible route.
High-titre infectivity
challenges
seemto
be
associatedwith bovine
brain,
retina, and spinal cord. Medium
infectivity
is
as-sociated
with
lymphoreticular
tissues.
Other
tissues,including
skeletal muscle_(meat),milk,
andblood,
hadno
detectableinfectivity.zt
The bovine prions
in
humans can
causesome
caseswhich differ in
several waysfrom
other casesof
CJD. These cases arebeing called variant Creutzfeldt-Jakob
disease
(vCJD) or
new
variant
CreutzfeldçJakob
dis-ease(nvCJD).
Thepatients
areyoung
(the age rangeis
between
16 and41),
andpresenting behavioral
chan-ges,ataxia,
andperipheral
sensory disturbances,while
progressive dementia developed later. The PrP
genesin all
cases analyzed
showed
no
mutation and
arehomozygous
for
methionine
atcodon
129 (the same asin
catttei.2oAlzheimer's
diseaseThe
hallmark of Alzheimer's
diseaseis
theaccumula-tion of
severalabnormal
proteins
in
thebrain,
such asP rion in neurode generative diseases
amyloid
pprotein,
pprotein
precursor,Apo
E, and PrP. Theseproteins
arepresent
in
the
muscle
fibers.
Themuscle
fibers
also
demonstrated
increased
PrPc
mRNA while
the abnormal brains
of
patients with
prion
diseasesdid
not
have increasedPrÊc mRNA.22
Diagnosis
of
prion
diseasesThe diagnosis
ofprion
diseases is based onclinical
andneuropathological
findings. The
typical
neuro-pathological
findings
of
these diseases arespongiform
change, astrocytosis,
andneuronal loss.
Of
these the mostspecific
is thespongiform
change,which
consistsof diffuse or focally
clustered, small, round
vacuolesthat may become
confluent.
In
some cases,there
areamyloid
plaques composedof
extracellular
accumula-tion
of PrP."
The definitive diagnosis can
only
be made byhistopathological examination
ofbrain biopsy
specimen.23'24Brain
biopsy, however,
placespatients
and
health personnel
atrisk
andmay miss the site
of
disease.23 ^
Most
cerebrospinal
fluid
proteins
in
CJD
patients
showed
two 30-kD proteins
detected
by
two-dimen-sional
electrophoresis and designated
asprotein
130and
131. Thesetwo proteins
are14-3-3 proteins.
The14-3-3
protein
was
abundantin
anextract of
normal
human brain, but
it
was notfound in normal
serum andin
serum
from
CJD patients.
In
humans and
other
mammals,
14-3-3 isanormal
neuronalprotein
consist-ing
severalisoforms,
andit
plays
apart
in
conforma-tional stabilization of
other proteins. The
presenceof
14-3-3
in
cerebrospinal
fluid
may
be
due
to
massiveneuronal
disruption
andthe
leakageof brain
proteins
into
cerebrospinalfluid.
Thequantity
of
74-3-3 presentin
cerebrospinal
fluid
may be
propotional
to
the
rate and the amountof
neuronal
destruction."
The l4-3-3
proteins
can be detectedby
animmunoas-say
method.
This
14-3-3
protein
can
alsobe found
in
cerebrospinal
fluid from
patients
with
herpessimplex
encephalitis
or recentinfarctions. Therefore,
it
should
be emphasized
that
the needto
usethe
14-3-3 marker
as a test isonly
usefulin
an appropriateclinical
setting.For
apatient
with
dementia, thedetection
of
14-3-3in
cerebrospinal
fluid
strongly
supports
a
diagnosis
of
CJD.23
^Some
other
studiesin
small numbers of patients
have suggested that thereis
anotherprotein in cerebrospinal
158
Kurniati and Wanandi Med J IndonesCONCLUSION
Prion
is
an infectious protein that
cause
neuro-diseases
in
humans
The
(PrP)
consistsof
two
rPc,oform,
andPrPst,
the
The
conversion
of
PrPc into PrPs"
is
a
posttranslational
Drocess
that involves
aconformational modification.
i{ow
PrPc
can beconverted
into PrPs"
remains to
beestablished
although
several
investigators have
sug-gested some
prion
multiplication
models.
Under-standing
the prion
propagation needs much
more
attention
becauseit
maycontribute
to thedevelopment
of
an effective therapy
for
these neurodegenerative
diseases.
The
development
of
premortem diagnostic test
of
prions is
needed, since adefinitive
diagnosis
ofprion
diseases
by histopathological examination
of
abrain
biopsy
specimenis
accompaniedby high
risks.
Thereare biochemical markers, such as neuron-specific
enolase
and 14-3-3 protein
in
cerebrospinal fluid,
which
canprovide
anobjective evidence
for
thediag-nosis
of
prion
diseases,especially the
Creutzfeldt-Jakob
disease. Thesemarkers
can be detected
by
animmunochemical examination.
However,
these
diag-nostic
testsshould
only
beconsidered
as amarker
in
appropriate
clinical findings
or
when other
diseaseshave been excluded.
The understanding
of prion
diseasesmight
open anew
and
emerging
areaof
investigation
in
molecular
biol-ogy,
cell biology,
genetics, andprotein chemistry.
REFERENCES
l.
Prusiner SB. Genetic and infectious prion disease. Arch Neurol 1993;50: I 129-53.2. Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982;216:136-M.
3.
Baldwin
MA,
CohenFE,
PrusinerSB. Prion
protein isoforms, a convergence of biological and structural inves-tigation. J Biol Chem 1995;270:19197-200.4. Prusiner SB. Prions and neurodegenerative disease. N Engl
J Med 1987;3 I 7:1571-81.
5. Estibiero JP. Multiple roles for PrP
in
the prion diseases.Trends Neurosci 1996;19:,257 -8.
6. Collinge
J,
SidleKCL,
MeadsJ,
IronsideJ,
Hill
AF.Molecular analysis of prion strain variation and the aetiol-ogy of 'new variant' CJD. Nature 1996;383:685-90. 7. Prusiner SB. Prion diseases and the BSE crisis. Science
1997:278:245-51.
8. Cohen FE, Pan
KM,
Huang Z, Baldwin M, Fletterick RJ,Prusiner SB. Structural clues to prion replication. Science
1994;264:530-1.
neuron-specific
enolase(NSE).
This
enzyme
is
a78-kD protein, localized
in
neurons
andneuroendocrine
cells,
synthesized
virtually
completely
within
the
central
nervous system. Raised
NSE levels
in
cerebrospinal
fluid
have
also been reported
in
other
neurological disorders, such
as
brain trauma, brain
tumors, subarachnoid haemorrhage,
and acute stroke.The
cut-off
value
of
cerebrospinal
fluid
NSE
is
35ng/ml.
This
value should
only
be considered ashighly
suggestive
of
CJD when other
diseases,such as
is-chaemic stroke or
brain
tumours
have beenexcluded.
NSE
in
cerebrospinal
fluid
appearsto be
a valuable
biochemical marker
in
casesof
advanced dementia
when
the
clinical
diaenosis
of
CJD cannot be
cor-roborated
by
electro-eiceph
alogra phy .2aTherapeutics
for
prion
diseasesThe most attractive therapeutic
target
is
interfering
with
the conversion
of
PrPu
into
PrPù".
There
areseveral
therapeutic
strategies tqat can be suggested, i.e.stabilizing
the structureof
PrP'
bybinding it
to adrug;
modifying
theaction
of
protein
X, which
might
func-tion
as
a molecular
chaperone.
The
PrPùcformation
seems
to
be
limited to
caveola-like
domains
so
thedrugs do
not
need to penetrate thecytosol
ofthe
cells,
but they must be able to enter the CNS. Drugs
thatdestabilize
the structure
of
PrPs"
might
also
prove
useful.T
The transformation
of
PrPc
into
PrPsc
requires
aninteraction
of
PrPc-PrPS".
Th"."
are some
sulfated
polyanion
compounds, such as pentosan sulfate,
dextran sulfate, heparin,
amyloid binding
dye
Congored, that can
irreversibly
inhibit PrP"" formation
andprion
syntherir.ll'25 Coughey
et
al., citated
by
Be-,r"n,l
I observed that PrPc canbind
directly
tosulfated
glycans, Congo red, and
endogenous
glycosamino-glycans, suggesting that sulfated
polyanions
maycom-petitively inhibit PrP'-PrP'"
interaction.
Shyng etal."
found that these
compounds
of
PrPL on the
surface
of
enhancing the rate
of
PrPc
glycans
may
also redistribute
a portion
of
PrP
molecules
to
an endocvtic comoartment that
is
un-favorable
for
the"onu"rrion
proàrr.t5
Understandins
how
PrPc unfolcts and refolds into
PrPs" may
oi"n
n"*
approach
to
deciphering
thecauses
of
and
developing effective
therapies
for
themore common neurodegenerative
diseases,ilcluding
Vol 8, No 3, July - September 1999
9. Collee JG, Bradley
R. BSE: a
decade on-partI.
Lancet 1997;349:636-41 .10. Prusiner SB. Molecular biology and pathogenesis of prion diseases. Trends Biochem Sci 1996;21:482-7.
ll.
BessenRA.
Neurodegenerative prion disease. Science&
Medicine 1996; 3:12-21.12. Mayer RJ, Landon M, Laszlo L, Lennox G, Lowe J. Protein processing
in
lysosomes: the new therapeutic target in neurodegenerative disease. Lancet 1992;340: I 56-9. 13. Huang Z, Gabriel JM, Baldwin MA, Fletterick RJ, PrusinerSB, Cohen FE. Proposed three-dimensional structure for the cellular
prion protein. Proc Natl Acad Sci USA
1994; 9l:7139-43.14. Martins VR, Graner E, Garcia-Abreu J, de Souza SJ, Mer-cadante AF, Viega SS, et al. Complementary hydropathy identifies
a cellular prion protein
receptor. Nature Med 199'1:3:1376-82.15. Rieger R, Edenhofer F, Lasmezas CI, Weiss. The human 37-kDa laminin receptor precursor interacts with the prion protein in eukaryotic cells. Nature Med 1997;3:1383-8. 16. Berg LJ. Insight into the role of the immune system in the
prion diseases. Proc Natl Acad Sci USA 1994;91:429-32. 17. Vogel G.
B
cells may propgate prions. Science 1997:278:2050.
18. Hegde RS, Mastrianni JA, Scott MR, DeFeaa KA, Tremblay P, Torchia
M, et al. A
transmembrane form of the prionPrion in neurodegenerative
diseases
159protein in neurodegenerative
disease. Science I998: 279:827-34.19. Epstein FH. Transmissible spongiform encephalopathies. N Engl J Med 1997;337:1821-8.
20. Agtzzi A. Neuro-immune connection in spread of prions in the body? Lancet 1997;349:742-3.
21. Collee JG, Bradley
R.
BSE: a decade on-part 2. Lancet 1997:349:715-21.22. Sarkozi E, Askansas V, Engel WK. Abnormal accumulation
of prion
protein mRNAin
muscle fibersof
patients with sporadic body myositis and hereditary inclusion-body myopathy. Am J Pathol 1994;145:1280-4.23. Hsich G, Kenney K, Gibbs CJ, Lee KH, Harrington MG. The l4-3-3 brain protein in cerebrospinal fluid as a marker for transmissible spongiform encephalopathies.
N
Engl J Med 1996:335:924-30.24.Zerr I, Bodemer M,
Racker S, GroscheS, Poser
S, Kretzschmar HA, e al. Cerebrospinal fluid concentration of neuron-specific enolasein
diagnosisof
Creutzfeldt-Jakob disease. Lancet 1995;345: I 609-10.