Site-directed mutagenesis of Cys-92 from the
a
-polypeptide of
Phaseolus
6
ulgaris
glutamine synthetase reveals that this highly
conserved residue is not essential for enzyme activity but it is
involved in thermal stability
M. Teresa Clemente, Antonio J. Ma´rquez *
Departamento de Bioquı´mica Vegetal y Biologı´a Molecular,Facultad de Quı´mica,Uni6ersidad de Se6illa,Apartado553,41080-Se6illa,Spain
Received 19 November 1999; received in revised form 5 January 2000; accepted 6 January 2000
Abstract
The residue Cys-92 from the a-polypeptide of Phaseolus 6ulgaris glutamine synthetase is a highly conserved residue in prokaryotic and eukaryotic glutamine synthetase genes. This cysteine residue was previously proposed as a good candidate for being essential for enzyme activity. We have examined through heterologous expression in Escherichia coli and site-directed mutagenesis the functional importance of this residue. We have found that the thiol group of Cys-92 is not essential either for glutamine synthetase biosynthetic or transferase enzyme activities. The characteristic inhibition byp-hydroxymercuribenzoate (a specific sulphydryl reagent) was not substantially altered as a consequence of replacement of Cys-92 by Ala. Immunoreactivity of the glutamine synthetase mutant protein, examined both under native and denaturing conditions, was similar to the wild-type, indicating that no significant conformational changes were produced as a consequence of the introduced mutation. However, the mutant enzyme C92A was considerably less stable than the wild-type. These results indicate that Cys-92 is not an essential residue for enzyme activity but it is important for stability of the glutamine synthetase protein. © 2000 Elsevier Science Ireland Ltd. All rights reserved.
Keywords: Glutamine synthetase; Plant nitrogen assimilation; Structure-function; Site-directed mutagenesis; Recombinant enzymes; Phaseolus 6ulgaris
www.elsevier.com/locate/plantsci
1. Introduction
Glutamine synthetase (GS) (E.C. 6.3.1.2) is a key enzyme involved in nitrogen metabolism, the reason why it has been thoroughly studied in many different groups of biological organisms. In particular, in higher plants this is the first enzyme in the main pathway of ammonium assimilation, both of ammonium derived from primary and secondary nitrogen/dinitrogen assimilation. Glu-tamine synthetase acts in conjunction with
gluta-mate synthase (GOGAT; E.C. 1.4.7.1 and E.C. 1.4.1.14) to convert ammonium ions and 2-oxoglu-tarate to glutamate, via glutamine, at the expense of ATP and reducing power. The glutamine and glutamate produced can then be used to synthesize all other nitrogen-containing compounds in the plant [1]. A distinctive feature of GS in higher plants is the existence within a single plant of a number of different cytosolic or chloroplastic isoenzymic forms produced as a result of the expression of a multigene family [2,3].
In spite of its importance, structure-function studies of individual amino acid residues in the plant enzyme have just been initiated [4 – 6] and are not available from the enzyme from other eukaryotic sources. Contrarily, the bacterial en-Abbre6iations: GS, glutamine synthetase; p-HMB,p
-hydroxymer-curibenzoate; U.S.E., unique site elimination; WT, wild-type.. * Corresponding author. Tel.: +34-95-4557145; fax: + 34-95-4626853.
E-mail address:[email protected] (A.J. Ma´rquez)
zyme has been the subject of three-dimensional structure determination for several bacterial sources such as Salmonella typhimurium [7,8] and Escherichia coli [9] as well as mechanistic models for the catalysis being proposed [10 – 12] which are consistent with results obtained by site-directed mutagenesis studies [9,13 – 15].
Prokaryotic and eukaryotic GS enzymes are quite different in their quaternary structure and regulatory properties [16,17]. However, the differ-ent GS genes sequenced, so far, indicate a strong degree of sequence conservation among different groups of organisms, even those distantly related in evolution, to the point that GS has been consid-ered as a good molecular clock in gene evolution [18,19]. In particular, alignment of eukaryotic GS sequences, both from animal and plant sources, indicates that they are very similar [20], as it is also their octameric quaternary structure. Therefore it seems interesting to use the site-directed mutagen-esis approach in order to test the functionality of conserved residues within the different GS polypeptides. Results obtained for the higher plant cytosolic enzyme could be possibly also extrapo-lated to animal and human GS.
An essential cysteine is reported to be located at the active site of mammalian GS [20,21] and there is evidence that there are two cysteine residues at the active sites of both cytosolic and chloroplast GS [22 – 25]. At least one of the cysteines [22], and possibly both [23], appear to be essential for en-zyme activity. Very recently, the two additional cysteine residues characteristic of plastidic GS have been examined for their role in the specific stimulation by thiol compounds of this particular isoenzyme [6]. As pointed out by Forde and Cul-limore [2] a particular cysteine residue, corre-sponding to Cys-92 of the a-polypeptide of P.
6ulgaris cytosolic GS, could be interesting to be analyzed in further detail as possibly essential for catalysis, since it is conserved in all organisms, and its bacterial homologue is located within the N-terminal domain adjacent to the ‘Trp-57’ loop that forms part of the cylindrical active site [7].
In the present work we have mutagenized an internal position within an a cDNA of P. 6ulgaris
GS in order to produce the replacement of Cys-92 by an alanine residue in the corresponding polypeptide. Also, the wild-type and mutant cD-NAs were heterologously expressed in E. coli, the recombinant enzymes being comparatively
charac-terized to probe whether the thiol group of Cys-92 is essential for catalysis.
2. Materials and methods
2.1. Heterologous expression of the gln a cDNA
from P. 6ulgaris in E. coli
The plasmid pcGS-a1 [26], containing a full length cDNA for P. 6ulgaris gln a gene, was
purified on a CsCl gradient. Later it was digested, first completely byBglII, then partially byHindIII (15 min at 0.5 U·mg−1 DNA). The 1.3 kb BglII – HindIII fragment was electroeluted from an agarose gel and inserted into the vector pUC18 previously digested withBamHI andHindIII. The ligation reactions were transformed into E. coli ET8894 (a glnAmutant host). Undetectable levels of GS enzyme activity in this bacterial host guar-anteed that activity measurements after transfor-mation could only be attributed to the higher plant GS. Competent cells were prepared by the method described by Hanahan [27]. Transfor-mants were selected on LB plates containing ampi-cillin. The recombinant expression plasmid (pcGS-18a) (4 kb in size) contained the first 12 codons of b-galactosidase fused in frame to the sixth codon of the GS a polypeptide. The con-struct was confirmed by restriction analysis and DNA sequencing.
Expression of the recombinant GS a polypep-tide from P.6ulgarisinE. coliwas achieved under the following optimized conditions for recovery of repetitive levels of GS enzyme activity: 100 ml cultures on LB medium containing ampicillin were inoculated with 50 – 100 ml from a frozen stock (an overnight culture which had been previously ini-tiated from a fresh plate). Cells were grown at 30°C for 22 – 26 h (4 – 8 h after reaching the sta-tionary phase), and were then harvested for en-zyme activity measurements.
2.2. Site-directed mutagenesis
Mutations were introduced into pcGS-18a es-sentially by the U.S.E. method as described by Deng and Nickoloff [28], making use of the com-mercial system from Pharmacia. The U.S.E.
selec-tion and mutagenic primers were
fol-lowing sequences: 5%
-GACTTGGTTGACGCGT-CACC AGTCACAG-3% and 5%
-CTTGTGATT-GCTGATGTTAC-3%. Underlined sequences show
nucleotides modified with respect to the original sequence of the plasmid. The process enabled the replacement of Cys-92 by Ala in the recombi-nantly expressed GS a polypeptide. Putative mu-tant colonies were confirmed by in situ and dot blot hybridizations using the g-32P labelled target mutagenic primer as a probe and a 42°C hy-bridization and washing temperature in 6×SSC buffer. Only 14% of the putative mutant colonies selected by the U.S.E. method gave positive hy-bridization signals. The expected mutations on the DNA were identified by DNA sequencing follow-ing the dideoxy method and were unique among a global reading of 2000 bp, thus confirming that undesired mutations were not present. A 2.9 kb HindIII –NcoI deletion fragment was subcloned for sequencing purposes using the forward univer-sal primer. The mutant plasmid DNA was finally transformed into the ET8894 E. coli host for ex-pression and analysis of the GS C92A mutant enzyme. Optimal conditions for expression were found to be similar to the wild-type as mentioned in the previous section. Other methods and details were as described by Sambrook et al. [29]. For comparative purposes to the mutant strain, we used a wild-type strain that had been submitted to the same U.S.E. procedure, except that only the selection primer, but not the mutagenic primer, were used. Kinetic, physicochemical and stability properties of the wild-type GS from this strain were identical to the standard wild-type.
2.3. Crude extract preparation and purification of wild-type and mutant recombinant GS
Crude extracts from recombinant bacteria were prepared (1 ml·g−1 FW) by sonication in 10 mM imidazole buffer, pH 7.5, containing 10 mM MnCl2 and 0.1% (w/v)b-mercaptoethanol (extrac-tion buffer).
Purified GS preparations were obtained by the following steps: (1) Streptomycin sulphate precipi-tation; (2) 35 – 55% ammonium sulphate fractiona-tion. Pellets were dissolved in a third of the original volume of the crude extract using 10 mM imidazole buffer, pH 7.5, containing 2.5 mM MnCl2 (I-Mn Buffer); (3) sephacryl S-300 gel filtration chromatography (16×700 mm) was run
in the same buffer; and (4) 2%,5%-ADP-Sepharose
4B affinity chromatography (10×90 mm). The column was equilibrated and washed with I – Mn buffer and GS enzyme activity eluted with buffer supplemented with 5 mM ADP. The final enzyme preparation was dialysed against 10 mM imidazole buffer, pH 7.5, containing 10 mM MgCl2 (I – Mg buffer), and subsequently used for physicochemi-cal and kinetic studies.
Total protein estimates in extracts were carried out by the method of Lowry [30] and/or Bradford [31], using bovine serum albumin as standard, and yielding similar results.
2.4. GS protein immunological analysis
SDS-polyacrylamide gel electrophoresis and Western blots were carried out as described by Pajuelo et al. [32] using separating gels with 12.5% (w/v) acrylamide and primary antibodies raised against GS purified from P. 6ulgaris root nodules
[33] (used at a final concentration of 1.8 mg protein·ml−1). Goat-antirabbit alkaline phos-phatase conjugates (Boehringer Mannheim) were used as secondary antibodies. The final images of the visualised blots were analysed by a Millipore Bioimage System densitometer in order to quan-tify the GS protein bands. Intensity of the mutant bands, after subtraction of the background, was referred to the amount of total protein loaded and expressed as % of the wild-type (further details in Fig. 1).
The immunological response of the GS mutant protein with respect to the wild-type was also examined under native (non-denaturing) condi-tions, both by enzyme-linked immunosorbent
as-say (ELISA) and Western blot following
plus two different anode solutions, accordingly to the manufacturer’s instructions for electrotransfer of high molecular weight proteins. Immunostain-ing of native GS protein bands was performed as described above for denaturing Western blots.
2.5. Enzyme assays
Transferase or biosynthetic assays were carried out with minor modifications of methods previ-ously described [35,36].
Standard reaction mixture for GS transferase assay contained in a final volume of 1 ml: 120 mmol MOPS buffer, pH 7.0; 90mmolL-glutamine;
2.4mmol MnCl2; 50 nmol ADP; 120 mmol hydrox-ylammonium chlorid neutralized with 60 mmol NaOH; 50 mmol Na2HAsO4 and 1-5 nkat of en-zyme. The reaction was incubated for 10 min at 37°C after which the product g-glutamyl hydroxa-mate was determined as described [35]. Blanks were similarly assayed, but omitting Na2HAsO4.
The standard GS biosynthetic assay contained in a final volume of 100ml: 10 mmol Tris – HCl 0.5 M, pH 7.5; 10 mmol L-glutamate; 5 mmol NH4Cl;
5 mmol MgCl2; 0.75mmol ATP and 10 – 70 pkat of enzyme. The assay mixture was incubated for 15 min at 37°C; the amount of inorganic ortophos-phate formed was later determined by the sensitive colorimetric green malaquite method [36,37]. Blanks were identical but omitted L-glutamate.
3. Results and discussion
3.1. Effect of the C92A mutation on GS enzyme acti6ities and GS protein
A C92A mutant was produced in which Cys-92 was replaced by alanine within the a-polypeptide of P. 6ulgaris GS recombinantly expressed in E. coli. This was done in order to probe whether the thiol group of cysteine was essential for catalysis. In this cysteine to alanine mutation, the thiol
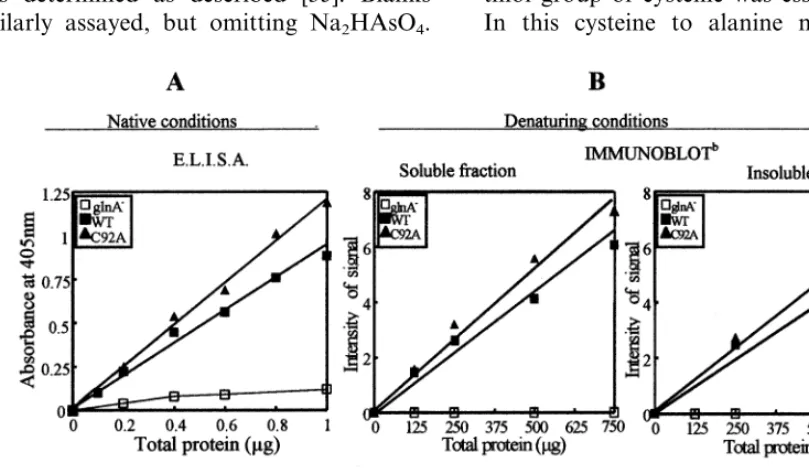
Fig. 1. Comparison of GS immunoreactivity in cell lysates using different immunochemical techniques and conditions. Comparative analysis of GS immunoreactivity in crude extracts of the WT and C92A mutant strains was carried out either by ELISA under native conditions (A) or by SDS-PAGE followed by immunoblot (denaturing conditions) of the soluble and insoluble fractions of the extracts (B). In all cases, GS immunoreactivity (determined by the absorbance at 405 nm of the ELISA assays or densitometry signals obtained from Western blots) was plotted against different amounts of total protein loaded. The extent of immunoreactive GS on each strain and condition, referred to the amount of total protein, was given by the slope of the linear representations obtained (correlation coefficients equal or higher than 0.989). For comparative purposes 100% was given for the WT on each particular case and condition. A control of no expression was included in the analysis, corresponding to results obtained for the host strain (gln A−) transformed with the original non-recombinant plasmid. The value obtained for this control
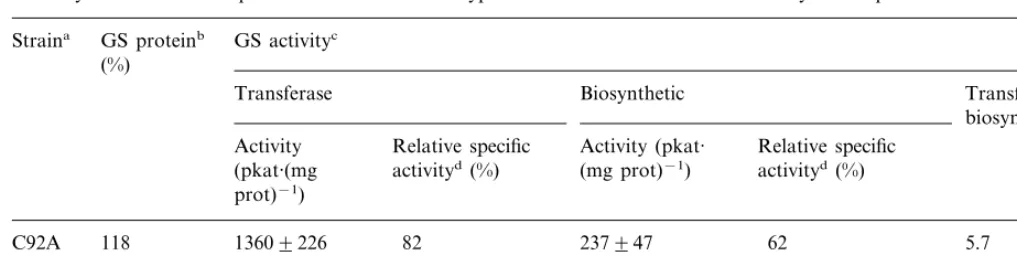
Table 1
GS enzyme activities and protein levels of the wild-type and C92A mutant strains in cell lysates supernatants
GS proteinb
Straina GS activityc (%)
Transferase Biosynthetic Transferase/
biosynthetic Activity Relative specific Activity (pkat· Relative specific
activityd (%)
(pkat·(mg (mg prot)−1) activityd(%) prot)−1)
13609226
C92A 118 82 237947 62 5.7
14009200 100 325971 100
100 4.3
WT
awild-type or mutant plasmids were used to transformE.coliET8894(glnA−) cells, lysed by sonication, assayed for GS activities
and protein.
bGS protein concentrations were determined from Western Blot under denaturing conditions using a scanning densitometer as described in Section 2, and expressed as relative to the wild type (100%).
cAssays were carried out as described in Section 2 using 25ml and 1ml of cell lysate supernatant for transferase and biosynthetic assays, respectively. Values in the table are expressed as the amount of product produced per minute and are the mean of three independent experiments9standard deviation. A control containing the expression vector (pUC18) without GS gene insert showed no detectable enzyme activity. Total protein concentrations in the lysates were 25–30 mg·ml−1.
dThe data in this column are the mean enzyme activities divided by specific GS protein concentrations, and were expressed as % of the value obtained for WT.
group of cysteine is replaced by hydrogen, result-ing in a side chain which lacks potential chemical reactivity and polar interactions. Replacement of the thiol group of a cysteine side chain with hy-drogen should completely abolish the residue’s ability to participate in enzyme catalysis, if this residue is essential.
The level of GS transferase and biosynthetic activity found in mutant C92A were similar to those of the wild-type. Transferase activity was almost identical, while biosynthetic activity was 27% lower in the mutant strain (Table 1).
Considering that enzyme activities for the wild-type and the mutant were similar, it was important to refer these values to the amount of GS protein able to produce these enzyme activities on each strain, in case variable levels of expression could be taking place between the wild-type and the C92A mutant. The level of GS protein was deter-mined immunologically directly in the crude ex-tracts, and resulted similar for the wild-type and C92A mutant. Thus, taking into account the level found for GS enzyme activities and GS protein, we obtained a ratio between both magnitudes which gave us the relative specific activity of the mutant enzyme as a % of the wild-type (Table 1). Since these measurements could be carried out directly in the crude extract, we excluded any
proteins were found very similar (Fig. 1A). Fig. 1(B) also shows that the proportion of immunore-active response between the soluble and insoluble fractions of the cell lysate was not significantly altered when examined under denaturing condi-tions, indicating that replacement of Cys-92 by Ala did not cause an increase in the level of GS protein disruption leading to denaturation. These data clearly indicate that the C92A mutation is not accompanied by global conformational changes in the GS protein. The immunological approach seems to be sufficient to support such conclusion, since it is well known that it is a powerful and specific tool to study conformational changes in proteins and it was successfully used to detect conformational changes for other types of mutations within the same polypeptide [5]. In ad-dition, we have also confirmed both by gel filtra-tion and non-denaturing western blots (not shown) that the size of the native homopolymeric C92A mutant protein (Mr 340 000) was identical to the wild-type and indicative of the characteristic octameric quaternary structure of eukaryotic GS. Bennet and Cullimore [38] also found an oc-tameric structure of the higher plant GS recombi-nantly expressed in E. coli, in spite of the natural GS from bacteria being dodecameric. Also, no change was detected in the behaviour of the C92A mutant protein, compared to the wild-type, with regard to other steps of the purification process (35 – 55% ammonium sulphate fractionation, affinity chromatography on 2%,5%ADP-Sepharose), in spite of being reported for other mutations [4,5]. Only a minor decrease was observed in the optimum pH and energy of activation of the mu-tant biosynthetic activity, changing respectively from 7.5 to 7.0, and from 48 to 34 kJ·mol−1. However, these changes were not reflected in mu-tant GS transferase activity, which maintained identical parameters to the wild-type. No changes were detected in the response of the enzyme to-wards inhibition by L-methionine sulfoximine, a
known transition analogue in structural studies of glutamine synthetase (result not shown).
Therefore we conclude that Cys-92 from the a-polypeptide of P. 6ulgaris GS, although highly
conserved in other biological organisms, does not seem to be an essential residue for enzyme activity or key structural features within the protein, since replacement of this residue does not lead to an important loss of function. This cysteine was
pre-viously postulated as a good candidate for being one of the conserved cysteines reported to be essential for enzyme activity [2,22 – 24], since the bacterial cysteine homologous to Cys-92 in P.
6ulgaris is close to Trp-57, which belongs to the
active site [7]. Unfortunately, no comparative study is available on the function of this particu-larly conserved cysteine in bacterial GS or the enzyme from other sources. Also, crystal models of higher plant GS are at present unavailable, making it difficult and not very reliable to confirm our results by computer modelling. However, the lack of essentiality of Cys-92 contrasts with results obtained in our laboratory for site-directed muta-genesis of other putatively conserved residues such as Asp-56 or Glu-297 from the same recombinant polypeptide, which were shown to be essential or crucially important for the level of transferase or biosynthetic enzyme activities, as well as for the immunoreactive response under native conditions of the protein [4,5].
3.2. Effect on the mutant GS enzyme of thiol group inhibitors
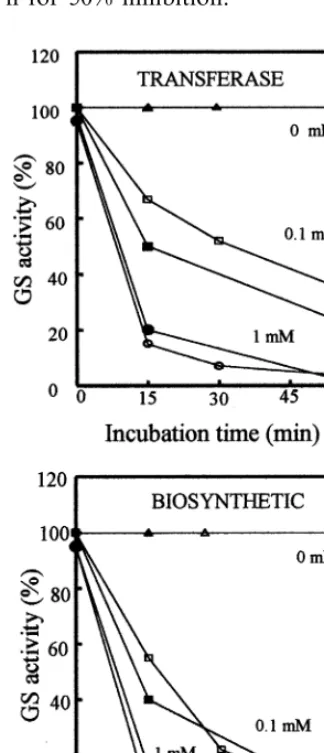
Incubation of the C92A mutant GS in the pres-ence of p-hydroxymercuribenzoate (p-HMB), a commonly used sulphydryl reagent, produced a strong inhibition of both the transferase and biosynthetic activities of the enzyme, similar to that obtained for the wild-type in a vast range of concentrations and incubation times (Fig. 2). This result indicates that Cys-92 from thea-polypeptide of P. 6ulgaris GS is not likely involved in the sensitivity of the enzyme towards p-HMB, thus confirming the lack of essentiality of this residue for enzyme activity, or at least that some other cysteine residues within the protein molecule must also be responsible for the inhibition produced by p-HMB.
3.3. Thermal instability of the C92A GS mutant protein
more unstable than the wild-type. Incubation of the enzyme at 4°C for long periods of time (Fig. 3 A) led to a loss of 100% of both biosynthetic and transferase enzyme activities in 72 h, while it was maintained almost 100% for the wild-type. Fig. 3 B confirms this result for shorter incubation peri-ods and at a higher temperature (22°C). Fifteen minutes were sufficient to completely inactivate both biosynthetic and transferase GS enzyme ac-tivities in the C92A mutant enzyme while the wild-type still retained almost 90% and required at least 1 h for 50% inhibition.
Fig. 3. Difference between thermal stability of WT and C92A mutant GSs. Purified GS preparations from WT and/or C92A mutant proteins were incubated either at 4°C (A) or 22°C (B) for different lengths of time, followed by both GS transferase and biosynthetic enzyme assays. Activity is expressed as % of the initial activity (1 nkat for transferase and 20 pkat for biosynthetic). Similar behaviour was obtained both for trans-ferase and biosynthetic GS enzyme activities, and therefore a single plot for each is presented. Results are the means of duplicate determinations that differed by less than 5%.
Fig. 2. Time-courses for inactivation of WT and C92A mu-tant GSs with different concentrations of p-HMB. 1 nkat of GS transferase and/or 20 pkat of biosynthetic enzyme activi-ties from WT and C92A mutant strains were incubated for different lengths of time in the presence of the indicated concentrations of the sulphydryl reagent p-HMB. Activity remaining at the end of each treatment was determined and plotted as % of the initial. WT, open symbols. C92A, closed symbols. Results are the means of duplicate determinations that differed by less than 5%.
Instability of the C92A GS mutant enzyme was also apparent after checking that 70% of the en-zyme activity was lost when 35 – 55% ammonium sulphate fractions of the crude extracts were stored at −70°C, while in these conditions 95% of the wild-type enzyme activity was recovered.
affinity constants and catalytic efficiencies of the purified mutant enzyme preparations for the dif-ferent substrates, as it has been possible to achieve for mutations in other residues [4,5]. In any case, any possible changes in these parameters would be more likely related to the aforementioned instabil-ity of the enzyme and, in our opinion, could not be reliably employed to further analyze the role of Cys-92 in the interaction of the enzyme with the substrates.
So far, we can say that the main consequence detected for the C92A mutation in the a-polypep-tide of P. 6ulgaris GS recombinantly expressed in E. coli is a pronounced decrease in stability of the mutant protein with regard to the wild-type. It is well known that plant GSs are highly sensitive to thermal inactivation [39]. Therefore we think that Cys-92 must be somehow involved in structural features leading to enzyme stability and this could be an important reason for this particular residue being highly conserved through evolution. In other enzyme systems it is common to show the presence of residues which are not essential for binding or catalysis but are important for the stability of the protein [40].
The present work, to our knowledge, is the first study of the role of individual cysteine residues within either prokaryotic or cytosolic eukaryotic GS. We hope that similar studies will be carried out with other GS genes in the near future, en-abling comparison with our results.
Acknowledgements
Financial support was provided by research project PB94-1192 from Direccio´n General de In-vestigacio´n Cientı´fica y Te´cnica (DGICYT, Spain), project PB97-0714 from Direccio´n General de Ensen˜anza Superior (DGES, Spain), project BIO95-1107-CE from Comisio´n Interministerial de Ciencia y Tecnologı´a (Spain), Junta de Andalucı´a (group CVI-0163) and the European Community Biotechnology Programme, as part of the Project of Technological Priority BIO2-CT93-0400 (1993-1997). We wish also to express our most sincere gratitude to Professor Brian G. Forde (Lancaster University, UK), for providing us with GS cDNA and antibodies, as well as to M.J. Cubas, P. Pajuelo and C. Mun˜oz for technical and secretar-ial assistance, and Diana Haun, Ph.D., for profes-sional English correction.
References
[1] B.J. Miflin, P.J. Lea, Ammonia assimilation, in: B.J. Miflin (Ed.), The Biochemistry of Plants, vol. 5, Aca-demic Press, New York, 1980, pp. 169 – 202.
[2] B.G. Forde, J.V. Cullimore, The molecular biology of glutamine synthetase in higher plants, Oxford Surv. Plant Mol. Cell Biol. 6 (1989) 247 – 296.
[3] H.-M. Lam, K.T. Coshigano, I.C. Oliveira, R. Melo-Oliveira, G.M. Coruzzi, The molecular genetics on nitro-gen assimilation into amino acids in higher plants, Annu. Rev. Plant Physiol. Plant Mol. Biol. 47 (1996) 569 – 593. [4] M.T. Clemente, A.J. Ma´rquez, Site-directed mutagenesis of Glu-297 from thea-polypeptide ofPhaseolus6ulgaris
glutamine synthetase alters kinetic and structural proper-ties and confers resistance to L-methionine sulfoximine,
Plant Mol. Biol. 40 (1999) 835 – 845.
[5] M.T. Clemente, A.J. Ma´rquez, Functional importance of Asp56 from thea-polypeptide ofPhaseolus6ulgaris glu-tamine synthetase, Eur. J. Biochem. 264 (1999) 453 – 460. [6] Y.A. Choi, S.G. Kim, Y.M. Kwon, The plastidic glu-tamine synthetase activity is directly modulated by means of redox change at two unique cysteine residues, Plant Sci. 149 (1999) 175 – 182.
[7] R.J. Almassy, C.A. Janson, R. Hamlin, N.-H. Xuong, D. Eisenberg, Novel subunit-subunit interactions in the structure of glutamine synthetase, Nature 323 (1986) 304 – 309.
[8] M.M. Yamashita, R.J. Almassy, C.A. Janson, D. Cascio, D. Eisenberg, Refined atomic model of glutamine syn-thetase at 3.5 A, resolution, J. Biol. Chem. 264 (1989) 17681 – 17690.
[9] S.H. Liaw, J.J. Villafranca, D. Eisenberg, A model for oxidative modification of glutamine synthetase, based on crystal structures of mutant H269N and the oxidized enzyme, Biochemistry 32 (1993) 7999 – 8003.
[10] T.D. Meek, J.J. Villafranca, Kinetic mechanism ofE.coli
glutamine synthetase, Biochemistry 19 (1980) 5513 – 5519. [11] S.H. Liaw, D. Eisenberg, Structural model for the reac-tion mechanism of glutamine synthetase, based on five crystal structures of enzyme-substrate complexes, Bio-chemistry 33 (1994) 675 – 681.
[12] S.H. Liaw, I. Kuo, D. Eisenberg, Discovery of the ammonium substrate site on glutamine synthetase, a third cation binding site, Protein Sci. 4 (1995) 2358 – 2365.
[13] W.M. Atkins, P.S. Stayton, J.J. Villafranca, Time-re-solved fluorescence studies of genetically engineered E.
coli glutamine synthetase. Effects of ATP on the tryp-tophan-57 loop, Biochemistry 30 (1991) 3406 – 3416. [14] W.M. Atkins, J.J. Villafranca, Time-resolved
fluores-cence studies of tryptophan mutants ofE.coliglutamine synthetase: conformational analysis of intermediates and transition-state complexes, Protein Sci. 1 (1992) 342 – 345. [15] M. Alibhai, J.J. Villafranca, Kinetic and mutagenic stud-ies of the role of the active site residues Asp-50 and Glu-327 ofE.coliglutamine synthetase, Biochemistry 33 (1994) 682 – 686.
glu-tamine synthetase and glutamate synthase, in: B.J. Miflin (Ed.), The Biochemistry of Plants, vol. 5, Academic Press, New York, 1980, pp. 271 – 327.
[17] S. Mc Nally, B. Hirel, Glutamine synthetase isoforms in higher plants, Physiol. Veg. 21 (1983) 761 – 774.
[18] G. Pesole, M.P. Bozzetti, C. Lanave, G. Preparata, C. Saccone, Glutamine synthetase gene evolution: a good molecular clock, Proc. Natl. Acad. Sci. USA 88 (1991) 522 – 526.
[19] J. Biesiadka, A.B. Legocki, Evolution of the glutamine synthetase gene in plants, Plant Sci. 128 (1997) 51 – 58. [20] R. Johnson, D. Piskiewicz, Primary structure of peptides
from bovine brain glutamine synthetase. Comparison with sequences of glutamine synthetases from other or-ganisms, Biochim. Biophys. Acta 827 (1985) 439 – 446. [21] D. Rajagopal Rao, K. Beyreuther, L. Jaenicke, A
com-parative study of pig and sheep-brain glutamine syn-thetases: tryptic peptides and thiol groups, Eur. J. Biochem. 35 (1973) 582 – 592.
[22] M.C. Ericson, S.A. Brunn, Cysteine residues at the active site of glutamine synthetase from spinach leaves, Biochem. Biophys. Res. Commun. 133 (1985) 527 – 531. [23] Z.G. Evstigneeva, A.V. Pushkin, N.P. Akentyeva, W.L.
Kretovich, Active site of glutamine synthetase from chloroplasts and cytosol of pea leaves, Physiol. Ve´g. 23 (1985) 861 – 868.
[24] N.P. Akente´va, N.A. Solove´va, A.V. Pushkin, Z.G. Evstigneeva, V.L. Kretovich, Secondary structure and functional groups of the active site of glutamine syn-thetase from pea chloroplast, Biokhimiya 48 (1983) 833 – 836.
[25] S.V. Tingey, G.M. Coruzzi, Glutamine synthetase of
Nicotiana plumbaginifolia. Cloning and in vivo expres-sion, Plant Physiol. 84 (1987) 366 – 373.
[26] C. Gebhardt, J.E. Oliver, B.G. Forde, R. Saarelainen, B.J. Miflin, Primary structure and differential expression of glutamine synthetase genes in nodules, roots and leaves of P.6ulgaris, EMBO J. 5 (1986) 1429 – 1435. [27] D. Hanahan, Techniques for transformation of E. coli,
in: D.M. Glover (Ed.), DNA cloning: a practical ap-proach, vol. 1, IRL Press, Oxford, 1985, pp. 109 – 135. [28] W.P. Deng, J.A. Nickoloff, Site directed mutagenesis of
virtually any plasmid by eliminating a unique site, Anal. Biochem. 200 (1992) 81 – 88.
[29] J. Sambrook, E.F. Fritsch, T. Maniatis, Molecular cloning. A Laboratory Manual, Cold Spring Harbor Laboratory Press, USA, 1989.
[30] O.H. Lowry, N.J. Rosebrough, A.L. Farr, R.J. Randall, Protein measurements with the Folin phenol reagent, J. Biol. Chem. 193 (1951) 265 – 275.
[31] M.M. Bradford, A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding, Anal. Biochem. 72 (1976) 248 – 254.
[32] E. Pajuelo, J.A. Borrero, A.J. Ma´rquez, Immunological approach to subunit composition of ferredoxin-nitrite reductase fromChlamydomonas reinhardtii, Plant Sci. 95 (1993) 9 – 21.
[33] J.V. Cullimore, B.J. Miflin, Immunological studies on glutamine synthetase using antisera raised to the two plants forms of the enzyme from P. 6ulgaris root nod-ules, J. Exp. Bot. 35 (1984) 581 – 587.
[34] P. Pajuelo, E. Pajuelo, B.G. Forde, A.J. Ma´rquez, Regu-lation of the expression of ferredoxin-glutamate synthase in barley, Planta 203 (1997) 517 – 525.
[35] A.R. Fergusom, A.P. Sims, Inactivation in vivo of glu-tamine synthetase and NAD specific glutamato dehydro-genase: its role in the regulatorion of glutamine synthetase in yeast, J. Gen. Microbiol. 69 (1971) 423 – 427.
[36] T.A.V. Rees, T.R. Larson, J.W.G. Heldens, F.G.J. Hun-ing, In situ glutamine synthetase activity in a marine unicellular alga. Development of a sensitive colorimetric assay and the effects of nitrogen status on enzyme activ-ity, Plant Physiol. 109 (1995) 1405 – 1410.
[37] P.A. Lanzeta, L.J. Alvarez, P.S. Reinach, O.A. Candia, An improved assay for nanomole amounts of inorganic phosphate, Anal. Biochem. 100 (1979) 95 – 97.
[38] M.J. Bennett, J.V. Cullimore, Expression of three plant glutamine synthetase cDNAs in E. coli. Formation of catalytically active isoenzymes, and complementation of a glnA mutant, Eur. J. Biochem. 193 (1990) 319 – 324. [39] B. Hirel, P. Gadal, Glutamine synthetase in rice, a
comparative study of the enzymes from roots and leaves, Plant Physiol. 66 (1980) 619 – 623.
[40] S.C. Schultz, J.H. Richards, Site-saturation studies of b-lactamase: production and characterization of mutant b-lactamases with all possible amino acid substitutions at residue 71, Proc. Natl. Acad. Sci. USA 83 (1986) 1588 – 1592.