PAPER
Neurofibromatosis
Disusun oleh:
PAVITRADEVI A/P N.KANNADHAS
NIM: 110 100 444
Supervisor:
dr. Bobby R.E. Sitepu, M. Ked (Oph)
Sp. M
PROGRAM PENDIDIKAN PROFESI DOKTER
DEPARTEMEN ILMU KESEHATAN MATA
FAKULTAS KEDOKTERAN
UNIVERSITAS SUMATERA UTARA
RSUP HAJI ADAM MALIK
MEDAN
KATA PENGANTAR
Puji syukur penulis panjatkan kepada Tuhan Yang Maha Esa karena telah melimpahkan berkatnya kepada penulis sehingga dapat menyelesaikan paper yang berjudul Neurofibromatosis ini.
Pada kesempatan ini, penulis ingin menyampaikan terima kasih yang sebesar-besarnya kepada dr. Bobby R.E. Sitepu, M. Ked (Oph) Sp. M selaku supervisor yang telah memberikan bimbingan dalam menyelesaikan paper ini.
Penulis menyadari masih banyak kekurangan dalam penulisan paper ini baik dari segi isi maupun sistematika penulisan karena keterbatasan kemampuan penulis. Oleh karena itu penulis sangat mengharapkan kritik dan saran dari semua pihak untuk menyempurnakan paper ini. Semoga paper ini dapat bermanfaat bagi kita semua.
Medan, Desember 2016
DAFTAR ISI
Halaman KATA PENGANTAR... i DAFTAR ISI... ii DAFTAR GAMBAR... iii BAB 1 PENDAHULUAN... 1.1 Latar Belakang... 1.2 Tujuan Penulisan... 1 1 2 BAB 2 TINJAUAN PUSTAKA...
2.1 Anatomi Mata... 2.1.1 Bagian Luar Mata... 2.1.2 Bagian Dalam Mata... 2.2 Neurofibromatosis... 2.2.1 Definisi... 2.2.2 Epidemiologi... 2.2.3 Etiologi... 2.2.4 Patofisiologi... 2.2.5 Manifestasi Klinis... 2.2.6 Diagnosa Banding... 2.2.7 Diagnosis... 2.2.8 Penatalaksanaan... 2.2.9 Komplikasi... 2.2.10 Prognosis... 3 3 3 4 9 9 9 10 10 11 16 17 18 19 20 BAB 3 KESIMPULAN... 21 DAFTAR PUSTAKA... 22 DAFTAR GAMBAR
Halaman Gambar 2.1. Bagian sagittal mata...3 Gambar 2.2. Lisch nodul pada iris mata... 12 Gambar 2.3. Wajah dengan neurofibroma plexiform ...13
BAB 1 PENDAHULUAN 1.1. Latar Belakang
Neurofibromatosis merupakan kelainan kongenital dari perkembangan neuroektoderm yang menyebabkan pembengkakan atau benjolan pada jaringan fibrosa. Neurofibromatosis dianggap sebagai phacomatosis karena menimbulkan bitnik-bintik berpigmen (cafe au lait), benjolan (tumor) yang berisi jaringan saraf dan bersifat jinak.1
Neurofibromatosis dibagi menjadi dua kelompok utama yaitu neurofibromatosis tipe 1 (NF-1), juga dikenal sebagai von Recklinghausen atau neurofibromatosis perifer dan neurofibromatosis tipe 2 (NF-2), juga dikenal sebagai neurofibromatosis akustik bilateral dan neurofibromatosis sentral.2
Neurofibromatosis tipe 1 menyumbang sekitar dari 1/2.000 sampai 1/5.000 populasi. Mutasi gen yang bervariasi ditemukan pada kasus neurofibromatosis tipe 1 namun tidak ada kasus rekuren yang dilapor dengan mutasi gen yang telah diidentifikasi.3 Neurofibromatosis tipe 2 terjadi pada 1 dari 37.000 kelahiran per
tahun, penderita neur dengan sekitar setengah dari individu yang terkena mewakili kasus pertama dalam keluarga sebagai akibat dari mutasi dominan yang baru.4
Manifestasi oftalmologi dari neurofibromatosis tipe 1 adalah lisch nodul, glioma saraf optik, tumor retina, dan neurofibroma plexiform. Manifestasi oftalmologi dari neurofibromatosis tipe 2 adalah katarak, fundus dan defek motor okular.2,5
Prognosis neurofibromatosis tergantung pada tipenya. Secara umum prognosis neurofibromatosis tipe 2 lebih buruk dibanding dengan neurofibromatosis tipe 1.6
1.2. Tujuan Penulisan
Tujuan penulisan paper ini adalah untuk mengetahui dan memahami tentang Neurofibromatosis. Selain itu, paper ini juga bertujuan untuk melengkapi persyaratan kepaniteraan klinik di bagian Ilmu Kesehatan Mata Fakultas Kedokteran Universitas Sumatera Utara.
TINJAUAN PUSTAKA 2.1 Anatomi Mata
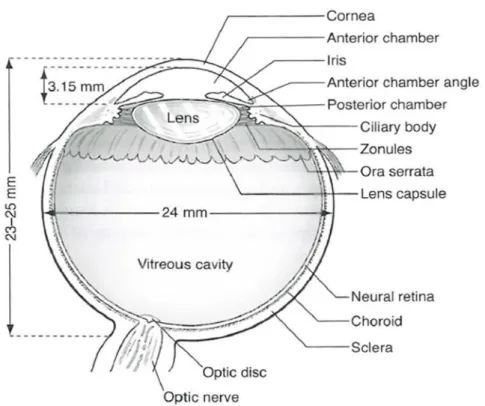
Gambar 2.1 : Bagian sagittal mata
Sumber : American Academy of Ophthalmology. Fundamentals and Principles of Ophthalmology. The Basic and Clinical Science Course, Section 2. 2014. p.37
2.1.1 Bagian Luar Mata
1. Palpebra (Kelopak mata)
Palpebra atau kelopak mata mempunyai fungsi melindungi bola mata, serta mengeluarkan sekresi kelenjar yang membentuk film air mata di depan kornea. Palpebra merupakan alat menutup mata yang berguna utuk melindungi bola mata terhadap trauma, trauma sinar, dan pengeringan bola mata. Palpebra mempunyai lapisan kulit yang tipis, longgar, dan elastik dengan sedikit folikel rambut serta tanpa lemak subkutan.7,8
Palpebra terdiri dari 4 lapisan jaringan utama yang dibagi dalam 2 lamela. Lamela anterior terdiri dari kulit dan otot orbicularis okuli, sedangkan lamella posterior terdiri dari lempeng tarsal dan konjungtiva palpebral.9
2. Kelenjar Lakrimal
Kelenjar lakrimal berbentuk seperti buah kenari dan terletak di fossa lakrimalis pada kuadran temporal di atas orbita. Kelenjar lakrimal menerima pasokan sensorik dari saraf lakrimal. Kelenjar air mata berfungsi untuk menghasilkan air mata untuk melembabkan mata, membersihkan mata dari debu dan membunuh kuman yang masuk ke mata.1
2.1.2 Bagian Dalam Mata
Kornea
Kornea adalah jaringan transparan, serat-serat kolagen, avaskular dengan ukuran 11-12 mm horizontal dan 10-11 mm vertical. Kornea berkontribusi besar untuk memberikan dioptric sebesar 74 % atau 43.25 D dari seluruh 58.6 D kekuatan dioptric manusia normal. Kornea mendapat nutrisi dari pembuluh-pembuluh darah limbus, humor akuos, dan air mata. Kornea terdiri atas 5 lapisan, yaitu : 5,8,10
a) Epitel
Tebalnya 50 μm, terdiri atas 5 lapis sel epitel tidak bertanduk yang saling tumpang tindih; satu lapis sel basal, sel poligonal dan sel gepeng.
Pada sel basal sering terlihat mitosis sel.
Sel basal menghasilkan membran basal yang melekat erat kepadanya. Bila terjadi gangguan akan mengakibatkan erosi rekuren.
b) Membran Bowman
Terletak di bawah membran basal epitel kornea yang merupakan kolagen yang tersusun tidak teratur seperti stroma dan berasal dari bagian depan stroma. Lapis ini tidak mempunyai daya regenerasi.
c) Stroma
Terdiri atas lamel yang merupakan susunan kolagen yang sejajar satu dengan lainnya, pada permukaan terlihat anyaman yang teratur sedang di bagian perifer serat kolagen ini bercabang.
d) Membrane descement
Merupakan membran aseluler dan merupakan batas belakang stroma kornea dihasilkan sel endotel dan merupakan membran basalnya.
Bersifat sangat elastik dan berkembang seumur hidup, mempunyai tebal 40 μm.
e) Endotel
Berasal dari mesotelium, berlapis satu, bentuk heksagonal, besar 20-40 μm. Endotel melekat pada membran descement melalui hemidesmosom dan zonula okluden.
Sklera
Sklera terbentuk dari fibril kolagen yang lebar yang dipertahankan oleh fibroblast. Ketebalan sklera 1mm sekitar kepala saraf optik dan 0.3mm pada posterior insersi otot. Otot bertanggung jawab untuk memindahkan bola mata yang melekat pada bola mata pada sklera.11
Konjungtiva
Konjungtiva merupakan membran mukosa yang transparan dan tipis yang membungkus permukaan posterior kelopak mata (konjungtiva palpebral) dan permukaan anterior sklera (konjungtiva bulbaris). Konjungtiva selain konjungtiva tarsal, berhubungan longgar dengan jaringan dibawahnya, oleh karenanya bola mata mudah digerakkan. Konjungtiva terdiri atas tiga bagian, yaitu :
a) Konjungtiva tarsal yang menututpi tarsus, konjungtiva tarsal sukar di gerakkan dari tasus.
b) Konjungtiva bulbi menututpi sklera dan mudah di gerakkan dari sklera di bawahnya.
c) Konjungtiva fornises atau forniks konjungtiva yang merupakan tempat peralihan konjungtiva tarsal dengan konjungtiva bulbi.
Konjungtiva bulbi dan forniks berhubungan dengan sangat longgar dengan jaringan di bawahnya sehingga bola mata mudah bergerak. Fungsi konjungtiva adalah sebagai proteksi pada sklera dan memberi pelumasan pada bola mata.8,12
Pupil
Pupil adalah lubang di tengah iris yang terletak depan lensa. Gerakan pupil dikendalikan oleh sistem saraf parasimpatis dan simpatis. Pupil akan konstriksi (miosis) saat mata menyala (aktivasi parasimpatis, relaksasi simpatik) dan melebarkan (midriasis) dalam gelap (aktivasi simpatik, relaksasi parasimpatis).11 Koroid
Koroid adalah segmen posterior uvea yang mempunyai ketebalan 0.25 mm, diantara retina dan sclera. Koroid terdiri atas 3 lapisan pembuluh darah, yaitu : 8,10
1. Lapisan paling dalam yaitu choriocapillaris 2. Lapisan tengah yaitu pembuluh darah kecil
3. Lapisan paling luar yaitu pembuluh darah besar
Iris
Iris adalah struktur datar, tipis, berbentuk cincin menempel ke ruang anterior. Iris mengandung banyak pembuluh darah dan jaringan ikat serta mengandung melanosit dan sel pigmen untuk memberi warna pada mata. Iris berupa permukaan pipih dengan apertura bulat yang terletak di tengah, yaitu pupil. Iris memisahkan bilik mata depan dari bilik mata belakang, yang berisi akuos humor. Di dalam stroma iris terdapat sfingter dan otot-otot dilator. Iris mengendalikan banyaknya cahaya yang masuk ke dalam mata.8,10
Korpus Siliaris
Korpus siliaris terdiri atas zona anterior yang berombak-ombak, pars plikata (2mm), dan zona posterior yang datar, pars plana (4mm). Prosessus siliaris berasal dari pars plikata, berfungsi sebagai pembentuk akuos humor. Muskulus siliaris berfungsi untuk mengubah tegangan pada kapsul lensa, sehingga lensa dapat mempunyai berbagai fokus baik untuk melihat objek dekat maupun jauh.8,18 Zonule (Ligamen suspensorium)
Zonule berasal dari lamina basal dari pars plana dan pars plicata korpus siliaris dan memegang lensa di tempatnya. Ligamen ini juga menghubungkan lensa ke badan siliaris dan memungkinkan lensa untuk berubah bentuk.10
Akueous humor
Akueous humor adalah suatu cairan transparan yang beredar di ruang anterior dan posterior. Ini menyediakan nutrisi untuk mendukung fungsi jaringannya pada segmen anterior, ekskresi sisa-sia metabolism, membantu mempertahankan tekanan intraocular dan bentuk bola mata.10
Fovea adalah depresi kecil pada retina dekat disk optik. Fovea memiliki konsentrasi tinggi cone. Ini adalah bagian dari retina di mana ketajaman visual yang terbesar.10
Vitreous humor
Vitreous humor suatu badan gelatin yang jernih dan avaskular yang membentuk dua pertiga volume dan berat mata. Vitreus sangat penting untuk metabolisme hasil metabolit dari jaringan intraocular seperti lensa, korpus siliaris dan retina. Vitreus jumlahnya sekita 4.0 ml dan mengandung air 99%. Sisa 1% meliputi dua komponen, kolagen dan asam hialuronat, yang memberi bentuk dan konsistensi mirip gel pada viterus karena kemampuannya mengikat banyak air.8 Lensa mata
Lensa adalah suatu struktur bikonveks, avaskular, tak berwarna, dan hampir transparan sempurna dan terletak di posterior chamber dan pupil. Tebalnya sekitar 4mm dan diameternya 9mm. Lensa ditahan ditempatnya oleh ligamentum suspensorium yang dikenal sebagai zonula zinni. Di sebelah anterior lensa terdapat akuos humor, disebelah posteriornya, vitreus. Lensa terdiri atas kapsul lensa, subkapsul, dan serat lensa. 65% lensa terdiri atas air, sekitar 35% nya protein.8,10
Retina
Retina adalah lembaran jaringan saraf berlapis yang tipis dan semitransparan yang melapisi bagian dalam dua pertiga posterior dinding bola mata. Lapisan-lapisan retina, mulai dari sisi dalamnya, adalah sebagai berikut: membran limitan interna, lapisan serat saraf, lapisan sel ganglion, lapisan pleksiform dalam, lapisan inti dalam, lapisan pleksiform luar, lapisan inti luar, membran limitans eksterna, lapisan fotoreseptor segmen dalam dan luar, dan epitel pigmen retina. Fotoreseptor ini dikenal sebagai cone (sel berbentuk kerucut) dan rod (sel berbentuk batang). Cone memungkinkan untuk mendeteksi warna sementara rod memungkinkan untuk melihat dalam cahaya yang kurang.8
Saraf Optik
Saraf optik berisi akson dari retina sel ganglion (sel-sel saraf retina) dan mengirimkan impuls dari retina ke otak. Pada akhirnya seseorang dapat melihat sebuah benda atau objek.11
2.2 Neurofibromatosis 2.2.1 Definisi
Neurofibromatosis merupakan kelainan kongenital dari perkembangan neuroektoderm yang menyebabkan pembengkakan atau benjolan pada jaringan fibrosa. Neurofibromatosis dianggp sebagai phacomatosis karena menimbulkan bitnik-bintik berpigmen (cafe au lait), benjolan (tumor) yang berisi jaringan saraf dan bersifat jinak.1
Ada dua bentuk utama neurofibromatosis, yaitu:
Tipe 1 (NF-1, Penyakit von Recklinghausen). Organ target utamanya adalah sistem saraf perifer, sistem saraf pusat (SSP), kulit, dan hampir tersebar luas.2
Tipe 2 (NF-2, sebelumnya dikenal sebagai neurofibromatosis akustik bilateral atau neurofibromatosis sentral) yaitu kondisi medis yang ditandai dengan terbentuknya tumor saraf pada sistem saraf pusat dan sumsum tulang belakang, kondisi ini bersifat herediter.2
2.2.2 Epidemiologi
Neurofibromatosis tipe 1 menyumbang sekitar dari 1/2.000 sampai 1/5.000 populasi. Mutasi yang bervariasi ditemukan pada kasus neurofibromatosis tipe 1 namun tidak ada kasus rekuren yang dilapor dengan mutasi yang telah diidentifikasi.3
Neurofibromatosis tipe 2 terjadi pada 1 dari 37.000 kelahiran per tahun, penderita neur dengan sekitar setengah dari individu yang terkena mewakili kasus
2.2.3 Etiologi
Neurofibromatosis cenderung berjalan dalam keluarga. Setiap anak dari orang tua yang terlibat memiliki kesempatan 50% dari mengembangkan neurofibromatosis. Dalam neurofibromatosis tipe 1, kromosom 17 bertanggung jawab untuk penyakit ini sementara kromosom 22 bertanggung jawab untuk neurofibromatosis tipe 2. Hampir 50% kasus tidak memiliki riwayat keluarga neurofibromatosis, sehingga penyebabnya adalah mutasi baru.13
Kedua neurofibromatosis tipe 1 dan 2 diperoleh melalui transmisi dominan warisan autosomal atau mutasi sporadis, dengan presentasi tipe 1 lebih umum daripada tipe 2. Dengan demikian, anggota keluarga yang sama dengan bawaan neurofibromatosis mungkin memiliki presentasi penyakit berbeda satu sama lain, karena mereka tidak selalu membawa mutasi gen yang sama. Ini dapat bervariasi dari penghapusan gen lengkap untuk penyisipan, berhenti dan mutasi pertukaran antar gen, membuat tingkat keparahan yang tepat dari penyakit sulit untuk di prediksi. Onset dari neurofibromatosis selama masa kanak-kanak biasanya menunjukkan khusus penyakit progresif yang lebih parah dapat diharapkan, tidak ada beda jenis kelamin atau predileksi ras.13
2.2.4 Patofisiologi
Neurofibromatosis tipe 1 terjadi setelah mutasi pada kromosom 17 yang mengkode protein yang disebut neurofibromin. Neurofibromin adalah tumor supresor gen yang berfungsi untuk menghambat onkoprotein p21 ras yang berperan dalam divisi sel.14
Neurofibromatosis tipe 2 disebabkan oleh mutasi pada kromosom 22 yang mengatur produksi merlin/schwnnomin protein yang berfungsi sebagai penekan tumor. Dalam tidak adanya kontrol penghambatan ini supresor tumor pada onkoprotein ras, terjadi proliferasi seluler tidak menentu dan tidak terkendali,
yang mengakibatkan proliferasi seluler dan perkembangan tumor yang tidak seimbang.15
2.2.5 Manifestasi Klinis Neurofibromatosis Tipe 1
Dua dari tujuh kriteria diagnostik berikut menandakan kehadiran neurofibromatosis tipe 1. Gejala tidak muncul saat kanak-kanak atau remaja dan menyulitkan diagnosis walaupun disuspek neurofibromatosis tipe 1. Tujuh kriteria klinis yang digunakan untuk mendiagnosis neurofibromatosis tipe 1 adalah : 13 Enam atau lebih cafe au lait makula lebih dari 5 mm diameter terbesar dalam
individu prapubertas dan lebih dari 15 mm diameter terbesar pada individu pasca pubertas
Dua atau lebih neurofibroma dari jenis apa pun atau satu plexiform neurofibroma
Bercak hitam di ketiak atau daerah inguinal
Optik glioma
Dua atau lebih nodul Lisch (iris hamartomas)
Sebuah lesi tulang khas seperti displasia sphenoid atau pseudarthrosis tibia
Saudara tingkat pertama (orang tua, saudara, atau keturunan) dengan neurofibromatosis tipe 1 seperti yang didefinisikan oleh kriteria di atas
Manifestasi Ophthalmologic dari neurofibromatosis tipe 1 adalah sebagai berikut: Lisch Nodul
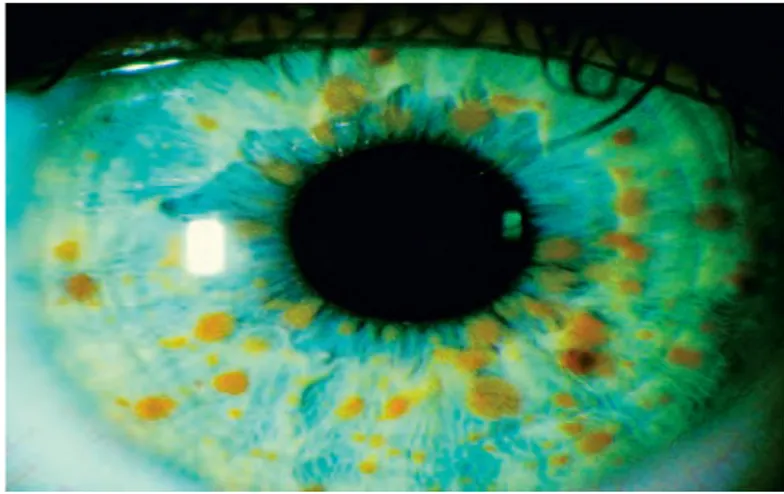
Lisch nodul adalah jenis yang paling umum dari keterlibatan okular di neurofibromatosis tipe 1. Nodul ini merupakan hamartomas melanositik. Nodul lisch muncul halus dan dapat dilihat dengan jelas dengan warna kuning kecoklatan pada pemeriksaan slit lamp. Seringkali, lesi asimtomatik hadir pada bagian belakang dan bilateral pada kedua mat. Namun, ada pula presentasi unilateral di pasien neurofibromatosis segmental. Lisch nodul jarang menimbulkan komplikasi okular dan pada pasien biasanya asimtomatik. Lisch nodul hadir di 94% dari pasien yang 6 tahun.14
Secara teoritis, cahaya ultraviolet adalah faktor dalam pembentukan nodul Lisch. Lisch nodul terdiri dari melanosit dan sel spindle, 2,3 dan sinar ultraviolet adalah mitogen dikenal melanosit. Lisch nodul memiliki karakteristik sebagai berikut:
Halus, biasanya bilateral, nodul terlihat menonjol
Biasanya muncul pada dekade pertama; hampir semua pasien dengan neurofibromatosis tipe 1 memiliki Lisch nodul pada usia 20 tahun
Merupakan hamartomas jinak, histologis identik dengan nevus iris.14
Gambar 2.2 : Lisch nodul pada iris mata
Sumber : 12. Bowling, Brad. Trauma. In : Kanski’s Clinical Ophthalmology: A Systematic Approach Eight Edition. 2016. Australia:
Elsevier. pg 844. Neurofibroma Plexiform
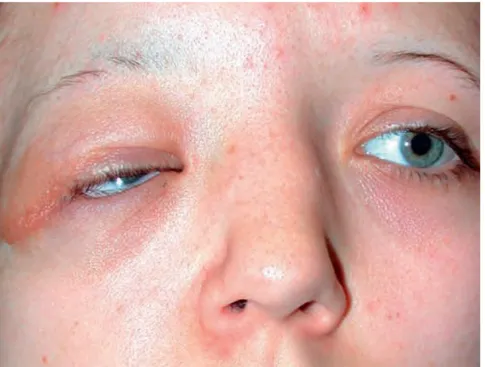
Neurofibroma plexiform adalah pembengkakan lembut dengan batas tidak jelas yang terletak di bawah kulit yang dapat menyusup ke orbit dan daerah temporal atau kelopak mata. Kelopak mata dengan neurofibroma biasanya merasa seperti 'kantong cacing' saat diraba. Neurofibroma orbital dapat menyebabkan strabismus atau proptosis, menyebabkan perubahan dalam panjang bola mata, dan telah dikaitkan dengan glaucoma infantil sekunder karena mempersempit sudut mata. Pasien lebih muda dari 10 tahun harus dipantau untuk amblyopia, yang dapat hasil dari ptosis atau anisometropia . Penyebab lain dari amblyopia hadir di 62% pasien neurofibromatosis termasuk kekeruhan lensa, kelainan retina dan pertumbuhan tumor intrakranial. Pada anak-anak, amblyopia mungkin akibat dari
menjauhkan sumbu visual yang sekunder untuk infiltrasi dan edema dari orbit dan kelopak mata. Bawaan glaukoma ipsilateral karena Neurofibroma plexiform telah digambarkan sebagai variasi dari gangguan perkembangan segmen anterior.14
Berikut ini adalah karakteristik dari neurofibroma plexiform kelopak mata:
Penebalan klopak mata atas
Deformitas berbentuk S
Perabaan seperti kantong cacing
Bawaan glaukoma ipsilateral untuk neurofibroma plexiform telah digambarkan sebagai variasi dari gangguan perkembangan segmen anterior.14
Gambar 2.3: Neurofibroma plexiform nodular pada palpebra Sumber : 12. Bowling, Brad. Trauma. In : Kanski’s Clinical Ophthalmology: A Systematic Approach Eight Edition. 2016. Australia:
Elsevier pg 845
Tumor Retina
Berikut ini adalah karakteristik dari tumor retina :
Hamartomas Astrocytic (tumor putih yang melibatkan saraf optik)
Hamartomas gabungan dari retina dan epitel pigmen retina
Hemangioma kapiler retina
Hamartomas astrocytic retina adalah tumor jinak pada lapisan saraf retina yang dapat terjadi bilateral yang melibatkan saraf optik dan kutub posterior retina, dengan beberapa lesi perifer yang luas ke retina anterior Jika saraf optik atau makula yang terlibat, pasien mungkin menunjukkan penurunan visus atau strabismus, sementara leukocoria dapat hadir jika tumor terletak di kutub posterior.14
Hamartoma Koroid
Berikut adalah karakteristik dari hamartoma koroid :
Biasanya dijumpai di bagian posterior.
Datar, lesi yang tidak jelas.
Mengandung neuronal dan komponen melanostik.14
Glioma Optik
Diperkirakan 15-40% anak dengan neurofibromatosis tipe 1 memiliki glioma saraf optik atau glioma jalur visual yang melibatkan saraf optik, Chiasma optikum, atau saluran optik. Beberapa lesi ini tidak menunjukkan gejala. Bilateral glioma saraf optik hampir patognomonik untuk neurofibromatosis tipe 1. Unilateral penurunan ketajaman dengan defek pupil aferen relatif (+/-) dan strabismus (+/-) dapat terjadi. glioma saraf optik muncul pada computed tomography (CT) scan atau gambar resonansi magnetik (MRI) sebagai dilatasi fusiform dari saraf optik.14
Glioma saraf optik secara lokal invasif dan lambat berkembang dengan potensi keganas yang rendah. Namun, glioma chiasmatic dapat menginvasi hipotalamus dan ventrikel ketiga, menyebabkan hidrosefalus obstruktif. Sekitar 10-38% dari pasien anak dengan glioma saraf optik memiliki neurofibromatosis tipe 1. Pada kebanyakan anak dengan glioma optik, gejala yang muncul mungkin proptosis yang tidak sakit dan penurunan ketajaman visual. Temuan fisik termasuk kehilangan penglihatan, kehilangan penglihatan warna, sebuah defek pupil aferen, dan pucat saraf optik atau atrofi. Sebuah lesi besar dapat memampatkan Chiasm optik, menyebabkan nystagmus atau gejala lainnya. gejala hipotalamus, seperti perubahan nafsu makan atau tidur, juga dapat terjadi. lesi besar dapat memampatkan ventrikel ketiga, sehingga hidrosefalus obstruktif disertai sakit kepala, mual, dan muntah.14
Optik jalur saraf glioma (OPGs) ini serius, tapi dapat disembuhkan, tumor otak yang muncul di dalam dan sekitar saraf optik. Setengah dari pasien dengan glioma saraf optik adalah pasien neurofibromatosis tipe 1. Neurofibromatosis tipe 1 terkait OPGs biasanya kurang agresif dibandingkan yang tidak terkait. Banyak pasien dengan OPGs tidak menunjukkan gejala. Chiasma atau keterlibatan otak yang berdekatan dapat menyebabkan gejala endokrin dan neurologis.14
Glioma saraf optik ini gejala biasanya hadir pada usia 6, dengan sebagian besar anak-anak didiagnosis pada usia 3 tahun. Seringkali, ketajaman visual dapat dinilai pada usia tiga tahun, penglihatan warna pada usia lima tahun dan bidang visual dengan usia delapan tahun.14
Neurofibromatosis Tipe 2
Diagnosis klinis neurofibromatosis tipe 2 mensyaratkan individu dengan setidaknya 1 dari persyaratan klinis berikut:
Bilateral schwannomas vestibular, yang relatif dikaitkan dengan neurofibromatosis tipe 2 dan unilateral schwannoma vestibular atau dua dari meningioma, schwannoma, glioma, neurofibroma, posterior subkapsular kekeruhan lenticular
Unilateral schwannoma vestibular dan dua dari meningioma, schwannoma, glioma, neurofibroma, posterior subkapsular kekeruhan lenticular. Beberapa meningioma dan unilateral schwannoma vestibular atau dua dari: schwannoma, glioma, neurofibroma, katarak
Namun, karena sekitar setengah dari kasus merupakan hasil dari mutasi baru, sejarah keluarga sering negatif.
Tidak seperti neurofibromatosis tipe 1, yang sering dikaitkan dengan sejumlah petunjuk diagnostik kulit, neurofibromatosis tipe 2 disertai dengan beberapa tanda-tanda eksternal. gejala yang muncul adalah sebagai berikut:
Gangguan pendengaran, dering di telinga, dan keseimbangan masalah yang terkait dengan lesi saraf vestibular
Defisit visual
Kelumpuhan saraf kranial.15
Manifestasi Ophthalmologic dari neurofibromatosis tipe 2 adalah sebagai berikut: Katarak
Katarak mempengaruhi sekitar dua-pertiga dari pasien. Kekeruhan pada lensa mengembangkan sebelum usia 30 tahun dan mungkin di bagian posterior subkapsular atau kapsuler, kortikal atau campuran.
Fundus
Membran epiretinal sering terjadi dan kombinasi hamartoma dari epitel pigmen retina relatif umum.
Defek Okular Motor
Hanya 10% yang terjadi dari keseluruhan.5
2.2.6 Diagnosa Banding 15,16
Neurofibromatosis tipe 1 Neurofibromatosis tipe 2
Brainstem Gliomas
Meningioma
Neurofibromatosis Tipe 2
Cauda Equina and Conus Medullaris Syndromes
Low-Grade Astrocytoma
Spinal Cord Hemorrhage
Spinal Cord Infarction
Spinal Epidural Abscess
Neurofibromatosis Tipe 1
Pediatric Ependymoma
2.2.7 Diagnosis a) Anamnesis
Riwayat keluarga keturunan pertama sangat penting untuk mendiagnosis neurofibromatosis tipe 1 sementara riwayat keluarga (tingkat pertama secara relatif) ditambah unilateral (pada satu sisi) schwannomas vestibular atau dua dari kondisi berikut penting dalam mendiagnosis neurofibromatosis tipe 2.13,15
b) Pemeriksaan Fisik
Inspeksi
Untuk neurofibromatosis tipe 1, harus amati apakah terdapat 2 atau lebih kriteria diagnostik yang dibahaskan di manisfestasi klinis.
Pada neurofibromatosis tipe 2 didapati paralisis nervus VII, gangguan visual dan pendengaran.,15,16
c) Pemeriksaan Penunjang
Pada neurofibromatosis tipe 1, dilakukan pemeriksaan tekanan darah dan pengambilan sampel melalui biopsi untuk mengetahui apakah ada sel-sel kanker atau tidak.17
Pada neurofibromatosis tipe 2, dilakukan tes darah untuk memeriksa apakah ada mutasi DNA. Tes ini kurang sensitif. Jadi bila hasilnya negatif, masih berisiko menderita neurofibromatosis tipe 2, pemeriksaan telinga untuk menilai kemampuan pendengaran dan pemeriksaan mata untuk menilai keberadaan katarak.18
Tes genetika dan MRI scan dilakukan untuk neurofibromatosis tipe 1 dan 2. Tes genetika untuk memeriksa ada atau tidaknya mutasi gen. Pemeriksaan ini dianjurkan bagi pasangan berisiko tinggi yang berencana untuk memiliki anak, begitu juga bagi ibu yang sedang hamil. Sampel bisa diambil embrio (calon janin), cairan ketuban, dan sel dari jaringan plasenta. •MRI scan dapat mendeteksi tumor di otak, telinga, dan saraf tulang belakang. MRI sering berguna dalam deteksi glioma intrakranial.17,18
2.2.8 Penatalaksanaan
Penanganan untuk neurofibromatosis tipe 1 dan 2 tergantung pada manifestasi klinis masing-masing.19,20
Neurofibromatosis tipe 1 Neurofibromatosis tipe 2 ADHD
Medikamentosa
Terapi perilaku
Gangguan keseimbangan
Latihan vestibular
Terapi fisik / terapi okupasi Lesi tulang (bony lesion)
Bracing
Observasi
Operasi
Gangguan pendengaran
Bahasa isyarat / pelatihan komunikasi alternative
BAER (Brainstem Auditory Evoked Response hearing test)
Alat bantu dengar
Medikamentosa (dalam penelitian klinis)
Hearing prosthetics
Implant koklea
Implan auditory brainstem Brain Tumor
Observasi
Operasi
Tinnitus
Terapi pelatihan ulang tinnitus
klinis)
Neurofibroma dermal
Observasi
Operasi
Medikamesntosa (dalam penelitian klinis) Nyeri Medikamentosa Akupuntur Intervensi perilaku Gangguan pendengaran IEP Medikamentosa Uji klinis Vestibular schwannomas Observasi Uji klinis Radioterapi Operasi Optik glioma Observasi Kemoterapi Neurofibroma plexiform Kemoterapi Terapi radiasi Operasi
Medikamentosa (dalam penelitian klinis) Nyeri Medikamentosa Akupuntur Intervensi perilaku 2.2.9 Komplikasi
Komplikasi yang mungkin terjadi dari neurofibromatosis tipe 1 adalah tumor pada saraf perifer, akar saraf dan plexi, kompresi spinal cord, ektasiasis dural, epilepsy, stroke, hydrocephalus, gangguan penglihatan, gangguan kardiovaskular, gangguan pendengaran dan sefalgia.21
Komplikasi dari neurofibromatosis tipe 2 adalah gangguan penglihatan, penumpukan cairan di otak, kelemahan pada ektremitas inferior dan superior.22
Prognosis untuk neurofibromatosis tipe 1 sangat bervariasi dalam manifestasinya dan kebanyakkkan orang menjalani kehidupan yang relatif panjang dan sehat namun ia dapat mengurangi harapan hidup.6
Neurofibromatosis tipe 2 umumnya memiliki prognosis yang lebih buruk. Sebagian besar morbiditas dari neurofibromatosis tipe 2 adalah hasil dari pengobatannya tersendiri. Deteksi dini dan perhatian yang cepat untuk komplikasi dapat mengurangi morbiditas dan mortalitas secara keseluruhan.6
BAB 3 KESIMPULAN
Neurofibromatosis adalah suatu kelainan genetik pada sistem saraf yang berpengaruh pada pertumbuhan dan perkembangan jaringan saraf, dimana neurofibroma muncul pada kulit dan bagian tubuh lainnya.
Gangguan ini dapat mempengaruhi semua ras, semua kelompok etnis dan jenis kelamin masing-masing dengan probabilitas yang sama. Neurofibromatosis telah, terlepas dari bentuk yang paling umum, jenis yang berbeda. Neurofibromatosis tipe 1, juga dikenal sebagai penyakit Reclkingshausen Von. Neurofibromatosis tipe 2 juga dikenal sebagai neurofibromatosis akustik bilateral dan neurofibromatosis sentral.
Diagnosa klinis ditegakkan berdasarkan anamnesa dan pemeriksaan fisik sesuai dengan manifestasi klinis yang ditimbulkan dari masing – masing jenis neurofibromatosis.
Terapi yang diberikan berupa pembedahan yang dapat bertujuan untuk kepentingan estetika maupun terapi pembedahan parsial pada neurofibromatosis tipe 2.
Penyakit ini merupakan penyakit yang berhubungan dengan herediter maka pencegahannya dapat berupa konsultasi genetik pada penderita yang merencanakan untuk memiliki keturunan.